

Abstract
Type IV pili mediate the initial interaction of many bacterial pathogens with their host cells. In Neisseria meningitidis, the causative agent of cerebrospinal meningitis, type IV pili-mediated adhesion to brain endothelial cells is required for bacteria to cross the blood-brain barrier. Here, Type IV pili-mediated adhesion of N. meningitidis to human brain endothelial cells was found to recruit the Par3/Par6/PKCζ polarity complex that plays a pivotal role in the establishment of eukaryotic cell polarity and the formation of intercellular junctions. This recruitment leads to the formation of ectopic intercellular junctional domains at the site of bacterial-cell interaction and a subsequent depletion of junctional proteins at the cell-cell interface with opening of the intercellular junctions of the brain-endothelial interface.
Keywords: Adaptor Proteins, Signal Transducing; metabolism; Antigens, CD; metabolism; Bacterial Adhesion; Blood-Brain Barrier; metabolism; microbiology; Brain; blood supply; cytology; microbiology; Cadherins; metabolism; Catenins; Cell Adhesion Molecules; metabolism; Cell Cycle Proteins; metabolism; Cell Line; Cell Polarity; Endothelial Cells; metabolism; microbiology; Endothelium, Vascular; metabolism; microbiology; ultrastructure; Fimbriae, Bacterial; physiology; Humans; Intercellular Junctions; metabolism; microbiology; ultrastructure; Membrane Proteins; metabolism; Neisseria meningitidis; pathogenicity; physiology; Phosphoproteins; metabolism; Protein Kinase C; metabolism; cdc42 GTP-Binding Protein; metabolism
Neisseria meningitidis is a commensal bacterium of the human nasopharynx that, after bloodstream invasion, crosses the blood-brain barrier (BBB) (1). Few pathogens have a tropism for the brain, indicating that N. meningitidis possess specific components to interact with the BBB. Meningeal colonization by invasive capsulated N. meningitidis is the consequence of the bacterial adhesion onto brain endothelial cells (2, 3) which is followed by bacterial division onto the apical surface of the cells (Movie S1). This process is mediated by Type IV pili (Tfp) (4–9). In addition, by powering a form of cell locomotion, reported as twitching motility (10), Tfp lead to the spread of the bacteria on the surface of the cells and the formation of microcolonies. Subsequent to the formation of these microcolonies, Tfp trigger the recruitment of cortical actin and signal transducing proteins leading to the formation of filopodia-like structures (2, 11–13). The crossing of the BBB by N. meningitidis implies that following Tfp mediated adhesion, the bacteria transcytose through the brain capillaries and/or open the brain endothelium.
To investigate whether adhesion of N. meningitidis affects the integrity of the adherens (AJ) and/or tight (TJ) junctions of human brain endothelial cells, the consequences of infection by N. meningitidis on the distribution of junctional proteins were analyzed using the human brain microvascular endothelial cell line hCMEC/D3 (14). After infection, components of the AJ (VE-cadherin, p120-catenin, β-catenin) and TJ (ZO1, ZO2, and claudin-5) were targeted underneath N. meningitidis colonies (Fig. 1A). At the site of N. meningitidis adhesion, these junctional proteins co-distributed with each other and with the actin honeycomb-like network. In non infected cells, the recruitment of junctional proteins usually occurs at the cell-cell interface and is controlled by several polarity proteins (Par3/Par6/PKCζ) (15–17). In infected monolayers, Par3 and Par6 were observed underneath N. meningitidis colonies (Fig. 1B). Thus, N. meningitidis triggers a signal leading to the formation of an ectopic domain containing filopodia-like structures and enriched in junctional proteins, thus resembling spot-like adherens junctions observed during early steps of junctional biogenesis. We refer to this domain as an “ectopic early junction-like domain” (18). Using isogenic derivatives, Tfp-induced signaling was shown to be responsible for the formation of these ectopic early junction-like domains (Fig. S1A and B). However, Tfp retraction through the PilT motor was not required for formation of the ectopic domains (Fig. S1D and E).
Figure 1. Neisseria meningitidis recruits ectopic junction-like domains beneath colonies.

(A) VE-cadherin (green), the main component of the endothelial AJ, co-localized with actin (red) beneath N. meningitidis colony (upper panel). Two other AJ components: p120-catenin and β-catenin, and three components of the TJ: ZO-1, ZO-2 and claudin-5 are recruited under N. meningitidis colonies (lower Panel). Arrow indicates a bacterial colony. Scale bars: 10μm. (B) YFP-tagged Par6 (par6-YFP) or myc-tagged Par3 (par3-myc), both green, are recruited underneath N. meningitidis colonies where they co-localize with actin (red). Areas outlined in white indicate the presence of a N. meningitidis colony. Scale bars: 10μm. The formation of these ectopic early junction-like domains is not found underneath all N. meningitidis colonies. Signaling underneath bacterial microcolonies required a minimal number of 20 bacteria per colony to be detected by immunofluorescence, with around 40–50% of microcolonies containing 40–50 bacteria. The average number of colonies signalling after 2 hours of infection is 40 %.
The small GTPase Cdc-42 is required for polarization of mammalian cells (19, 20). The role of this component in the recruitment of the polarity complex by N.meningitidis was investigated. Transfection of a dominant negative mutant of Cdc42 or knockdown of Cdc42 by RNAi inhibited the recruitment of Par6, Par3 (Fig. 2A, S2A), VE-cadherin, p120-catenin and actin (Fig. 2B, S2B, S3). These results link the Cdc42/polarity complex pathway with the formation of the ectopic early junction-like domains.
Figure 2. The Cdc42-Par3/Par6/PKCζ pathway controls the formation of ectopic early junction-like domains.
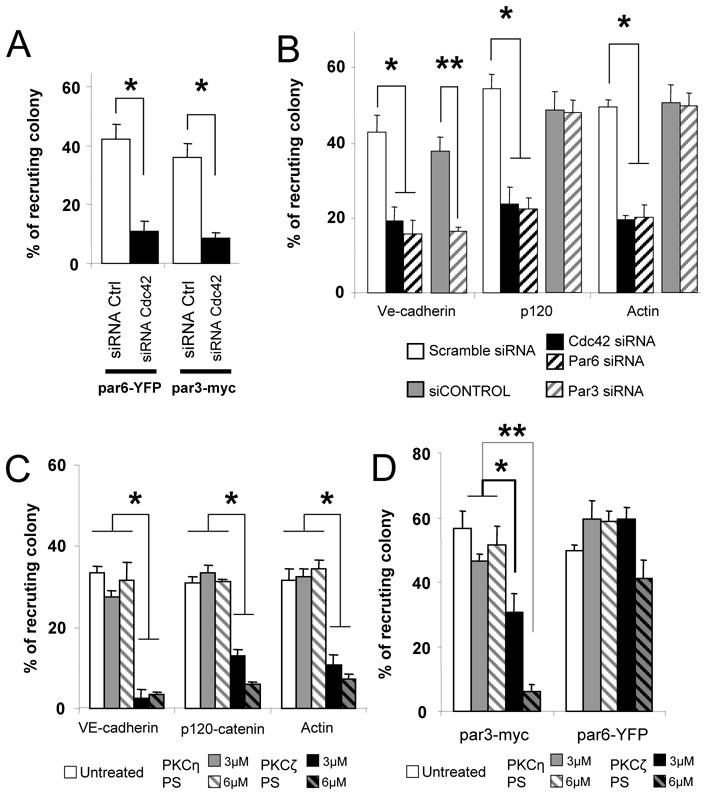
(A) Knockdown of Cdc42 was performed using specific siRNA duplexes (Cdc42 siRNA). Cells were cotransfected with par6-YFP or par3-myc. Knockdown of Cdc42 by RNAi reduced the recruitment of par6-YFP and par3-myc by 4 fold. * t test (p<0.005). (B) Knockdown of Cdc42, Par6 and Par3 were performed as described (27) (Cdc42 siRNA, Par6 siRNA and Par3 siRNA). Scramble siRNA and siCONTROL were used as control for Cdc42/Par6 and Par3 knockdown, respectively. Knockdown of Cdc42 by RNAi reduced the recruitment of VE-cadherin, p120-catenin and actin by 2.2 fold, 2.3 fold and 2.5 fold, respectively. See also figure S3. Knockdown of Par6 by RNAi reduced the recruitment of VE-cadherin, p120-catenin and actin by 2.7 fold, 2.4 and 2.4 fold, respectively. Knockdown of Par3 by RNAi reduced the recruitment of VE-cadherin by 2 fold. * t test (p<0.01), ** t test (p<0.002). (C, D) HCMEC/D3 cells were either incubated with 3μM or 6μM of PKCζ pseudosubstrate inhibitor (PKCζ-PS) or PKCη-PS (control), or left untreated. (C). PKCζ-PS (6μM) reduced VE-cadherin, p120-catenin and actin recruitment by 8.5 fold, 5 fold and 4.9 fold, respectively. * t test (p<0.001). (D) HCMEC/D3 cells were transfected with either par6-YFP or par3-myc. Six μM PKCζ-PS reduced par3-myc recruitment by 9 fold, but par6-YFP recruitment was not affected (* t test (p<0.001), ** t test (p<0.01)). Data are expressed as mean +/− SEM.
The role of the polarity complex in the recruitment of junctional proteins was further explored by studying the inhibition of Par3 and Par6 using either dominant negative mutants or knockdown by RNAi. PKCζ inhibition was assessed using a PKCζ pseudosubstrate inhibitor (PKCζ-PS) (21). Inhibition of Par6 and PKCζ reduced the recruitment of p120-catenin, VE-cadherin and actin (Fig. 2B, 2C, S2C, S3) and that of Par3 (Fig. 2D, S2E), consistent with the finding that the Par6/PKCζ complex recruits Par3 at intercellular junction domains (22). On the other hand, inhibition of Par3 reduced only the recruitment of VE-cadherin (Fig. 2B, S2D, S3), consistent with Par3 being specifically needed for junctional proteins targeting at early cell-cell junctions (23). These observations confirmed the role of the polarity complex in the recruitment of the junctional proteins by N. meningitidis.
The sequence of events leading to the targeting of AJ proteins at the cell-cell junctions during cellular polarization remains unknown. To get insight into this process, we engineered a VE-cadherin knockdown of hCMEC/D3 cells by stable expression of a VE-cadherin shRNA (VEC shRNA) (Fig. 3A, 3B, S4A). In this cell line, p120-catenin and actin were still recruited beneath N. meningitidis colonies, whereas recruitment of β-catenin was dramatically reduced. On the other hand, down-regulation of p120-catenin using RNAi (Fig. 3C, S4B) resulted in inhibition of VE-cadherin and of actin recruitment. Consistent with a previous report, cortactin and Arp2/3 were not recruited by the bacterial colonies in p120-catenin knockdown cells (24) (Fig. S4C). Furthermore, inhibition of Src kinase, which phosphorylate cortactin and is activated following the formation of the cortical plaque (25) did not modify p120-catenin recruitment but inhibited VE-cadherin and actin recruitment (Fig. S4D, S4E). Taken together, these results strongly suggest that p120-catenin-mediated recruitment of actin and VE-cadherin requires the recruitment and phosphorylation of cortactin by the Src kinase. In summary, Cdc42, via the polarity complex, organizes this ectopic early junction-like domain, mainly by the initial recruitment of p120-catenin.
Figure 3. P120-catenin is key to the recruitment of both actin and AJ proteins.
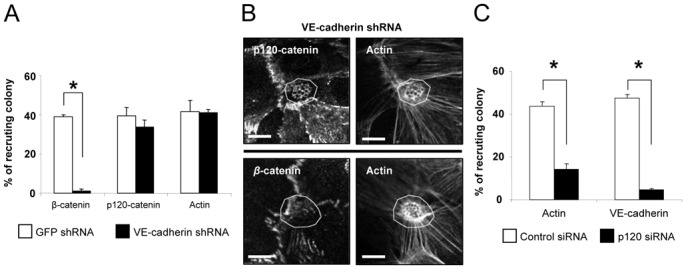
(A, B) VE-cadherin silencing was performed by stable expression of a VE-cadherin shRNA (VEC shRNA). (A) Recruitment of β-catenin, p120-catenin and actin was determined by immunofluorescence. Knockdown of VE-cadherin had no effect on the recruitment of p120-catenin and actin but reduced β-catenin recruitment by 20 fold. * t test (p<0.001). (B) In VEC-shRNA expressing cells, p120-catenin was still recruited beneath N. meningitidis colonies where it colocalized with actin (upper panel) while β-catenin was no longer recruited (lower panel). Areas outlined in white indicated the location of a N. meningitidis colony. Scale bars: 10μm. (C) Silencing of p120-catenin was performed using a specific siRNA duplex (p120 siRNA). Recruitment of VE-cadherin and actin was determined by immunofluorescence. Knockdown of p120-catenin reduced VE-cadherin and actin recruitment by 10 fold and 4 fold, respectively. * t test (p<0.001). Data are expressed as mean +/− SEM.
We asked whether the signal triggered by Tfp and leading to the formation of these ectopic early junction-like domains destabilized intercellular junctions, especially by redirecting a recycling pool of junctional proteins to the N. meningitidis adhesion site. First, inhibition of protein synthesis did not prevent recruitment of VE-cadherin (Fig. S5A). Second, inhibition of clathrin coated pit formation blocked VE-cadherin recruitment (Fig. S5B and S5C) suggesting that VE-cadherin internalization is required for its targeting underneath N. meningitidis colonies. Third, when monolayers were tagged before infection with a VE-cadherin monoclonal antibody, antibodies are relocalized beneath colonies in infected monolayers (Fig. S6). Thus the VE-cadherin delocalized by the bacteria was coming from the intercellular junctions. This redistribution of the AJ proteins was associated with a reduction of the amount of tagged VE-cadherin at the intercellular junction (Fig. S6, Movie S2). Thus the junctional VE-cadherin is internalized and then mistargeted at the site of bacterial cell interactions.
Depletion of intercellular junction proteins from the cell-cell interface could open a paracellular route for bacterial spread. Indeed, N. meningitidis was shown to increase permeability to Lucifer Yellow (LY) a compound which mark passive paracellular diffusion (Fig. 4A) (26). Moreover, this increase relied on PKCζ activity and bacterial piliation (Fig. 4A). This modification of permeability was associated with the formation of gaps between infected cells (Fig. 4B). The number of gaps increased over time and was reduced by the PKCζ pseudosubstrate inhibitor (Fig. 4B and 4C). Gaps did not form when cells were infected with a non piliated strain, showing that these gaps are due to Tfp-mediated signaling (Fig. 4C). Indeed, piliated strain cross the monolayer at a higher rate than non-piliated isogenic derivatives or a piliated strain in the presence of PKCζ PS (Fig. 4D). Thus the signaling induced by N. meningitidis Tfp leading to the recruitment of the polarity complex is associated with large alterations of the intercellular junctions sufficient for the bacteria to cross the brain endothelial cell monolayer.
Figure 4. N. meningitidis induced PKCζ activity facilitates cell-cell junction opening.
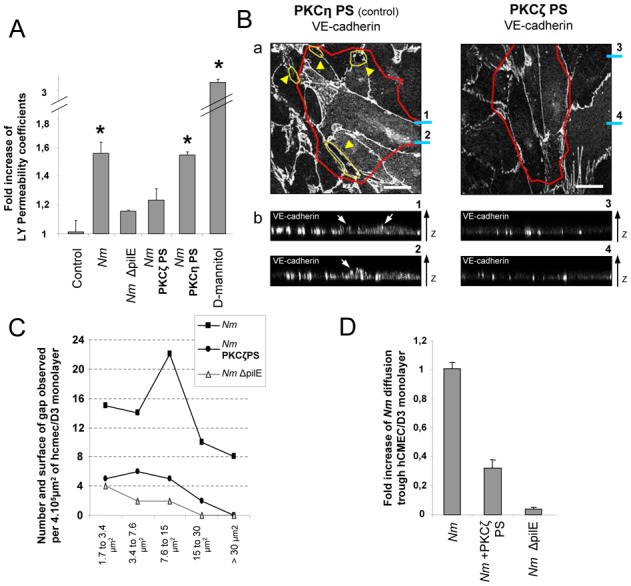
(A) The permeability coefficient of Lucifer Yellow was measured 4h post-infection by N. meningitidis (Nm) or its non piliated isogenic strain (Nm ΔpilE), or following treatment by PKCζ-PS or PKCη-PS (6μM). N. meningitidis induced a 1.55 fold increase compare to control. D-mannitol, which disrupts all cell-cell junctions, induced a 3.1 fold increase. * t test (p<0.001). (B) HCMEC/D3 cells were incubated with 6μM of PKCζ-PS or of PKCη-PS (control). (a) VE-cadherin localization was analyzed on the baso-lateral cross-section of N. meningitidis infected cells. Yellow arrow heads and areas outlined in yellow indicate gaps between cells. Areas outlined in red indicate the presence of N. meningitidis colonies. Blue bars marked 1–4 refer to Z-axis reconstruction image 1–4 on the lower panel. Scale bars: 20μm. (b) Z-axis reconstructions from stack of 0.12μm interval images show that VE-cadherin is apically relocalized underneath N. meningitidis colonies (white arrows) only in cells treated with PKCη-PS (control). (C) HCMEC/D3 cells grown on 3.0μm pore size inserts were treated or not with PKCζ-PS and incubated with N. meningitidis (Nm) or its non piliated isogenic strain (N.meningitidis ΔpilE). Size and quantity of gaps observed 4h after infection are calculated as described (27). (D) Diffusion of N. meningitidis (Nm) through a hCMEC/D3 monolayer, 4h post-infection, is 3.2 fold higher than diffusion of N. meningitidis in presence of 6μM PKCζ-PS and 16.5 fold higher than diffusion of its non-piliated derivative (Nm ΔpilE). The rate of N. meningitidis internalization, determined by gentamicin protection assay, is very low (1CFU in 3,5.105), identical to that of a control without cells, thus excluding a possible transcytosis of bacteria. Data are expressed as fold increase of N. meningitidis diffusion and calculated as described (27). Data from B, C and D are one representative experiment of three independent duplicate. Data are expressed as mean +/− SEM.
In summary, N. meningitidis microcolonies trigger via type IV pili a signal resembling the one responsible for the formation of AJ at cell-cell junctions. This leads to the formation of ectopic early junction-like domains (Fig. S7), thus disorganizing the cell-cell junctions and opening the paracellular route allowing N. meningitidis to cross the BBB and to invade the meninges.
Supplementary Material
Acknowledgments
The authors thank M. Drab, P. Martin, I. Allemand and N. Simpson for reviewing the manuscript. The authors are grateful to M. Garfa-Traore and N. Goudin for technical support. Mathieu Coureuil was funded by “la Fondation pour la Recherche Medicale” (FRM).
References and Notes
- 1.van Deuren M, Brandtzaeg P, van der Meer JW. Clin Microbiol Rev. 2000 Jan;13:144. doi: 10.1128/cmr.13.1.144-166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pron B, et al. J Infect Dis. 1997 Nov;176:1285. doi: 10.1086/514124. [DOI] [PubMed] [Google Scholar]
- 3.Mairey E, et al. J Exp Med. 2006 Aug 7;203:1939. doi: 10.1084/jem.20060482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Virji M, et al. Mol Microbiol. 1991 Aug;5:1831. doi: 10.1111/j.1365-2958.1991.tb00807.x. [DOI] [PubMed] [Google Scholar]
- 5.Nassif X, et al. Mol Microbiol. 1993 May;8:719. doi: 10.1111/j.1365-2958.1993.tb01615.x. [DOI] [PubMed] [Google Scholar]
- 6.Rudel T, Scheurerpflug I, Meyer TF. Nature. 1995 Jan 26;373:357. doi: 10.1038/373357a0. [DOI] [PubMed] [Google Scholar]
- 7.Kallstrom H, Liszewski MK, Atkinson JP, Jonsson AB. Mol Microbiol. 1997 Aug;25:639. doi: 10.1046/j.1365-2958.1997.4841857.x. [DOI] [PubMed] [Google Scholar]
- 8.Kirchner M, Heuer D, Meyer TF. Infect Immun. 2005 May;73:3072. doi: 10.1128/IAI.73.5.3072-3082.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merz AJ, So M, Sheetz MP. Nature. 2000 Sep 7;407:98. doi: 10.1038/35024105. [DOI] [PubMed] [Google Scholar]
- 10.Mattick JS. Annu Rev Microbiol. 2002;56:289. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- 11.Merz AJ, Enns CA, So M. Mol Microbiol. 1999 Jun;32:1316. doi: 10.1046/j.1365-2958.1999.01459.x. [DOI] [PubMed] [Google Scholar]
- 12.Mikaty G, et al. PLoS Pathog. 2009 Feb;5:e1000314. doi: 10.1371/journal.ppat.1000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eugene E, et al. J Cell Sci. 2002 Mar 15;115:1231. doi: 10.1242/jcs.115.6.1231. [DOI] [PubMed] [Google Scholar]
- 14.Weksler BB, et al. FASEB J. 2005 Nov;19:1872. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 15.Muller HA, Wieschaus E. J Cell Biol. 1996 Jul;134:149. doi: 10.1083/jcb.134.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamanaka T, et al. Genes Cells. 2001 Aug;6:721. doi: 10.1046/j.1365-2443.2001.00453.x. [DOI] [PubMed] [Google Scholar]
- 17.Hurd TW, Gao L, Roh MH, Macara IG, Margolis B. Nat Cell Biol. 2003 Feb;5:137. doi: 10.1038/ncb923. [DOI] [PubMed] [Google Scholar]
- 18.Vasioukhin V, Bauer C, Yin M, Fuchs E. Cell. 2000 Jan 21;100:209. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- 19.Joberty G, Petersen C, Gao L, Macara IG. Nat Cell Biol. 2000 Aug;2:531. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- 20.Koh W, Mahan RD, Davis GE. J Cell Sci. 2008 Apr 1;121:989. doi: 10.1242/jcs.020693. [DOI] [PubMed] [Google Scholar]
- 21.Etienne-Manneville S, Manneville JB, Nicholls S, Ferenczi MA, Hall A. J Cell Biol. 2005 Sep 12;170:895. doi: 10.1083/jcb.200412172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki A, Ohno S. J Cell Sci. 2006 Mar 15;119:979. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- 23.Ooshio T, et al. J Cell Sci. 2007 Jul 15;120:2352. doi: 10.1242/jcs.03470. [DOI] [PubMed] [Google Scholar]
- 24.Boguslavsky S, et al. Proc Natl Acad Sci U S A. 2007 Jun 26;104:10882. doi: 10.1073/pnas.0702731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann I, Eugene E, Nassif X, Couraud PO, Bourdoulous S. J Cell Biol. 2001 Oct 1;155:133. doi: 10.1083/jcb.200106148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madgula VL, Avula B, Reddy VLN, Khan IA, Khan SI. Planta Med. 2007 Apr;73:330. doi: 10.1055/s-2007-967137. [DOI] [PubMed] [Google Scholar]
- 27.Described in the Supporting Online Material: Materials and methods section.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.