

Abstract
Background
In addition to hypertension control, direct renin inhibition has been shown to exert direct beneficial effects on the heart in post-infarction cardiac remodeling. This study elucidates the possible contribution of mitochondria to the anti-hypertrophic effects of the direct renin inhibitor aliskiren in post-infarction heart failure complicated with diabetes in rats.
Methods
Diabetes was induced in male Sprague-Dawley rats by a single injection of streptozotocin (IP, 65 mg/kg body weight). After 7 days, the animals were randomly assigned to 4 groups: sham, heart failure, sham+aliskiren, and heart failure+aliskiren. Post-infarction HF was induced by coronary artery ligation for 4 weeks.
Results
showed that heart failure reduced ejection fraction and cardiac output by 41% (P<0.01) and 42% (P<0.05), respectively, compared to sham-operated hearts. Cardiac dysfunction was associated with suppressed state 3 respiration rates and respiratory control index in mitochondria, and increased mitochondrial permeability transition pore (PTP) opening. In addition, heart failure reduced expression of the major mitochondrial sirtuin, SIRT3 and increased acetylation of cyclophilin D, a regulatory component of the PTP. Aliskiren significantly improved cardiac function and abrogated mitochondrial perturbations.
Conclusion
Our results demonstrate that aliskiren attenuates post-infarction remodeling which is associated with its beneficial effects on mitochondria.
Keywords: Post-myocardial infarction, Heart failure, Renin angiotensin system, Mitochondria
Introduction
Activation of the renin-angiotensin system (RAS) plays a pivotal role in the pathogenesis of cardiac hypertrophy, remodeling, and heart failure (HF). The end-effector of the RAS is angiotensin II (AngII) which has detrimental effects on cell metabolism through activation of AngII type I receptors (AT1R) [1, 2]. Consequently, RAS blockade at the level of AngII synthesis (angiotensin converting enzyme inhibitors) or blockade at the level of action (AT1R blockers) attenuates ventricular remodeling and improves cardiac function in animal models of myocardial infarction [1, 3, 4]. Tissue levels of AngII can also be reduced by inhibiting the enzymatic activity of renin, which converts angiotensinogen to AngI, the initial and rate-limiting step of the RAS. The newly developed direct renin inhibitor aliskiren demonstrated high antihypertensive activity and prevented ventricular remodeling, hypertrophy, and apoptosis after myocardial infarction in mice [5]. Furthermore, the beneficial effects of aliskiren on cardiac function were independent of blood pressure [5]. The anti-remodeling effect of aliskiren might be due to inhibition of local synthesis and action of AngII in the myocardium. In fact, aliskiren prevented cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis, which was associated with inhibition of intracellular AngII production in diabetic hearts [6]. However, the molecular and cellular mechanisms underlying the direct positive outcomes of renin inhibition on the heart remain to be elucidated.
The protective effects of aliskiren on cardiac remodeling can be mediated through the prevention of mitochondrial dysfunction. Initially, adaptive remodeling after myocardial infarction seems to be beneficial; however, over time it becomes deleterious, eventually leading to HF. Mitochondrial dysfunction occurs in both the ischemic and the non-ischemic ventricular walls where mitochondria are energetically uncoupled [7, 8]. Heart failure induces impairments in the Krebs cycle and electron transport chain leading to reduced ATP [9]. Mitochondria become a major source of reactive oxygen species (ROS) due to inhibition of complexes I and III leading to accumulation of superoxide anion in failing hearts [10, 11]. In addition, stimulation of AT1R by AngII in cardiac remodeling enhances ROS generation through activation of NADPH oxidase [12, 13]. Oxidative stress accompanied by Ca2+-overload, ATP depletion and elevated Pi in the failing myocardium can induce formation of the mitochondrial permeability transition pores (PTP) in the mitochondrial inner membrane. Previous studies demonstrated that in vivo renin inhibition by aliskiren attenuates oxidative stress and myocardial remodeling in the transgenic rats with chronically elevated tissue AngII levels [14]. In the present study, we elucidated the possible contribution of mitochondria to anti-remodeling effects of the direct renin inhibitor aliskiren during post-infarction HF in rats. Our results demonstrate that aliskiren suppresses cardiac hypertrophy and improves heart function in post-infarcted HF. The anti-remodeling action of aliskiren is associated with its beneficial effects on mitochondria.
Materials and Methods
Male Sprague-Dawley rats weighing 175-200 g were purchased from Charles River (Wilmington, MA). All experiments were performed according to protocols approved by the University Animal Care and Use Committee and conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Models of experimental diabetes and myocardial infarction
Experimental diabetes was induced by injection of streptozotocin (IP, 65 mg/kg body weight) dissolved in 0.1 M sodium citrate (pH 4.5). Seven days after injection, the animals with blood glucose =15 mM were further used to induce post-infarction remodeling. Animals were randomly assigned to the following four treatment groups: 1) sham surgery (sham); 2) sham surgery with aliskiren (sham+Als); 3) myocardial infarction (HF); or 4) MI with aliskiren (HF+Als). The experimental design is depicted in Fig. 1. The surgical procedure was performed as previously described [15]. Briefly, rats were anesthetized, intubated, and artificially ventilated with room air. The animal’s respiration rate was 65 to 70 breaths per minute and body temperature was maintained at 37°C. A lateral thoracotomy was performed and the heart was gently exposed. The left main coronary artery was ligated ~3 mm from its origin by using a firmly tied silk suture (7-0) to induce myocardial infarction. For the sham procedure the ligature was placed in an identical fashion but not tied. The animals were then followed for 4 weeks post-surgery. Treatment with aliskiren (50 mg/kg per day, IP) started immediately after surgery and continued during 4 weeks. Aliskiren was generously provided by Novartis Pharma AG (Basel, Switzerland). On the day of the experiment, cardiac function was analyzed by echocardiography, then the rats were sacrificed and their hearts were quickly removed for isolation of mitochondria.
Fig. 1.
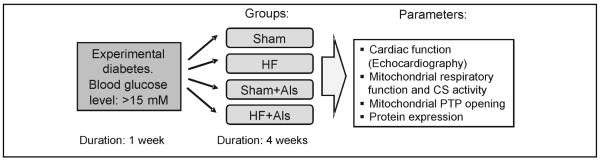
Experimental design. Sham surgery or CAL were performed in rats one week after experimental diabetes (blood glucose: >15 mM) had been induced by streptozotocin. Experimental groups: Sham: hearts subjected to sham surgery; HF: hearts subjected to CAL; Sham+Als: hearts subjected to sham surgery and were treated with aliskiren (Als); CAL+Als: hearts subjected to CAL and were treated with Als.
Echocardiography
Echocardiographic measurements were performed by a technician blinded to the treatment groups. Rats were anaesthetized, and the M-mode and 2D echocardiography images were obtained with a high-frequency 8-4 MHz 10-mm broadband phased P10 probe attached to a digital portable ultrasound system Micromaxx (Sonosite Inc.) with advanced image processing capabilities using Sitelink Image Manager (Sonosite Inc.). Diastolic and systolic measurements of left ventricle (LV) dimensions (LVIDd, LVIDs), LV end-systolic and end-diastolic volumes (LVESV, LVEDV), the thickness of the interventricular septum (IVSs, IVSd) and posterior wall (LVPWs, LVPWd), and heart rate (HR) were recorded. Stroke volume (SV), cardiac output (CO), LV fractional shortening (FS %) and ejection fraction (EF%) were calculated as SV = LVEDV–LVESV, CO=(SV*HR)/1000, FS = (LVIDd-LVIDs)/LVIDd × 100% and EF=((LVEDV-LVESV)/LEDV)*100, respectively.
Isolation of mitochondria
To isolate mitochondria, the ventricles were cut, weighed and homogenized using a Polytron homogenizer in 5 ml of ice-cold sucrose buffer containing 300 mM sucrose, 10 mM Tris-HCl, and 2 mM EGTA; pH 7.4. Mitochondria were isolated from the homogenate by centrifugation for 2 min at 2,000 × g at 40C to remove cell debris, followed by centrifugation of the supernatant at 10,000 × g for 5 min. The pellet was then washed two times at 10,000 μ g for 5 min. The final pellet was resuspended in 300 μl of sucrose buffer and used for measurement of mitochondrial respiratory function, PTP opening, protein expression, and citrate synthase activity.
Measurement of the respiration rates in isolated cardiac mitochondria
Measurement of mitochondrial respiration was performed at 30°C using a YSI Oxygraph (Yellow Springs) model 5300 equipped with a Clark-type oxygen electrode [15]. Mitochondria were suspended in a buffer containing (in mM): 125 KCl, 20 MOPS, 10 Tris, 0.5 EGTA, and 2 KH2PO4, pH 7.2, supplemented with either of 2.5 mM 2-oxoglutarate and 1 mM L-malate or 2.5 mM succinate and 1 μM rotenone for complex I and complex II, respectively. Respiration rates were measured in the absence (state 2) and presence (state 3) of 1 mM ADP. Additionally, 20 μM cytochrome c was added at state 3 to measure the cytochrome c-induced stimulation of mitochondrial respiration. Citrate synthase activity was assayed by measuring coenzyme A formation at 412 nm [16].
Measurement of PTP opening in isolated mitochondria
Swelling of de-energized mitochondria as an indicator of PTP opening in the presence or absence of calcium was determined by monitoring the decrease in light scattering at 520 nm. Mitochondria were incubated at 25°C in 3 ml buffer containing (in mM): 150 KSCN, 20 MOPS, 10 Tris, and 2 nitrilotriacetic acid, supplemented with 0.5 μM rotenone, 0.5 μM antimycin and 2 μM A23187.
SDS-PAGE and Western blotting
Equal amounts of homogenate or mitochondrial protein were resolved by SDS-PAGE. The membranes were immunoblotted with actin, acetylated lysine, voltage dependent anion channel (VDAC), cytochrome c oxidase IV subunit (COXIV), SIRT3 (Cell Signalling), cyclophilin D (CyP-D), adenine nucleotide translocase (ANT), AT1R, AT2R, SIRT4, SIRT5 (Santa Cruz) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Sigma-Aldrich) antibodies followed by secondary antibodies.
Co-immunoprecipitation
Acetylated lysine was immunoprecipitated from mitochondrial extracts. The immunoprecipitates were then separated by SDS-PAGE, blotted onto nitrocellulose membranes and the Western blots were developed using antibody against CyP-D or ANT (Santa Cruz) [17]. Immunoprecipitation assays with ANT and CyP-D were performed as controls.
RNA isolation and reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from frozen myocardial samples (80-100 mg) using the Trizol reagent according to the manufacturer instructions (Invitrogen). The reaction was carried out in one-step format using Power SYBR Green PCR Master Mix (Applied Biosystems), and Multiscribe Reverse Transcriptase (Applied Biosystems) in the presence of RNase Inhibitor (Applied Biosystems). The reaction was performed in a Step One Plus (Applied Biosystems) realtime PCR machine. For analysis of atrial natriuretic peptide (ANP), the primers used were: forward (5′- ccg aga cag caa aca tca ga-3′) and reverse (5′-tgt tgg aca ccg cac tgt at-3′). Normalization was carried out against ribosomal protein S11 (RPS11), for which expression was detected using QIAGEN QuantiTect Primer Assay (Qiagen). Fold change in expression was calculated using the 2ΔΔCt method [18].
Statistical analysis
Data are presented as means±SEM of 10-12 experiments per group. Statistical significance was evaluated using two-way ANOVA, followed by Tukey’s multiple comparison post-hoc test or an unpaired 2-tailed Student’s t-test. Differences were considered to be statistically significant when P<0.05.
Results
Aliskiren attenuates cardiac hypertrophy induced by post-infarction remodeling
To assess the possible effect of aliskiren on cardiac hypertrophy due to remodeling, myocardial infarction was induced in rats which were treated with aliskiren and compared with untreated rats. Sham procedure and left main coronary artery ligation induced lethality in 7% and 14% of rats, respectively. In addition, no significant difference in survival rates was seen between aliskiren-treated and untreated animals. Noteworthy, animals in all groups had no significant changes in blood glucose levels at 4 weeks after surgery compared to pre-surgery values (Fig. 2A). Furthermore, post-infarction remodeling with or without aliskiren treatment had no effect on glucose levels in diabetic rats. The heart weight-to-body weight ratio (HW/BW) was significantly increased in HF group compared to sham-operated animals. However, there was no difference in HW/BW values between the sham and HF groups treated with aliskiren during 4 weeks indicating that aliskiren could be involved in attenuation of cardiac hypertrophy induced by myocardial infarction (Fig. 2B). In addition, post-infarction remodeling increased expression of the hypertrophy gene marker, ANP, in the heart 3.4 times (P<0.05) compared to sham-operated rats. However, aliskiren-treated hearts contained reduced levels of ANP mRNA confirming the anti-hypertrophic effect of the direct renin inhibitor on the heart (Fig. 2C). Analysis of the expression of AT1R and AT2R in heart homogenates revealed that post-infarction remodeling slightly increased AT1R expression (P=N.S.) with no effect on AT2R (Fig. 3A,B). Interestingly, the AT1R level in post-infarcted hearts treated with aliskiren was reduced by 25% (P<0.05) compared to the untreated hearts (Fig. 3A). In addition, we examined whether mitochondria contain AngII receptors. Protein expression of AT1R and AT2R was determined in Percoll-purified mitochondria isolated from healthy (untreated) rats. As shown in Fig. 3C, western blot analysis demonstrated that both receptors are present in mitochondria. Notably, the expression of AT2R is much higher than AT1R.
Fig. 2.
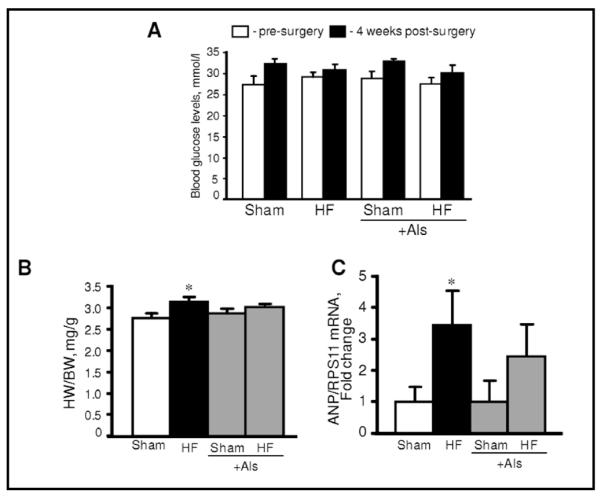
Blood glucose levels (A), heart-to-body weight ratio (HW/BW, B) and expression of atrial natriuretic peptide (ANP, C) in sham-operated and post-infarcted hearts treated with or without aliskiren (Als). ANP gene expressions were normalized to ribosomal protein S11 (RPS11) mRNA. *P<0.05 HF vs. Sham.
Fig. 3.
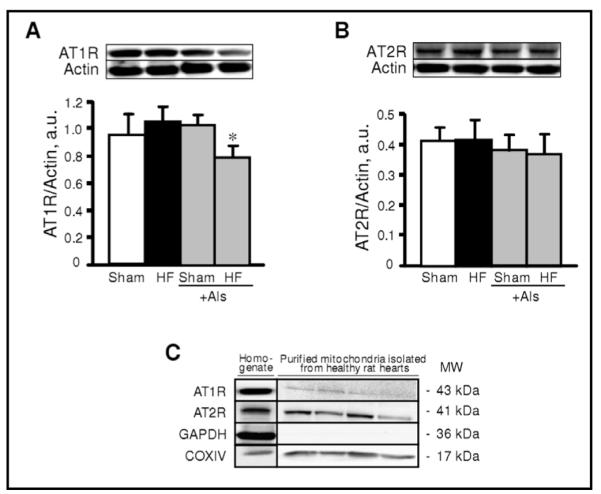
Protein expression of angiotensin II type 1 (AT1R, A) and type 2 (AT2R, B) receptors in sham-operated and post-infarcted hearts treated with or without aliskiren (Als), and in Percoll-purified mitochondria isolated from control hearts (C). Homogenate (A, B) and mitochondrial (C) samples were resolved by SDS-PAGE and immunoblotted with AT1R or AT2R antibodies. Actin and GAPDH were used as the housekeeping proteins for cytosol whereas COXIV expression is shown as a mitochondrial marker. *P<0.05 HF+Als vs. HF.
Aliskiren treatment improves cardiac function in post-infarction heart failure
In the next set of experiments, we examined whether aliskiren improves cardiac function in post-infarction HF. It is important to note that the purpose of our study was to investigate if acute diabetes can compromise the anti-remodeling effects of aliskiren. Indeed, rats with an acute (7 days) model of diabetes do not develop all characteristics of diabetes except hyperglycemia. Accordingly, cardiac function of sham-operated diabetic rats exhibit normal cardiac function (ejection fraction: 78%, cardiac output: 128 μl/min). Cardiac remodeling at 4 weeks was associated with significant impairment of heart contractility. The hearts with post-infarction HF exhibited significantly less EF (41%, P<0.01), CO (42%, P<0.05) and LVDFS (52%, P<0.01) compared to sham-operated hearts. Furthermore, HF reduced LVDs and increased LVESV at 4 weeks after ligation. However, post-infarction HF rats treated with aliskiren demonstrated improved cardiac function compared to the untreated HF group. As shown in Fig. 4, EF in rats treated with aliskiren (HF+Als) was 43% (P<0.05) higher than that in HF group. The EF recovery was associated with improved values of SV, LVDFS, LVDs and LVESV. Noteworthy, aliskiren had no effect on cardiac function in sham-operated rats. Thus, these data demonstrate that aliskiren abrogates cardiac dysfunction in the failing heart induced by myocardial infarction.
Fig. 4.

The effect of aliskiren (Als) on cardiac output (CO, A), ejection fraction (EF%, B) and left ventricle (LV) fractional shortening (FS%, C) of hearts 28 days after sham-procedure or CAL. Calculations of functional parameters of the heart are given in Materials and Methods. *P<0.05 and **P<0.01 HF vs. Sham; +P<0.05 HF+Als vs. HF.
Aliskiren improves respiratory function of mitochondria
Respiration rates of mitochondria were measured using the substrates for complexes I and II of the electron transport chain. Respiration of mitochondria with complex II substrates was not affected by HF. However, the maximal ADP-stimulated respiration rate (state 3) with substrates for complex I was 27% (P<0.05) less in HF compared to sham-operated rats (Fig. 5A). The respiratory control index (RCI), which indicates the efficiency of respiratory coupling decreased by 38% (P<0.05) in the HF group (Fig. 5B). To further establish whether the integrity of the outer mitochondrial membrane is affected by HF, we measured the state 3 respiration rate driven by complex I substrates in the presence of exogenous cytochrome c when added directly to mitochondria in the oxygraph chamber. Cytochrome c stimulated state 3 in mitochondria isolated from failing hearts by 71% (P<0.01 vs sham group) (Fig. 5C). The low mitochondrial respiration rate and high cytochrome c-induced stimulation of state 3 were associated with reduced activity of citrate synthase, a mitochondrial marker enzyme, in ligated hearts. These experiments confirm that the structural integrity of mitochondria was affected by HF (Fig. 5D). Release of cytochrome c due to damage to the outer mitochondrial membrane could result in dysfunction of cytochrome c oxidase and decrease mitochondrial respiration at complex IV. The state 3 respiration rate with complex I substrates as well as RCI of the complex in aliskiren-treated hearts (HF+Als group) were significantly higher than in the HF group (Fig. 5A,B). Aliskiren-treated hearts demonstrated less stimulation of state 3 by exogenous cytochrome c than HF animals, indicating a preserved structural integrity of mitochondria (Fig. 5C). Altogether, these findings suggest that treatment with aliskiren attenuates mitochondrial dysfunction in post-infarcted hearts.
Fig. 5.

The effect of aliskiren (Als) on state 3 respiration rate (A), respiratory control index (RCI, B), cytochrome c-stimulated respiration (C) and citrate synthase activity (D) of mitochondria isolated from sham-operated and post-infarcted hearts treated with or without aliskiren (Als). Cytochrome c (cyt c, 20 μM) was added directly to the cuvette to measure cytochrome c stimulation of respiration with complex I substrates in the presence of 1 mM ADP (state 3). Data are shown as mean ± SEM of 10-12 hearts in each group. *P<0.05 and **P<0.01 HF vs. Sham; +P<0.05 HF+Als vs. HF.
Mitochondria isolated from aliskiren-treated hearts demonstrate less PTP opening
The extent of mitochondrial PTP opening was measured in two sets of experiments on the basis of Ca2+-induced swelling, measured as light scattering. Sanglifehrin A, a strong inhibitor of PTP opening, was used as a positive control in these experiments. In the first set, PTP opening was assessed in mitochondria isolated from sham and HF hearts treated or untreated with aliskiren. Results for this first set demonstrated that mitochondria isolated from the hearts with HF are more susceptible to Ca2+-induced swelling due to increased PTP formation (Fig. 6). However, aliskiren-treated hearts revealed less rate of mitochondrial swelling and hence, less PTP opening than untreated post-infarcted hearts. In the second set, we evaluated the possible direct effect of aliskiren on Ca2+-induced swelling. Aliskiren added to mitochondrial suspensions isolated from the untreated post-infarcted hearts failed to have a direct inhibitory effect on Ca2+-induced PTP opening (Fig. 6). These studies provide strong evidence indicating that aliskiren-treated post-infarcted hearts are more resistant to PTP opening although the effect of the drug on PTP components is apparently indirect.
Fig. 6.

Rate of Ca2+-induced swelling as an indicator of PTP opening in mitochondria isolated from sham-operated or post-infarction hearts treated with aliskiren (Als). Measurement of the PTP under de-energized conditions was performed by monitoring the calcium-induced decrease in light scattering (A520). (A) Original traces are shown for one mitochondrial preparation derived from hearts of sham, HF, sham+Als or HF+Als groups. Additionally, mitochondria isolated from hearts of HF group were treated in vitro with 10 μM Als (HF+Als in vitro) or 0.6 μM sanglifehrin A (HF+SfA in vitro). To measure the effect of Als or SfA on PTP opening in vitro, the inhibitors were added directly to the cuvette and swelling of mitochondria was monitored. Matrix swelling was induced by 200 μM Ca2+. Maximal swelling was induced by 1 mM Ca2+. (B) Rate of swelling of mitochondria. Data are expressed relative to the maximum rate determined at 1 mM Ca2+ and were determined by differentiation of the traces shown to obtain the maximum rate of change of A520. *P<0.05 shows the level of significance between the compared groups.
Aliskiren enhances SIRT3 expression and protein acetylation in the heart
The results shown in the previous sections provide evidence that the anti-remodeling effect of aliskiren may be mediated through mitochondria by inhibition of PTP formation. CyP-D is known as an essential regulator of PTP formation, and recent studies have shown that CyP-D activity can be regulated by mitochondrial sirtuins through its acetylation/deacetylation [19, 20]. Sirtuins are characterized as class III histone deacetylases and depend on NAD+ for their activity; the deacetylase activity of sirtuins is elevated by increased NAD+ levels and reduced by high nicotinamide and/or NADH levels [21]. Of the seven isoforms of sirtuins, only SIRT3, SIRT4, and SIRT5 are localized in mitochondria. They have been shown to regulate energy metabolism through acetylation/deacetylation of proteins involved in the electron transport chain and oxidative phosphorylation. Among these mitochondrial sirtuins, SIRT3 exhibits the highest deacetylase activity [22]. We evaluated the levels of protein acetylation in HF and the effect of aliskiren in this regulatory event. Results demonstrated that HF increases acetylation of mitochondrial proteins in post-infarcted hearts (Fig. 7A). SIRT3-mediated cell survival under physiological conditions may include deacetylation of CyP-D leading to prevention of PTP formation. Accordingly, hyperacetylation of CyP-D due to downregulation of SIRT3 can instigate PTP formation and mitochondria-mediated cell death. The observed increase of protein acetylation was associated with the reduction of SIRT3 expression with no remarkable changes in SIRT4 and SIRT5 protein levels (Fig. 7B). However, treatment with aliskiren restored the SIRT3 level in the failing hearts (Fig. 7C). Furthermore, results of immunoprecipitation demonstrated that HF increased the acetylated CyP-D level in mitochondria which was attenuated by treatment with aliskiren. Thus, these data demonstrate the possible role of the SIRT3/CyP-D-mediated pathway in the inhibition of PTP opening by aliskiren.
Fig. 7.

The effects of post-infarction HF on mitochondrial total protein, CyP-D acetylation, and sirtuin levels. (A, B,C) Mitochondrial proteins were resolved by SDS-PAGE and probed with anti-acetylated lysine (Ac-Lys), SIRT3, SIRT4 or SIRT5 antibodies. The same membranes were stripped and reprobed with COXIV as a housekeeping protein for mitochondria. (D) Immunoprecipitation (IP) of mitochondrial proteins with Ac-Lys followed by immunoblotting (IB) for CyP-D and ANT was performed to assay CyP-D acetylation. In addition, IP was performed with CyP-D and ANT antibodies as controls.
Discussion
The present study examines whether anti-hypertrophic effects of the direct renin inhibitor aliskiren are associated with mitochondria in post-infarcted rat hearts with acute diabetes. Our results demonstrated that: i) aliskiren diminishes hypertrophy and improves cardiac function with no effect on blood glucose in post-myocardial infarction diabetic rats, ii) the anti-remodeling effect of aliskiren is associated with improved respiratory function of mitochondria, and iii) the effect of aliskiren on mitochondria is mediated, at least in part, through inhibition of PTP opening possibly due to mitochondrial SIRT3-induced CyP-D deacetylation.
Recent studies provide strong evidence on the pleiotropic effects of RAS inhibitors on diabetes and cardiovascular disease beyond blood pressure control [2, 23, 24]. The beneficial effects of aliskiren to improve mitochondrial function in the post-infarcted heart could be mediated through its direct and/or indirect effects on the organelles. Analysis of mitochondrial respiratory function revealed efficient protection of aliskiren against mitochondrial remodeling induced by post-infarction HF. Our in vitro experiments revealed no changes in Ca2+-induced swelling in mitochondria when aliskiren was directly added to the mitochondrial suspension (Fig. 6). These results provide strong evidence in favor of an indirect action of aliskiren on PTP formation. Aliskiren could prevent “mitochondrial remodeling” at least through two different mechanisms including AngII-dependent and/or AngII-independent pathways. Inhibition of renin activity by aliskiren diminishes AngI production which is the first and rate-limiting process of the RAS. As a result, reduced levels of AngII could attenuate the hypertrophic response of the heart to myocardial infarction. We found that aliskiren reduced the expression of AT1R in post-infarcted hearts (Fig. 3). Pro-hypertrophic signals of AngII are mediated via these receptors, whose overexpression induces cardiac hypertrophy and remodeling [25]. Accordingly, downregulation of the receptor by aliskiren could reduce autocrine/paracrine effects of AngII and prevent cardiac remodeling resulting from myocardial infarction. Moreover, since components of the intracellular RAS system have been found in the heart [26, 27], and aliskiren could also inhibit local production of AngII, and its intracellular effect in the failing heart. Intracellular synthesis of renin was shown in cardiomyocytes [6, 28], although other studies were unable to confirm existence of the intracellular renin [29]. Previous studies demonstrated that aliskiren reduced intracellular levels of AngII and renin in cardiomyocytes isolated from diabetic rats, further suggesting a role for an intracrine mechanism of AngII in hyperglycemic conditions [6]. The AngII-independent effect of aliskiren could be mediated through the prorenin/renin receptor. Cardiomyocytes have been previously shown to contain a functional prorenin/renin receptor stimulation of which can increase the efficiency of angiotensinogen cleavage by membrane-bound renin [30]. Binding of renin to the prorenin/renin receptor activated ERK1/2 and p38 MAPKs and upregulated profibrotic genes independent of AngII generation [31]. Aliskiren could be involved in inhibition of the rennin receptor internalization and, as a consequence, prevent AngII-independent signaling pathway and its deleterious consequences. Most recent studies demonstrated that in combination with the inhibitor of angiotensin converting enzyme or AngII receptor blocker, aliskiren exerted additional synergistic protective effects against ventricular remodeling following myocardial infarction in rodents [32, 33] although clinical trials demonstrated no additional benefit of RAS blockade with aliskiren in post-infarcted patients [34]. The beneficial effects of combined therapy could indicate existence of AngII-independent pathways through which aliskiren would affect cell metabolism.
It is not clear how AngII-dependent and AngII-independent beneficial effects of aliskiren could converge on mitochondria. Aliskiren has been previously shown to reduce the number of smaller mitochondria and their structural abnormalities in the heart of transgenic rats with chronically elevated tissue AngII levels, although function of mitochondria was not determined in these studies [14]. The intracellular mechanisms of the effects of aliskiren on mitochondria may include abrogation of AngII-induced ROS generation and Ca2+-overload that together with ATP depletion compromise mitochondrial function and induce PTP opening. Consistent with this hypothesis, the present study revealed improved mitochondrial respiration (state 3) and reduced extent of PTP opening in aliskiren-treated hearts following myocardial infarction. Aliskiren reduced cytochrome c release from mitochondria to the cytoplasm in failing hearts (Fig. 5) indicating that the drug preserves the integrity of the outer mitochondrial membrane. Attenuation in the release of cytochrome c and other pro-apoptotic proteins from the intermembrane space may prevent mitochondria-mediated apoptosis. Furthermore, preservation of cytochrome c could enhance the cytochrome c oxidase activity and improve respiration at complex IV. Inhibition of PTP opening would attenuate depolarization and matrix swelling of mitochondria ultimately preventing mitochondria-mediated cell death. The inhibitory effect of aliskiren on the mitochondrial PTP can be mediated through prevention of SIRT3, the main mitochondrial sirtuin isoform, in the failing heart. SIRT3 plays a central role in FAO and ATP synthesis in cells [35, 36], and its decreased expression is associated with aging, and neurodegenerative and metabolic diseases. We found that cardiac remodeling following myocardial infarction for 4 weeks markedly reduced expression of SIRT3 with no effect on SIRT4 and SIRT5 expression. However, mitochondria isolated from aliskiren-treated post-infarcted hearts exhibited preserved SIRT3 expression compared to untreated rats (Fig. 7). Recent studies provide strong evidence that CyP-D, the main regulatory protein of the PTP complex, can be acetylated. SIRT3-mediated cell survival under physiological conditions may include deacetylation of CyP-D leading to prevention of PTP formation. Accordingly, hyperacetylation of CyP-D due to downregulation of SIRT3 can instigate PTP formation and mitochondria-mediated cell death. CyP-D acetylation facilitates its interaction with adenine nucleotide translocase (ANT) thereby inducing PTP formation in the inner mitochondrial membrane [19, 20]. Our data are consistent with this hypothesis showing that under normal conditions, SIRT3 targets CyP-D maintaining it in a deacetylated state (Fig. 8). However, in the failing heart SIRT3 is downregulated, promoting PTP opening. In aliskiren-treated hearts, PTP formation was prevented by preservation of SIRT3 keeping CyP-D deacatylated. These findings suggest that the reduction of cardiac hypertrophy could be secondary to the abrogation of mitochondrial dysfunction induced by the SIRT3/CyP-D/PTP pathway. Similar observations have been reported previously for the heart with hemodynamic overload [37]. Anti-hypertrophic effects of SIRT3 can be mediated through deacetylation and nuclear translocation of FoxO3, where it promotes transcription of anti-oxidant enzymes [38].
Fig. 8.
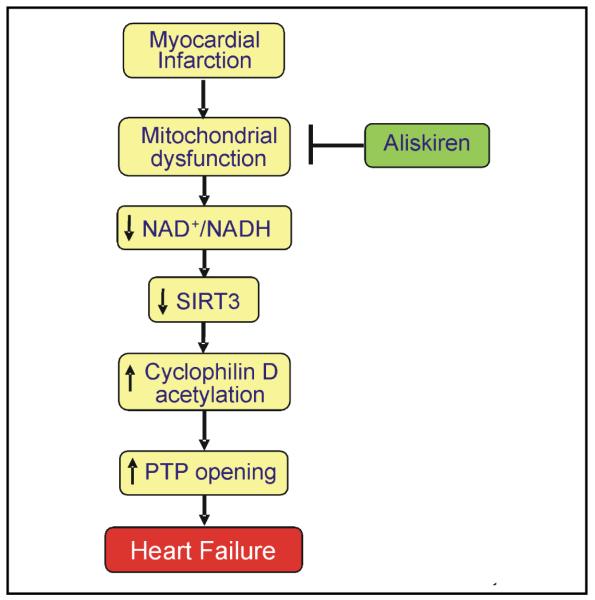
Proposed mechanism of the mitochondria-mediated anti-remodeling effect of aliskiren in post-infarction heart failure.
In summary, the present study demonstrates the direct renin inhibitor aliskiren exerts anti-remodeling effects in HF resulting from myocardial infarction in rats, and the effects are mediated, at least in part, through improvement in mitochondrial function. The beneficial effects of aliskiren on mitochondria may include, among others, attenuation of CyP-D acetylation due to preservation of mitochondrial SIRT3 in post-infarcted hearts. Furthermore, the present study does not exclude the possible effect of aliskiren on mitochondria through AngII receptors present in mitochondria. Purified mitochondria isolated from healthy rat hearts revealed expression of AT1R and AT2R in mitochondria (Fig.3C). These data coincide with the recent study demonstrating expression of mitochondrial AT1R and AT2R in kidney [39]. Further studies are required to clarify the role of the intracellular RAS in mediating both AngII-dependent and AngII-independent effects of aliskiren to reduce cardiac remodeling.
Acknowledgements
The authors thank Ms. Miriam Castro for her invaluable technical assistance in performing of experiments. This work was supported by Novartis Pharma AG (Basel, Switzerland), and in part, by the RCMI grant G12RR-03051 from the National Center for Research Resources, National Institutes of Health.
Footnotes
Fax +41 61 306 12 34 karger@karger.ch www.karger.com
References
- 1.Wollert KC, Drexler H. The renin-angiotensin system and experimental heart failure. Cardiovasc Res. 1999;43:838–849. doi: 10.1016/s0008-6363(99)00145-5. [DOI] [PubMed] [Google Scholar]
- 2.Dorn GW. Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol. (2nd) 2009;6:283–291. doi: 10.1038/nrcardio.2009.12. [DOI] [PubMed] [Google Scholar]
- 3.Schieffer B, Wirger A, Meybrunn M, Seitz S, Holtz J, Riede UN, Drexler H. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin ii type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation. 1994;89:2273–2282. doi: 10.1161/01.cir.89.5.2273. [DOI] [PubMed] [Google Scholar]
- 4.Wollert KC, Studer R, von Bulow B, Drexler H. Survival after myocardial infarction in the rat. Role of tissue angiotensin-converting enzyme inhibition. Circulation. 1994;90:2457–2467. doi: 10.1161/01.cir.90.5.2457. [DOI] [PubMed] [Google Scholar]
- 5.Westermann D, Riad A, Lettau O, Roks A, Savvatis K, Becher PM, Escher F, Jan Danser AH, Schultheiss HP, Tschope C. Renin inhibition improves cardiac function and remodeling after myocardial infarction independent of blood pressure. Hypertension. 2008;52:1068–1075. doi: 10.1161/HYPERTENSIONAHA.108.116350. [DOI] [PubMed] [Google Scholar]
- 6.Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin ii production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57:3297–3306. doi: 10.2337/db08-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: Potential for therapeutic interventions. Heart Fail Rev. 2002;7:115–130. doi: 10.1023/a:1015320423577. [DOI] [PubMed] [Google Scholar]
- 8.Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. doi: 10.1007/s10741-007-9079-1. [DOI] [PubMed] [Google Scholar]
- 9.Sheeran FL, Pepe S. Energy deficiency in the failing heart: Linking increased reactive oxygen species and disruption of oxidative phosphorylation rate. Biochim Biophys Acta. 2006;1757:543–552. doi: 10.1016/j.bbabio.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 11.Sam F, Kerstetter DL, Pimental DR, Mulukutla S, Tabaee A, Bristow MR, Colucci WS, Sawyer DB. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail. 2005;11:473–480. doi: 10.1016/j.cardfail.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing nadph oxidase in angiotensin ii-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 13.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, Cave AC, Shah AM. Contrasting roles of nadph oxidase isoforms in pressure-overload versus angiotensin ii-induced cardiac hypertrophy. Circ Res. 2003;93:802–805. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- 14.Whaley-Connell A, Habibi J, Cooper SA, Demarco VG, Hayden MR, Stump CS, Link D, Ferrario CM, Sowers JR. Effect of renin inhibition and at1r blockade on myocardial remodeling in the transgenic ren2 rat. Am J Physiol Endocrinol Metab. 2008;295:E103–109. doi: 10.1152/ajpendo.00752.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Javadov S, Huang C, Kirshenbaum L, Karmazyn M. NHE-1 inhibition improves impaired mitochondrial permeability transition and respiratory function during postinfarction remodelling in the rat. J Mol Cell Cardiol. 2005;38:135–143. doi: 10.1016/j.yjmcc.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Srere PA. Citrate synthase. Methods Enzymol. 1969;13:3–11. [Google Scholar]
- 17.Javadov S, Rajapurohitam V, Kilic A, Zeidan A, Choi A, Karmazyn M. Anti-hypertrophic effect of NHE-1 inhibition involves GSK-3beta-dependent attenuation of mitochondrial dysfunction. J Mol Cell Cardiol. 2009;46:998–1007. doi: 10.1016/j.yjmcc.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(−delta delta c(t)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin d induces dissociation of hexokinase ii from the mitochondria. J Cell Sci. 2010;123:894–902. doi: 10.1242/jcs.061846. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Shulga N, Pastorino JG. Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-d mediated by sirtuin-3. J Cell Sci. 2010;123:4117–4127. doi: 10.1242/jcs.073502. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Guarente L. Sirtuins as potential targets for metabolic syndrome. Nature. 2006;444:868–874. doi: 10.1038/nature05486. [DOI] [PubMed] [Google Scholar]
- 22.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr., Weissman S, Verdin E, Schwer B. Mammalian sir2 homolog sirt3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verdecchia P, Angeli F, Mazzotta G, Gentile G, Reboldi G. The renin angiotensin system in the development of cardiovascular disease: Role of aliskiren in risk reduction. Vasc Health Risk Manag. 2008;4:971–981. doi: 10.2147/vhrm.s3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ismail H, Mitchell R, McFarlane SI, Makaryus AN. Pleiotropic effects of inhibitors of the raas in the diabetic population: Above and beyond blood pressure lowering. Curr Diab Rep. 2010;10:32–36. doi: 10.1007/s11892-009-0081-y. [DOI] [PubMed] [Google Scholar]
- 25.Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M. Overexpression of angiotensin ii type i receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci U S A. 2000;97:931–936. doi: 10.1073/pnas.97.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dostal DE, Baker KM. The cardiac renin-angiotensin system: Conceptual, or a regulator of cardiac function? Circ Res. 1999;85:643–650. doi: 10.1161/01.res.85.7.643. [DOI] [PubMed] [Google Scholar]
- 27.Miyazaki M, Takai S. Tissue angiotensin ii generating system by angiotensin-converting enzyme and chymase. J Pharmacol Sci. 2006;100:391–397. doi: 10.1254/jphs.cpj06008x. [DOI] [PubMed] [Google Scholar]
- 28.Singh VP, Le B, Bhat VB, Baker KM, Kumar R. High-glucose-induced regulation of intracellular ang ii synthesis and nuclear redistribution in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H939–948. doi: 10.1152/ajpheart.00391.2007. [DOI] [PubMed] [Google Scholar]
- 29.Krop M, Danser AH. Circulating versus tissue renin-angiotensin system: On the origin of (pro)renin. Curr Hypertens Rep. 2008;10:112–118. doi: 10.1007/s11906-008-0022-1. [DOI] [PubMed] [Google Scholar]
- 30.Connelly KA, Advani A, Kim S, Advani SL, Zhang M, White KE, Kim YM, Parker C, Thai K, Krum H, Kelly DJ, Gilbert RE. The cardiac (pro)renin receptor is primarily expressed in myocyte transverse tubules and is increased in experimental diabetic cardiomyopathy. J Hypertens. 2011;29:1175–1184. doi: 10.1097/HJH.0b013e3283462674. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen G, Muller DN. The biology of the (pro)renin receptor. J Am Soc Nephrol. 2010;21:18–23. doi: 10.1681/ASN.2009030300. [DOI] [PubMed] [Google Scholar]
- 32.Ye Y, Qian J, Castillo AC, Perez-Polo JR, Birnbaum Y. Aliskiren and Valsartan reduce myocardial AT1 receptor expression and limit myocardial infarct size in diabetic mice. Cardiovasc Drugs Ther. 2011;25:505–515. doi: 10.1007/s10557-011-6339-z. [DOI] [PubMed] [Google Scholar]
- 33.Higashikuni Y, Takaoka M, Iwata H, Tanaka K, Hirata Y, Nagai R, Sata M. Aliskiren in combination with valsartan exerts synergistic protective effects against ventricular remodeling after myocardial infarction in mice. Hypertens Res. 2012;35:62–69. doi: 10.1038/hr.2011.136. [DOI] [PubMed] [Google Scholar]
- 34.Solomon SD, Shin SH, Shah A, Skali H, Desai A, Kober L, Maggioni AP, Rouleau JL, Kelly RY, Hester A, McMurray JJ, Pfeffer MA. Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur Heart J. 2011;32:1227–1234. doi: 10.1093/eurheartj/ehq522. [DOI] [PubMed] [Google Scholar]
- 35.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. Sirt3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mptp by sirt3-mediated deacetylation of cypd at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O’Rourke B, Walston JD. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci USA. 2011;108:14849–14854. doi: 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]