

Summary
This study explored the anti-leukaemic efficacy of novel irreversible inhibitors of the major nuclear export receptor, chromosome region maintenance 1 (CRM1, also termed XPO1). We found that these novel CRM1 antagonists, termed SINE (Selective Inhibitors of Nuclear Export), induced rapid apoptosis at low nanomolar concentrations in a panel of 14 human T-cell acute lymphoblastic leukaemia (T-ALL) cell lines representing different molecular subtypes of the disease. To assess in vivo anti-leukaemia cell activity, we engrafted immunodeficient mice intravenously with the human T-ALL MOLT-4 cells, which harbour activating mutations of NOTCH1 and NRAS as well as loss of function of the CDKN2A, PTEN and TP53 tumour suppressors and express a high level of oncogenic transcription factor TAL1. Importantly, we examined the in vivo anti-leukaemic efficacy of the clinical SINE compound KPT-330 against TALL and acute myeloid leukaemia (AML) cells. These studies demonstrated striking in vivo activity of KPT-330 against T-ALL and AML cells, with little toxicity to normal murine haematopoietic cells. Taken together, our results show that SINE CRM1 antagonists represent promising “first-in-class” drugs with a novel mechanism of action and wide therapeutic index, and imply that drugs of this class show promise for the targeted therapy of T-ALL and AML.
Introduction
The treatment of acute lymphoblastic leukaemia (ALL) has improved over the last few decades as a result of the combination of intensive chemotherapy, radiotherapy and stem cell transplantation. However, T-cell acute lymphoblastic leukaemia (T-ALL) remains fatal in approximately 25% of children and in 50–70% of adults, prompting the need to develop new therapies (Pui and Evans 2006, Pui, et al 2008). In this study, we explored selective inhibition of nuclear-cytoplasmic trafficking as a new anti-T-ALL therapeutic strategy and demonstrate striking anti-leukaemic efficacy of novel inhibitors of nuclear exporter CRM1 (exportin 1 (CRM1 homolog, yeast); XPO1) in preclinical models of T-ALL.
Nuclear-cytoplasmic transport is a fundamental property of eukaryotic cells, mediated in part by the karyopherin family of proteins, which transport proteins and ribonucleic acids between the nucleus and the cytoplasm (Siddiqui and Borden 2012, Xu, et al 2010). The major nuclear exporter protein CRM1, one of seven exportins, mediates the transport of approximately 220 proteins (Xu, et al 2012a) and several mRNAs. Interestingly, CRM1 is the sole nuclear exporter of the major tumour suppressor and growth regulatory proteins p53 (TP53), p73 (TP73), FOXO (FOXO1; counteracts PI3K/AKT), IκB/NF-κB (NFKB1), Rb (RB1), p21 (CDKN1A, and NPM (NPM1) (Fornerod, et al 1997, Fukuda, et al 1997, Ossareh-Nazari, et al 1997, Turner, et al 2012). CRM1 is upregulated in a range of solid tumours and haematological malignancies and its overexpression is correlated with poor prognosis, suggesting that alterations in nuclear-cytoplasmic trafficking, and hence mislocalization of tumour suppressor proteins, cell cycle regulators, and/or pro-apoptotic proteins, could lead to oncogenesis and resistance to chemotherapy (Huang, et al 2009, Noske, et al 2008, Shen, et al 2009, van der Watt, et al 2009, Yao, et al 2009).
CRM1 recognizes export cargos that contain short leucine–rich nuclear export signal (NES) consensus sequences (Dong, et al 2009, Guttler, et al 2010, Monecke, et al 2009, Xu, et al 2012b). Extensive studies with well-established natural product CRM1 inhibitors leptomycin B, ratjadone, anguinomycin, and goniothalamin, and recently developed small molecule inhibitors of CRM1, such as, N-azolylacrylates, KOS-2464, and CBS9106 (Bonazzi, et al 2010, Daelemans, et al 2002, Kudo, et al 1999, Meissner, et al 2004, Mutka, et al 2009, Sakakibara, et al 2011, Van Neck, et al 2008, Wach, et al 2010) have clearly demonstrated the requirement of CRM1 nuclear export activity for the growth and survival of cancer cells. Blockade of CRM1 transport by these inhibitors has been shown to induce cancer cell death, possibly by promoting the forced nuclear retention of tumour suppressor proteins that are normally inactivated by cytoplasmic mislocalization. Moreover, interference with CRM1-directed nuclear export by these inhibitors has been shown to promote nuclear localization of topoisomerase IIα and to sensitize multiple myeloma cells to the topoisomerase II inhibitors etoposide and doxorubicin (Turner, et al 2009). However, despite the ability of existing CRM1 inhibitors to counteract the CRM1-mediated nuclear export and to promote anti-proliferative and apoptotic signalling pathways in cancer cells, these compounds exhibit extensive toxic effects against normal cells, apparently due to both on-target and possibly off-target activities (Mutka, et al 2009, Sakakibara, et al 2011). These caveats clearly emphasize the need for the development of CRM1 inhibitors with increased selectivity for cancer cells and reduced toxicity to normal cells as a prerequisite for their translation into clinical use. We and others have recently reported the striking anti-AML activity and high selectivity of a new class of drug-like, small molecule CRM1 antagonists called Selective Inhibitors of Nuclear Export, or SINE (Etchin, et al 2012, Ranganathan, et al 2012). SINE drugs were developed based on an in silico molecular modelling strategy, in which a structural model of the NES groove of CRM1 is used as a framework for selection and optimization of virtual library of irreversible CRM1 inhibitors (Etchin, et al 2012, Turner, et al 2012).
Recently, the first ever clinical trials of an oral SINE compound, KPT-330, were initiated, with two trials running in parallel: one includes patients with advanced solid tumours whose disease has progressed after at least one prior therapy for metastatic disease (NCT01607905); the second includes patients with advanced haematological malignancies including chronic lymphocytic leukaemia, non-Hodgkin lymphoma, multiple myeloma, and Waldenstrom macroglobulinaemia whose disease has relapsed after standard therapies (NCT01607892). Patients with AML will be eligible in future clinical trials once the tolerability profile of KPT-330 has been established.
The present study showed that the SINE compounds are highly active against human TALL cells carrying different genetic alterations. These compounds induce rapid apoptosis in TALL cells in vitro and promote striking growth suppression of T-ALL cells engrafted into immunodeficient mice. Importantly, our data demonstrate that KPT-330 is very active in preclinical models of T-ALL as well as AML, with minimal toxicity to normal blood cells both in the periphery and in the bone marrow. These data indicate that KPT-330 is a promising drug for the treatment of T-ALL as well as AML, and support the ongoing and future development of this novel class of agents.
Materials and Methods
SINE CRM1 Antagonists
KPT-185, KPT-251 (Etchin, et al 2012), and KPT-330 (molecular weight of 443.31, chemical formula: C17H11F6N7O) are structurally similar, selective CRM1 inhibitors with distinct pharmacokinetic (PK) properties (Karyopharm Therapeutics Inc., Natick, MA). KPT-251 and -330 are suitable for in vivo use; KPT-185 is the most potent CRM1 inhibitor but has very poor PK properties making it unsuitable for in vivo use.
Cell lines and cell viability assay
T-ALL cell lines (HPB-ALL, DU528, Jurkat, MOLT-4, SKW-3, KARPAS-45, HSB-2, KOPTK1, PF-382, CCRF-CEM, SUPT7, MOLT-16, P12-ICHIKAWA, LOUCY) were cultured in RPMI 1640 medium (GIBCO, Grand Island, NY, USA), supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cell Titer Glo assay (Promega, Madison, WI, USA) was used to assess cell viability upon treatment with either dimethyl sulfoxide (DMSO) or KPT-185. Cells were plated at a density of 10,000 cells per well in a 96-well plate and incubated with DMSO or increasing concentrations of KPT-185. The cell viability was measured after 72 h exposure to KPT-185 and reported as a percentage of DMSO control cells. Jurkat cells that overexpress BCL2 were generated using MSCV-IRES-GFP retroviral expression system. Jurkat cells infected with BCL2 or control vector viruses were sorted by flow cytometry and the expression of BCL2 confirmed by Western blot analysis using BCL2 antibody (Cell Signaling, Danvers, MA, USA).
Apoptosis Analysis
Jurkat and MOLT-4 cells were incubated with either DMSO control or KPT-185 for 6 h or 13 h, washed with phosphate-buffered saline (PBS), and co-incubated with Annexin V- fluorescein isothiocynate (FITC) and propidium iodide (PI) from MEBCYTO Apoptosis Kit (MBL Co., Ltd., Nagoya, Japans). Cells were analysed by two-colour FACS cytometry (BD FACS Canto, BD Biosciences, San Jose, CA, USA) and the percentage of Annexin V and PI positive cells was determined based on the dot plots of FITC vs. PI.
Mitochondrial Sensitivity in permeabilized whole cells
2 × 104 cells/well of Jurkat cells were used. 15 μl of 100 μM peptide in T-EB (300 mM Trehalose, 10 mM HEPES-KOH pH 7.7, 80 mM KCl, 1 mM EGTA, 1 mM EDTA, 0.1% bovine serum albumin, 5 mM succinate) were deposited per well in a black 384-well plate (BD Falcon no. 353285). One volume of the 4x single cell suspension was added to one volume of a 4x dye solution (4 μM JC-1, 40 μg/ml oligomycin, 0.02% digitonin, 20 mM 2-mercaptoethanol) in T-EB. This 2x cell/dye solution was incubated for 5–10 min at room temperature to allow permeabilization and dye equilibration. 15 μl of the cell/dye mix was then added to each treatment well of the plate and the fluorescence at 590 nm monitored every 5 min at room temperature. Percentage loss of Ψm was calculated by normalization to the solvent only control DMSO (0%) and the positive control FCCP (Ryan, et al 2010).
Cell cycle analysis
Jurkat and MOLT-4 cells were incubated with serial dilutions of KPT-185 for 24 h, washed with PBS, fixed with 70% ethanol, and incubated overnight at −20°C. The cells were then washed with PBS, stained with PI/RNase staining buffer (BD Biosciences), and analysed by flow cytometry using BD FACS Canto (BD Biosciences). The DNA histograms of Jurkat and MOLT-4 cells were analysed using FCS Express 4 Flow Cytometry cell cycle analysis software (De Novo Software, Los Angeles, CA, USA) and ModFit LT cell cycle analysis software (Verity Software House, Topsham, ME, USA).
Orthograft mouse models
T-ALL orthograft mouse model
MOLT-4 cells (3 × 106) expressing luciferase were injected into 7-week-old female NOD-SCID-IL2Rcγnull (NSG) mice (The Jackson Laboratory, Bar Harbor, ME, USA) via tail-vein injections. The leukaemia burden was established by bioluminescence imaging (BLI) using an IVIS Spectrum system (Caliper Life Sciences, Hopkinton, MA, USA) every 3–5 days. After onset of leukaemia, mice were divided into 3 groups (n=8) and treated by oral gavage either with vehicle control (Pluronic F-68/PVP-K29/32), KPT-251 (50 mg/kg on days 1, 4, 6; 75 mg/kg on days 8, 11, 13, 15, 25, and 27 or until mice became moribund), or KPT-330 (20 mg/kg for days 1, 4, 6; and 25 mg/kg on days 8, 11, 13, 15, 25, 27, 29, 32, 34, and 36 or until mice became moribund) 3 times per week.
AML orthograft mouse model
Luciferase-expressing MV4-11 cells (2×106) were intravenously injected into 7-week-old female NSG mice. After leukaemia progression was established by BLI, mice were split into 2 groups of 9 mice and treated with either vehicle (Pluronic F-68/PVP-K29/32) or KPT-330 3 times per week at 20 mg/kg (days 1–7) and 25 mg/kg (days 8–35). Following 5 weeks of treatment, femur from one mouse from the treatment group was fixed in 10% formalin, sectioned, and paraffin-embedded. Slides were stained with haematoxylin and eosin and photographed using an Olympus BX41 microscope with Q-color5 digital camera (Olympus, Center Valley, PA).
For T-ALL and AML in vivo studies, peripheral blood counts were analysed using Hemavet 950 F instrument (Drew Scientific, Dallas, TX, USA). Survival of the KPT-treated mice was measured as the time from the start of treatment until moribund state. Survival benefit was assessed by Kaplan-Meier survival analysis.
All animal studies were performed using protocols approved by the Dana-Farber Cancer Institute Institutional Care and Use Committee.
Results
Novel SINE CRM1 antagonists promote rapid apoptosis in T-ALL cell lines in vitro
To assess the anti-leukaemic activity of SINE compounds against T-ALL cells, we tested the effects on the cell viability of KPT-185, one of the three structurally highly related SINE compounds (KPT-185, KPT-251, and KPT-330). The growth of 14 T-ALL lines, which harbour different genetic aberrations, was dramatically reduced in response to treatment with KPT-185, with 50% inhibitory concentration (IC50) values of 16–395 nM after 72 h of exposure (Fig 1A and Table I). No particular genetic abnormality was associated with sensitivity to KPT-185. To determine whether the observed decrease in cell viability is due to apoptosis, we measured the effects of KPT-185 on two sensitive T-ALL lines, Jurkat and MOLT-4, with the early apoptosis marker, Annexin V. For these experiments, Jurkat and MOLT-4 cells were co-stained with propidium iodide (PI) to monitor for the appearance of the late-apoptotic and/or necrotic events. Analysis of Annexin V-staining showed a dose-dependent increase in the percentage of apoptotic Jurkat and MOLT-4 cells upon incubation of cells with 30, 60, or 120 nM KPT-185 for 6 or 13 h when compared to DMSO-treated cells (Fig 2). Strikingly, both Jurkat and MOLT-4 cells demonstrated increased fractions of early apoptotic cells upon incubation with KPT-185 for only 6 h. For example, 69% of Jurkat cells exhibited positive Annexin V staining following incubation with 30 nM of KPT-185 for 6 h (Fig 2B). Apoptotic MOLT-4 cells were detected in response to treatment with 120 nM KPT-185 for 6 h with pronounced levels of apoptosis being observed after 13-h exposure. Specifically, 48% of MOLT-4 cells stained positive for Annexin V following treatment with 120 nM KPT-185 for 13 h (Fig 2A).
Figure 1. KPT-185 promotes cell death in T-ALL cells.

(A) Cell viability of T-ALL cell lines after treatment with KPT-185 for 72 h (Cell Titer Glo assay, Promega). (B) Relative cell viability of Jurkat cells that either overexpress BCL2 or carry control MSCV vector. Cell viability was measured using Cell Titer Glo assay following treatment with KPT-185 for 24 h. (C) Mitochondrial sensitivity of Jurkat cells that either overexpress BCL2 (Jurkat BCL2) or carry control MSCV vector (Jurkat) in presence of 0.03 μM Bim peptide following 24-hour exposure with 1 or 10 nM KPT-330. Mean +/− standard deviation values for three biological replicates are shown in A, B, and C.
Table I.
Sensitivity of 14 T-ALL cell lines to KPT-185.
| CELL LINE | Oncogene group | Major Chromosomal Rearrangement | NOTCH1 | FBXW7 | MYB | PTEN | KRAS/NRAS | TP53 | other genes | IC50 at 72 h (nM) |
|---|---|---|---|---|---|---|---|---|---|---|
| HPB-ALL | TLX3 | MUT | WT | WT | JAK1 E966V | 16 | ||||
| DU.528 | TAL1 | TAL1-TRD@ | WT | MUT | GAIN | WT | 17 | |||
| JURKAT | TAL1 | MUT | MUT | NULL | MUT | CREBBP MUT | 19 | |||
| MOLT-4 | TAL1 | MUT | WT | GAIN | NRAS G12C | MUT | 23 | |||
| SKW-3/KE-37 | MYC-TRA@ | MUT | GAIN | 29 | ||||||
| KARPAS-45 | TAL1 | MLL-FOXO4 fusion | MUT | MUT | MUT | 32 | ||||
| HS-B2 | TAL1 | LCK-TRB@, STIL-TAL1 | WT | MUT | RB1 deletion | 46 | ||||
| KOPT-K1 | LMO2-TRG@ | MUT | WT | WT | MUT | 50 | ||||
| PF-382 | TAL1 | MUT | WT | WT | NULL | NRAS G12S | MUT | 59 | ||
| CCRF-CEM | TAL1 | STIL-TAL1 | MUT | MUT | GAIN | NULL | KRAS G12D | MUT | 74 | |
| SUP-T7 | LYL1 | LYL1-TRB@ | MUT | WT | 88 | |||||
| MOLT-16 | TAL1 | MYC-TRA@, STIL-TAL1 | WT | WT | WT | WT | MUT | 112 | ||
| P12-ICHIKAWA | LMO2 | MUT | MUT | NULL | NRAS G12D | MUT | 132 | |||
| LOUCY | ETP | WT | WT | WT | MUT | 395 |
References: Catalog of Somatic Mutations in Cancer (Forbes, et al 2008, Forbes, et al 2011), Maser, et al 2007, O’Neil, et al 2007, Palomero, et al 2007, Weng, et al 2004.
IC50, 50% inhibitory concentration; MUT, mutated; WT, wild-type.
Figure 2. KPT-185 induces rapid apoptotic cell death in T-ALL cells.

Dot plot analysis of Annexin V and propidium iodide (PI) staining of the MOLT-4 (A) and Jurkat (B) cells upon treatment with either DMSO or different concentrations of KPT-185 for 6 or 13 h. Mean +/− standard deviation percentages of Annexin/PI positive cells are graphed in A and B. FITC, fluorescein isothiocyanate.
KPT-330 has exhibited similar effects on the viability of T-ALL cells. Treatment with clinical compound KPT-330 produced results very similar to KPT-185 and also reduced cell growth in MOLT-4, Jurkat, HBP-ALL, KOPTK-1, SKW-3, and DND-41 cell lines, with IC50 values of 34–203 nM after 72 h of exposure (Supplementary Fig 1A). Like KPT-185, KPT-330 elicited rapid apoptotic response in T-ALL cells (Supplementary Fig 2). These data clearly indicate that the KPT-SINE compounds promote rapid apoptosis at low nanomolar concentrations in T-ALL cells treated in vitro. Furthermore, overexpression of the anti-apoptotic protein BCL2 in Jurkat cells markedly decreased the sensitivity of cells to KPT-185 and KPT-330, pointing to the involvement of intrinsic (mitochondrial) signalling pathway in the SINE-induced apoptosis (Fig 1B and Supplementary Fig 1B). BCL2 levels remained unchanged upon treatment of Jurkat cells with KPT-330 for 12 h (Supplementary Fig 1C).
We have further explored the involvement of intrinsic apoptosis in triggering cell death in response to KPT-330 by measuring the population of cells that undergo mitochondrial outer membrane permeabilization (MOMP) upon treatment with KPT-330. For this experiment, Jurkat cells were treated with KPT-330 and the mitochondrial sensitivity was assessed using Bim peptide (Del Gaizo Moore and Letai 2012). Treatment by KPT-330 of Jurkat cells increased the priming of cells that ultimately were killed by the drug, consistent with KPT-330 killing via the mitochondrial pathway of apoptosis (Fig 1C). Importantly, overexpression of BCL2 protects against apoptosis, as demonstrated by the dramatic decrease in priming in BCL2-overexpressing Jurkat cells in response to treatment with KPT-330 (Supplementary Fig 1C).
SINE promote cell cycle arrest in G1 phase
We next performed cell cycle analysis to examine whether KPT SINE compounds alter cell cycle progression of T-ALL cells. Cell cycle distribution of Jurkat and MOLT-4 was determined by PI staining after treatment of cells with either DMSO control or increasing concentrations of KPT-185 for 24 h (Fig 3). Our data clearly demonstrated cell cycle arrest at G1 phase in the MOLT-4 cell line in response to treatment with KPT-185 as shown by an increase in the G1 fraction from 54% in the control DMSO-treated cells to 81% in KPT-treated cells (Figs 3A and B). Interestingly, the treatment with KPT-185 induced a much less prominent increase in cells in the G1 phase of the cell cycle in Jurkat cells (Figs 3C and D). Similar effects on cell cycle progression were obtained upon treatment of MOLT-4 and Jurkat cells with KPT-330 compound (Supplementary Fig 3).
Figure 3. Treatment with KPT-185 leads to cell cycle arrest in G1 phase.

(A and B) PI (DNA content) histograms of MOLT-4 cells treated with DMSO or KPT-185 (30 and 60 nM) for 24 h. (B) Mean +/− standard deviation percentages of MOLT-4 cells in each cell cycle stage are shown. (C) PI histograms of Jurkat cells upon incubation with either DMSO or KPT-185 (15 and 30 nM) for 24 h. (D) Mean +/− standard deviation percentages of Jurkat cells in each cell cycle stage are shown. The DNA histograms were analysed using FCS Express software (De Novo Software).
SINE exhibit remarkable growth suppression of T-ALL cells in vivo
To assess in vivo efficacy of KPT-SINE against human T-ALL cells, we engrafted MOLT-4 cells expressing luciferase into NOD-SCID-IL2Rgnull (NSG) mice, enabling quantification of leukaemia cells by serial BLI. For this experiment, 3×106 MOLT-4 cells were engrafted into mice and monitored for leukaemia burden using BLI of the mice following D-luciferin injection. Once leukaemia development and progression were established (Supplementary Fig 4), mice were divided into control and treatment groups and administered orally either vehicle control, KPT-251 (50 mg/kg on days 1, 4, 6; 75 mg/kg on days 8, 11, 13, 15, 25, and 27 or until mice became moribund), or KPT-330 (20 mg/kg for days 1, 4, 6; and 25 mg/kg on days 8, 11, 13, 15, 25, 27, 29, 32, 34, and 36 or until mice became moribund). After 15 days from the start of treatment, the vehicle-treated mice demonstrated logarithmic expansion of the leukaemia burden, became moribund and were sacrificed (Figs 4 and 5A and B). Remarkably, the KPT-treated mice showed striking suppression of the leukaemia cell growth, as indicated by low and relatively unchanged BLI values (Figs 4 and 5A and B). These mice were taken off treatment for one week to allow them to regain body weight, after which the treatment with SINE was reinstated (Figs 5A and C). The Kaplan-Meier survival analysis showed a significant survival benefit for the mice treated with either KPT-251 or KPT-330 as compared to vehicle-treated animals (Fig 5B). These findings establish the efficacy of novel SINE CRM1 antagonists against T-ALL cells in vivo.
Figure 4. SINE compounds dramatically suppress the growth of MOLT-4 cells engrafted into NSG mice.
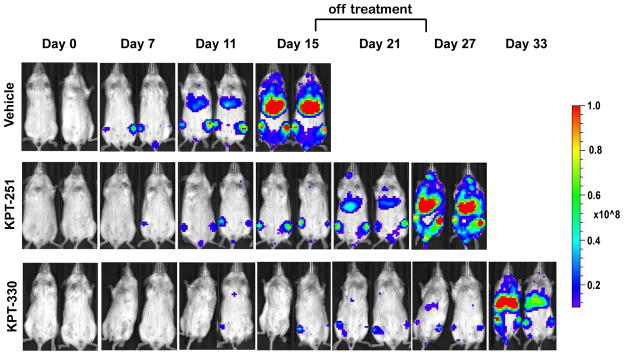
Bioluminescent images of two representative mice treated with either vehicle control, or SINE compounds, KPT-251 and KPT-330, for the indicated number of days. Scale bar shows the bioluminescence intensities. Mice were not dosed on days 18, 20, and 22 to allow for weight recovery.
Figure 5. SINE compounds inhibit growth of MOLT-4 cells in vivo and provide a significant survival benefit.

(A) Mean bioluminescence of mice treated with vehicle, KPT-251, and KPT-330 during the 36 days of treatment or until mice became moribund and were sacrificed (n=8) All treatments significantly delayed leukaemia growth by two-way ANOVA analysis (p<0.0001). (B) Kaplan-Meier survival analysis of mice treated with vehicle control, KPT-251 and KPT-330 (n=8; p< 0.0001). (C) Weights of mice treated with vehicle, KPT-251, or KPT-330 from the start of treatment until day 21 are displayed.
KPT-330 demonstrates high activity against AML cells in vivo
We next examined the anti-leukaemic activity of the clinical compound KPT-330 against AML cells in vivo, following up on our previous studies of the tool compound KPT-251 in this model (Etchin, et al 2012). For these studies, we intravenously injected AML MV4-11 cells that express luciferase into NSG mice and monitored for the development of leukaemia. Following leukaemia onset and progression, mice were orally administered KPT-330 at 20 mg/kg (days 1–7) and 25 mg/kg (days 8–35) three times a week for 5 weeks. As shown in Figure 6, treatment with KPT-330 dramatically suppressed the growth of MV4-11 cells with minimal net growth of leukaemia cells during the treatment period (Fig 6A). Survival analysis of KPT-330 demonstrated significant survival benefit in animals treated with KPT-330 as compared to mice treated with vehicle (Fig 6B; vehicle data reference from our previous study (Etchin, et al 2012)).
Figure 6. KPT-330 suppresses growth of MV4-11 cells in vivo.

(A) Mean bioluminescence of mice treated with KPT-330 for 35 days (n=9). Representations of the mean and standard error bioluminescence of vehicle-treated mice that we reported previously are shown for comparison (Etchin, et al 2012) (shaded area). (B) Kaplan-Meier survival analysis of mice treated with either vehicle control or KPT-330 (n=8; p< 0.0001). Vehicle-treated mice were moribund and were all sacrificed at the end of treatment on day 35.
SINE spare normal haematopoietic cells
To determine the selectivity of SINE against T-ALL cells, we assessed the effect of KPT-SINE on normal mouse haematopoietic cells. We obtained peripheral counts of white blood cells (WBC), neutrophils, and platelets, and measurement of haematocrit for animals treated with KPT-251 or KPT-330 after 26 days from the start of therapy. Strikingly, the treatment of mice with these compounds resulted in only minimal toxicity to circulating blood cells (Fig 7). The lack of toxicity was also observed in circulating blood counts of mice engrafted with AML MV4-11 cells after treatment with KPT-330 for 31 day (Fig 7). Furthermore, the bone marrow biopsy of mice engrafted with MV4-11 and treated with KPT-330 for 35 days demonstrated normal haematopoietic cell morphology and cellularity within the bone marrow (Fig 8), as we have previously shown for the tool compound KPT-251 (Etchin, et al 2012).
Figure 7. SINE compounds spare normal haematopoietic cells.

White blood cell (WBC; A), neutrophil (B), and platelet (C) counts, and haematocrit reading (D) after treatment of mice engrafted with MOLT-4 with either KPT-251 or KPT-330 for 26 days and mice engrafted with MV4-11 for 31 days. Mean and standard error ranges for vehicle-treated mice from our previous work (Etchin, et al 2012) are shown for reference.
Figure 8. Bone marrow of mice treated with KPT-330 show normal haematopoietic cell morphology.
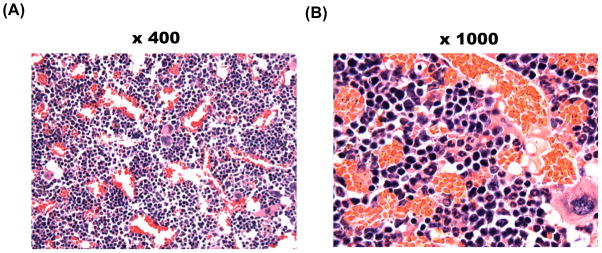
4-μm sections of bone marrow isolated from mice engrafted with MV4-11 and treated with KPT-330 for 35 days. Haematoxylin and eosin staining of bone marrow at x400 (A) and x1000 (B) magnification are shown.
Discussion
CRM1 coordinates the nuclear-cytoplasmic export of ~220 proteins (Xu et al., 2012) and several RNAs, including mediators of proliferative and prosurvival signalling pathways, and has been shown to be required for the survival of cancer cells (Turner, et al 2012). the present study established the antileukaemic effects of SINE inhibitors in T-ALL cells, both in vitro and in pre-clinical orthograft models of the disease. Our data show that low nanomolar concentrations of SINE induce rapid apoptosis of T-ALL cell lines. These compounds dramatically reduce viability in 14 T-ALL cell lines in vitro and suppress growth of human MOLT-4 T-ALL cells engrafted into immunocompromised mice. We also show that orally administered KPT-330, the clinical compound of this class, is very active in vivo against AML cells as well as T-ALL cells, with little toxicity to normal haematopoietic cells.
T-ALL lines tested in the KPT-SINE sensitivity assays represent different T-ALL subsets. These include T-ALL lines characterized by aberrant expression of TAL1 (DU528, Jurkat, MOLT-4, HSB-2, PF-382, CCRF-CEM, and MOLT-16), LYL1 (SUP-T7), TLX3 (HPB-ALL), MYC-TRA@ (SKW3), and MLL-FOXO4 (KARPAS-45), which define different molecular pathways leading to T-ALL (Armstrong and Look 2005, Ferrando and Look 2003). These lines also harbour a variety of genetic alterations in NOTCH1, NRAS, and FLT3, and the tumour suppressor genes, PTEN and TP53. Interestingly, T-ALL lines that were most sensitive to treatment with KPT-SINE, such as MOLT-4, Jurkat, HPB-ALL, DU.528, SKW-3, and KARPAS-45, do not cluster into the same oncogene group or display a similar pattern of genetic abnormalities (Table 1). For example, MOLT-4, HPB-ALL, and SKW-3 lines belong to different oncogenic subgroups, due to their expression of TAL1, TLX3, and MYC-TRA@, respectively, and yet the three lines show similar IC50 values in response to treatment with KPT-SINE. This observation suggests that common vulnerabilities exist in different oncogenic subclasses of TALL as well as AML, presumably based on the shared dependence of the transformed cells on the nuclear-cytoplasmic balance of CRM1 cargos that are broadly required for leukaemia cell survival.
Our findings demonstrated remarkable selectivity of the KPT-SINE compounds in inducing apoptosis in T-ALL as well as AML cells without causing toxicity to normal haematopoietic cells. This is probably due to the advanced design of SINE based on structure-based modelling and the in silico screening of a virtual inhibitor library, which greatly reduced the off- and on-target toxicity that has characterized leptomycin B and other CRM1 inhibitors. In this molecular modelling strategy, the structure of the NES groove of CRM1 was used as a framework for a docking-and-binding mode analysis of a small virtual library of compounds. The crystal structure of the CRM1-Ran-RanBP1 complex bound to KPT-251 shows that KPT-251 penetrates deep into the NES groove to outcompete CRM1 protein cargo and block nuclear export (Etchin, et al 2012).
The three KPT-SINE compounds (KPT-185, KPT-251, and KPT-330) presented in our study inhibit CRM1 nuclear export activity by covalent modification of the essential Cys528 residue of the NES groove of CRM1. These three compounds are structurally similar, but differ in the pharmacokinetic parameters. KPT-251 and KPT-330 display high Tmax, area under the curve, and bioavailability as compared to in vitro tool compound, KPT-185. In the toxicology studies, the primary effects of orally administered KPT-SINE compounds were dose-dependent reductions in food intake with consequent reductions in body weights, with minimal clinical gastrointestinal (GI) symptoms (no or mild non-bloody diarrhoea, no or minimal vomiting, etc.), and associated with relatively modest or no GI atrophy. These side effects can be minimized by reducing the dosing frequency of SINE to 2–3 times/week with at least 48 h between dosing and by providing the animals with food supplements. This schedule maintained in vivo activity of the KPT-SINE compounds (see Figures 5 and 6).
The mechanism underlying the ability of CRM1 inhibition to selectively eliminate cancer cells and not normal cells remains to be elucidated. Nearly all of the major tumour suppressor proteins (TSPs) such as TP53, TP73, FOXO1, -3 and -4, p21, p27, RB, BRCA1 and -2, APC, IκB, NPM, PAR4, and others are exported from the nucleus exclusively by CRM1, despite the fact that there are six other known nuclear export proteins. Therefore, CRM1 inhibition leads to the forced nuclear retention, upregulation, and activation of multiple TSPs. Restoration/reactivation of TSPs is known to selectively kill tumour cells (Martins, et al 2006, Ventura, et al 2007). Because multiple TSPs are activated at once, tumouricidal activity is largely independent of the underlying oncogenic growth drivers responsible for maintaining the neoplastic cell. One hypothesis is that TSPs initiate a “genome fidelity survey”, a survey which cancer cells will fail, leading to their selective elimination. CRM1 inhibition by SINE compounds in normal cells leads to transient cell cycle arrest without cytotoxicity, followed by fast recovery once the drug is removed. This was shown recently in vitro for normal B-cells, normal T and NK cells (Lapalombella, et al 2012). Our group has shown no toxicity of normal haematopoietic cells in mice treated with KPT-330. These results were confirmed in Good Laboratory Practice toxicology studies that formed the basis for the ongoing Phase 1 clinical trials with KPT-330 in haematological and solid tumour malignancies.
Given the rapidity of apoptosis induction observed in neoplastic leucocytes, it is possible that cancer cells are reliant on the proactive maintenance of growth-promoting and/or anti-apoptotic signalling pathways that are mediated by CRM1 nuclear export. Presumably normal cells do not share the same level of pathway dependence, leading to a greater level of resistance to inhibition of CRM1 by KPT-330. Therefore, forced nuclear sequestration of CRM1 cargos following treatment with SINEs results in the shift of balance of the pro- and anti- survival signals leading to cancer cell death, but is tolerated by normal cells, implying a differential dependence on one or more CRM1-mediated pathways. Future investigations will explore the unique mechanisms underlying the hyperactive apoptotic signalling in response to treatment with SINE compounds.
Our study findings demonstrate the clinical relevance of targeting T-ALL cells with SINE compounds, as has already been shown for AML (Etchin, et al 2012, Ranganathan, et al 2012). Importantly, we show that SINE drugs induce the rapid suppression of T-ALL cells as well as AML cells in vivo in orthograft models established in NSG immunosuppressed mice. These and other preclinical studies have provided the basis for the recent initiation of Phase I clinical trials testing the oral SINE KPT-330 in both solid tumours and haematological malignancies. These trials will reveal the maximum tolerated dose, which, based on our study, is more likely to involve anorexia, which can be mitigated by food supplements and that is highly reversible upon cessation of dosing, than it is to be based on toxicities affecting normal haematopoietic cells. Because many drugs currently used to treat leukaemia have profound haematological toxicity, if KPT-330 shows similar selective antineoplastic activity in early trials against human haematological malignancies, then it may work well in combination with existing chemotherapy regimens.
Supplementary Material
Acknowledgments
The research was supported by William Lawrence and Hughes Blanche Foundation (J.E. and T.S.), Karyopharm Therapeutics Incorporated, Alex’s Lemonade Stand (J.E.), the Leukemia and Lymphoma Society Translational grant (A.T.L.), National Cancer Institute (1K99CA157951; T.S.), the Children’s Leukemia Research Association (T.S.) and the Japan Society for the Promotion of Science (T.S.), the Kay Kendall Leukaemia Fund, UK (M.R.M.), and NIH-K08CA160660 (A.K.).
Footnotes
Conflict of Interest
S.S, M.K., and D.M. are employees of Karyopharm Therapeutics Incorporated and receive compensation and hold equity in the Company.
Author Contributions
J.E. designed experiments, analysed data, and wrote the paper. T.S., M.R.M, and A.K. helped design experiments, analyse data, and edit the manuscript. A.L.C. helped design and perform xenograft mouse experiments. S.J.R. carried out histological analysis. S.S., M.K., and D.M. designed KPT-SINE and analysed data. A.L.K. designed mouse xenograft studies and analysed the results of in vivo mouse experiments. A.T.L. guided the research presented in the paper, analysed data, and wrote the paper.
References
- Armstrong SA, Look AT. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol. 2005;23:6306–6315. doi: 10.1200/JCO.2005.05.047. [DOI] [PubMed] [Google Scholar]
- Bonazzi S, Eidam O, Guttinger S, Wach JY, Zemp I, Kutay U, Gademann K. Anguinomycins and derivatives: total syntheses, modeling, and biological evaluation of the inhibition of nucleocytoplasmic transport. J Am Chem Soc. 2010;132:1432–1442. doi: 10.1021/ja9097093. [DOI] [PubMed] [Google Scholar]
- Daelemans D, Afonina E, Nilsson J, Werner G, Kjems J, De Clercq E, Pavlakis GN, Vandamme AM. A synthetic HIV-1 Rev inhibitor interfering with the CRM1-mediated nuclear export. Proc Natl Acad Sci U S A. 2002;99:14440–14445. doi: 10.1073/pnas.212285299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Letai A. BH3 profiling - Measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett. 2012 doi: 10.1016/j.canlet.2011.12.021. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, Chook YM. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, Mansour MR, Barcelo C, McCauley D, Kauffman M, Shacham S, Christie AL, Kung AL, Rodig SJ, Chook YM, Look AT. Anti-leukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2012 doi: 10.1038/leu.2012.219. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando AA, Look AT. Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin Hematol. 2003;40:274–280. doi: 10.1016/s0037-1963(03)00195-1. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC) Curr Protoc Hum Genet. 2008;Chapter 10(Unit 10):11. doi: 10.1002/0471142905.hg1011s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M. CRM1 is responsible for intracellular transport meditted by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- Guttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Gorlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- Huang WY, Yue L, Qiu WS, Wang LW, Zhou XH, Sun YJ. Prognostic value of CRM1 in pancreas cancer. Clin Invest Med. 2009;32:E315. [PubMed] [Google Scholar]
- Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y, Goettl V, Mahoney E, Berglund C, Gupta S, Farmer A, Mani R, Johnson AJ, Lucas D, Mo X, Daelemans D, Sandanayaka V, Shechter S, McCauley D, Shacham S, Kauffman M, Chook YM, Byrd JC. Selective inhibitors of nuclear export (SINE) show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120:4621–4634. doi: 10.1182/blood-2012-05-429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, Lin E, Mani V, Jiang S, McNamara K, Zaghlul S, Edkins S, Stevens C, Brennan C, Martin ES, Wiedemeyer R, Kabbarah O, Nogueira C, Histen G, Aster J, Mansour M, Duke V, Foroni L, Fielding AK, Goldstone AH, Rowe JM, Wang YA, Look AT, Stratton MR, Chin L, Futreal PA, DePinho RA. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–971. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner T, Krause E, Vinkemeier U. Ratjadone and leptomycin B block CRM1-dependent nuclear export by identical mechanisms. FEBS Lett. 2004;576:27–30. doi: 10.1016/j.febslet.2004.08.056. [DOI] [PubMed] [Google Scholar]
- Monecke T, Guttler T, Neumann P, Dickmanns A, Gorlich D, Ficner R. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science. 2009;324:1087–1091. doi: 10.1126/science.1173388. [DOI] [PubMed] [Google Scholar]
- Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB, Murli S. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009;69:510–517. doi: 10.1158/0008-5472.CAN-08-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noske A, Weichert W, Niesporek S, Roske A, Buckendahl AC, Koch I, Sehouli J, Dietel M, Denkert C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer. 2008;112:1733–1743. doi: 10.1002/cncr.23354. [DOI] [PubMed] [Google Scholar]
- O’Neil J, Tchinda J, Gutierrez A, Moreau L, Maser RS, Wong KK, Li W, McKenna K, Liu XS, Feng B, Neuberg D, Silverman L, DeAngelo DJ, Kutok JL, Rothstein R, DePinho RA, Chin L, Lee C, Look AT. Alu elements mediate MYB gene tandem duplication in human T-ALL. J Exp Med. 2007;204:3059–3066. doi: 10.1084/jem.20071637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, Castillo M, Nagase S, Cordon-Cardo C, Parsons R, Zuniga-Pflucker JC, Dominguez M, Ferrando AA. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–1210. doi: 10.1038/nm1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, Walker A, Klisovic R, Blum W, Caligiuri M, Croce CM, Marcucci G, Garzon R. Pre-clinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012;120:1765–1773. doi: 10.1182/blood-2012-04-423160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci U S A. 2010;107:12895–12900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara K, Saito N, Sato T, Suzuki A, Hasegawa Y, Friedman JM, Kufe DW, Vonhoff DD, Iwami T, Kawabe T. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood. 2011;118:3922–3931. doi: 10.1182/blood-2011-01-333138. [DOI] [PubMed] [Google Scholar]
- Shen A, Wang Y, Zhao Y, Zou L, Sun L, Cheng C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery. 2009;65:153–159. doi: 10.1227/01.NEU.0000348550.47441.4B. discussion 159–160. [DOI] [PubMed] [Google Scholar]
- Siddiqui N, Borden KL. mRNA export and cancer. Wiley Interdiscip Rev RNA. 2012;3:13–25. doi: 10.1002/wrna.101. [DOI] [PubMed] [Google Scholar]
- Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–1032. doi: 10.1016/j.bcp.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JG, Marchion DC, Dawson JL, Emmons MF, Hazlehurst LA, Washausen P, Sullivan DM. Human multiple myeloma cells are sensitized to topoisomerase II inhibitors by CRM1 inhibition. Cancer Res. 2009;69:6899–6905. doi: 10.1158/0008-5472.CAN-09-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Watt PJ, Maske CP, Hendricks DT, Parker MI, Denny L, Govender D, Birrer MJ, Leaner VD. The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer. 2009;124:1829–1840. doi: 10.1002/ijc.24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Neck T, Pannecouque C, Vanstreels E, Stevens M, Dehaen W, Daelemans D. Inhibition of the CRM1-mediated nucleocytoplasmic transport by N-azolylacrylates: structure-activity relationship and mechanism of action. Bioorg Med Chem. 2008;16:9487–9497. doi: 10.1016/j.bmc.2008.09.051. [DOI] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Wach JY, Guttinger S, Kutay U, Gademann K. The cytotoxic styryl lactone goniothalamin is an inhibitor of nucleocytoplasmic transport. Bioorg Med Chem Lett. 2010;20:2843–2846. doi: 10.1016/j.bmcl.2010.03.049. [DOI] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Xu D, Farmer A, Chook YM. Recognition of nuclear targeting signals by Karyopherin-beta proteins. Curr Opin Struct Biol. 2010;20:782–790. doi: 10.1016/j.sbi.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargos. Mol Biol Cell. 2012a;23:3673–3676. doi: 10.1091/mbc.E12-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Farmer A, Collett G, Grishin NV, Chook YM. Sequence and structural analyses of nuclear export signals in the NESdb database. Mol Biol Cell. 2012b;23:3677–3693. doi: 10.1091/mbc.E12-01-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Dong Y, Lin F, Zhao H, Shen Z, Chen P, Sun YJ, Tang LN, Zheng SE. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep. 2009;21:229–235. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.