

Abstract
Application of headspace solid-phase microextraction (HS-SPME) coupled with high-resolution gas chromatographic (HRGC) analysis was studied for determining lactones in wines. Six different SPME fibers were tested, and the influence of different factors such as temperature and time of desorption, ionic strength, time of extraction, content of sugar, ethanol, tannins and anthocyanins, and pH and influence of SO2 were studied. The proposed HS-SPME-GC method is an appropriate technique for the quantitative analysis of γ-butyrolactone, γ-hexalactone, trans-whiskey lactone, γ-octalactone, cis-whiskey lactone, γ-nonalactone, γ-decalactone, δ-decalactone, and γ-undecalactone in wines. Method reproducibility and repeatability ranged between 0.6 and 5.2% for all compounds. Detection limit for γ-butyrolactone was 0.17 mg/L and a few μg/L for the rest of the compounds. The optimized method has been applied to several wine samples.
1. Introduction
Among volatile components of wine, lactones and particularly the γ-lactones and whiskey lactones play an important role in terms of their contribution to the aroma.
γ-Lactones series, δ-lactones, and whiskey lactones are the most abundant lactones in wines and the more sensory important lactones.
Lactone smell is usually described as “fruity” or “coconut-like, fruity” (γ-hexalactone); “coconut-like” (γ-octalactone); “peach-like, milky” (γ-decalactone), or “fruity, sweet floral” (γ-dodecalactone). These compounds are formed by cyclisation of the corresponding γ-hydroxycarboxylic acids [1].
Lactones are among the most important compounds contributing to the sensory characteristics of wines aged in oak wood. Whiskey lactones and some volatile phenols coming directly from wood have been recognized as important odor active compounds in Madeira wines [1, 2].
They are already present in natural oak and their content increases due to ageing. From an organoleptic point of view, they are the most important lactones extractable from oak casks. Furthermore, they have been reported as potential aging markers in Madeira wines [3, 4].
Oak species, geographical origin, silvicultural treatment of tree, and processing of wood have influence on volatile composition of barrel wood. These volatile compounds are susceptible to migrate from oak wood to wine. Although the volatile composition of wine undergoes an evolution during bottle aging, this is carried out in such a way that the most important characteristics spread into wine from wood and remain until the end of bottle aging. These conclusions emphasize the importance of species and geographical origin of oak wood in the volatile composition of wines during aging [5].
Other less studied lactones such as pantolactone and the 4-carbethoxy-ç-butyrolactone have been found in samples submitted to oxidative ageing specially in barrel-aged wine [6].
Chemical structure defines their sensorial and chemical properties [6, 7]. Lactones aromatic descriptors are influenced by the type of aromatic ring, functional groups, and substituent length chain [8]. Synergic effects can exist due to smell similarities [9].
cis-Whiskey-lactone isomer is a stronger odorant than the trans-isomer reflected by its olfaction threshold and is an important contribution to wine aroma [10]. The rest of lactones can influence wines like Zinfandel, Pinot Noir, Merlot, and Cabernet Sauvignon, particularly, γ-nonalactone and δ-decalactone with smell values higher than threshold [8].
γ-Butyrolactone is considered as an inductor of physical and psychic addictions and has been classified as psychotropic by FDA (US Food and Drug Administration) [11].
Different methods have been proposed for extraction of wine. Analytical methods for gas chromatography determination of lactones need a previous concentration step due to the low concentrations existing in wine [6, 10, 12]. However, these methodologies are not free from artefacts [13], and none of them has been optimized for a wide group of lactones.
A great number of wine aroma compounds have been characterised as lactones, many of them extracted by SPME. Published works account for particular compounds like diacetyl [14], sotolon [15], families of compounds such as sulphides and disulphides [16, 17], aldehydes [18–20], esters [21], alcohols [22], butyltin compounds [23], terpenes [24, 25], acetals [26], fatty acids [27, 28], or wide sets of different volatile compounds [29, 30].
The aim of this work was to apply the GC-MS technique combined with automatic headspace (HS) SPME to develop a new method to determine a set of lactones in wine (γ-butyrolactone, γ-hexalactone, γ-octalactone, whiskey-lactone, γ-nonalactone, γ-decalactone, δ-decalactone, and γ-undecalactone) and to apply the method to determine lactone content in white and red wines samples.
2. Experimental
2.1. Chemicals and Reagents
The following lactones were studied (CAS number in brackets): γ-butyrolactone [96-48-0], γ-hexalactone [695-06-7], γ-octalactone [104-50-7], cis- and trans-whiskey-lactones [39212-23-2], γ-nonalactone [104-61-0], γ-decalactone [706-14-9], δ-decalactone [705-86-2], and γ-undecalactone [104-67-6]. γ-Heptalactone [105-21-5] and 3,4-dimethylphenol [95-65-8] were used as internal standards (IS). These standards with purity above 99% were supplied by Aldrich (Steinheim, Germany, and Milwaukee, WI, USA) and Fluka (Buchs, Switzerland). Sodium chloride [7647-14-5] supplied by Merck was used to control ionic strength. Ethanol (analytical reagent grade; Merck, Darmstadt, Germany) [64-17-5] and Milli-Q water (Millipore, Bedford, USA) were used as solvents. Commercial tannins and anthocyanins were purchased from Agrovin (Alcazar de San Juan, Spain); sucrose [57-50-1], tartaric acid [87-69-4], potassium disulfite [16731-55-8], and sodium hydroxide [1310-73-2] (analytical reagent grade) were purchased from Panreac (Barcelona, Spain).
Individual stock standard solution in ethanol of γ-butyrolactone, γ-hexalactone, γ-heptalactone, γ-octalactone, γ-nonalactone, γ-decalactone, δ-decalactone, and γ-undecalactone and 3,4-dimethylphenol (5000 mg/L) and whiskey-lactone (10000 mg/L) were prepared. Concentrated synthetic wine solution containing L(+)-tartaric acid [87-69-4] (11 g/L) and ethanol (13%) was prepared and adjusted to pH 3.2 with sodium hydroxide and was used to prepare all synthetic test solutions. Stock solution of potassium disulfite (5.55 g/L) in water and tannins (25.39 g/L) with anthocyanins (127.00 g/L) in ethanol (Agrovin, Ciudad Real, Spain) were also prepared. Individual internal standard stock solution containing γ-heptalactone (13.12 mg/L) and 3,4-dimethylphenol (12.93 mg/L) were prepared using ethanol (13%). The rest of the solutions were prepared by mixture and dilution of these stock solutions.
All parameters have been optimized using a synthetic wine solution containing concentrations of lactones. Table 1 shows those concentrations and the low and high level concentrations used for recoveries calculations.
Table 1.
Concentrations of synthetic solutions containing lactones.
| Compound | Concentrations (μg/L) | ||
|---|---|---|---|
| Low level | High level | Optimization | |
| γ-Butyrolactone | 14400 | 36200 | 20300 |
| γ-Hexalactone | 146 | 365 | 98.9 |
| γ-Heptalactone | 494 | ||
| Whiskey-lactone I | 48.3 | 144 | 100 |
| γ-Octalactone | 3.96 | 9.90 | 9.9 |
| Whiskey-lactone II | 48.3 | 144 | 100 |
| γ-Nonalactone | 19.8 | 59.5 | 100 |
| γ-Decalactone | 3.83 | 9.56 | 9.9 |
| δ-Decalactone | 124 | 310 | 96.8 |
| γ-Undecalactone | 1.92 | 4.80 | 9.9 |
| 3,4-Dimethylphenol | 491 | ||
Either individual stock standard solutions or real wine samples were prepared in 2 mL vials adding 0.77 mL of sample and 0.03 mL of internal standard solution. The vials were tightly capped with PTFE-lined cap and shaken for 10 min at 200 min−1.
2.2. Equipment
Regularly verified pipettes and class A volumetric flasks were used in solution preparation. A precision balance (Sartorius BP 210-S), a pH meter (WTW, pH 197-S), Milli-Q gradient A10 (Millipore), and a mechanical shaker (Selecta, Rotabit) were used in the study.
2.3. SPME Fibers
Six fibers coated with different stationary phases and various film thicknesses were purchased from Supelco (Bellefonte): polydimethylsiloxane 100 μm (PDMS/100), carboxen-polydimethylsiloxane 75 μm (CAR/PDMS), polydimethylsiloxane-divinylbenzene 65 μm (PDMS/DVB), polyacrylate 85 μm (PA), Carbowax-divinylbenzene 65 μm (CW/DVB), and divinylbenzene-carboxen-polydimethylsiloxane 50/30 μm (DVB/CAR/PDMS). All fibers were conditioned according to manufacturer recommendations.
2.4. Chromatography
The analyses were carried out on a 3800 GC gas chromatograph equipped with an 8200 Standalone autosampler, a 1079 split/splitless injector, and a mass spectrometry detector Saturn 2000 (Varian, Walnut Creek, CA, USA). Injections were performed in splitless mode, using a 0.75 mm I.D. liner which improved GC resolution. Ionization mode used was electronic impact.
Separations were performed using a DB-WAXETR capillary column (60 m, 0.25 mm I.D., 0.5 μm film thickness) (J&W Scientific) with an injector temperature of 250°C (valid for all fibers) and an oven temperature program of 100°C (5 min), 8°C/min, 240°C, and 240°C (7.5 min). Carrier gas was helium at 2 mL/min flow. Peak identification was accomplished using retention time and experimental spectra obtained from individual standard solutions and confirmed using the NIST mass spectra database (Standard Reference Data of National Institute of Standards and Technology, USA).
The GC-MS transfer line temperature was 240°C. The MS operated in electron impact mode at 70 eV and collected data at a rate of 1.0 scans/s over a mass range of m/z 25–350. The ion source temperature was 200°C, the detector voltage was set to 1500 V, and the detector temperature was 300°C.
2.5. Experimental Design
Chromatographic conditions have been set using both synthetic solutions and wine samples to ensure a good chromatographic resolution and no coelution of compounds.
Extraction time and reproducibility, extraction temperature, desorption time and temperature, and ionic strength were optimized as a need of establishing basic instrumental parameters and simultaneously for selecting the appropriate SPME fiber.
The following steps are designed to reveal and correct possible matrix effect due to specific parameters such as phenolics, sugar, pH, sulphur dioxide, and ethanol.
3. Results and Discussion
3.1. Extraction Time
Figure 1 shows an example chromatogram of a spiked wine sample for the chromatographic conditions cited above. It shows no peak coelution and the mixture is eluted in 25 min.
Figure 1.
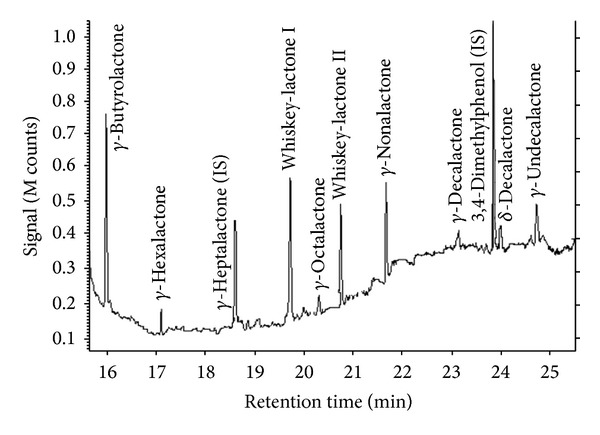
TIC chromatogram obtained with a PA fiber for a synthetic wine spiked with the different analytes.
Table 2 shows retention time, molecular weight, and enthalpy of vaporization of analytes.
Table 2.
| Compound | Retention time (min) | Molecular weight (g/mol) | ΔH
V
(KJ/mol) |
|---|---|---|---|
| γ-Butyrolactone | 15.97 | 86.04 | 54.40 ± 0.40 |
| γ-Hexalactone | 17.09 | 114.14 | 57.20 ± 0.30 |
| γ-Heptalactone (IS) | 18.60 | 128.17 | 62.30 ± 0.30 |
| Whiskey-lactone I (trans-whiskey-lactone) |
19.73 | 156.22 | 48.36 |
| γ-Octalactone | 20.31 | 142.20 | 66.46 ± 0.40 |
| Whiskey-lactone II (cis-whiskey-lactone) |
20.74 | 156.22 | 48.36 |
| γ-Nonalactone | 21.67 | 156.22 | 70.30 ± 0.20 |
| γ-Decalactone | 23.14 | 170.25 | 75.60 ± 0.30 |
| 3,4-Dimethylphenol (IS) | 23.86 | 122.16 | 85.70 ± 0.10*** |
| δ-Decalactone | 24.01 | 170.25 | 74.20 ± 0.25 |
| γ-Undecalactone | 24.73 | 184.28 | 79.51 ± 0.40 |
***Enthalpy of sublimation.
γ-Lactones show a clear direct relation between molecular weight, enthalpy of vaporization, and retention time. Compounds with higher molecular weight are less volatile and elute later. cis- and trans-Whiskey lactones elute earlier and have lower vaporization enthalpy than their C9 isomer γ-nonalactone probably due to structural differences, although δ-decalactone shows a similar behaviour as γ-decalactone.
Because of the kinetic nature of the extraction process, it is heavily influenced by fiber type and extraction time. Optimization of both parameters is the first step when building a microextraction method. Since the final aim of this work is to determine the analytes in sweet wines, which can have a high content in sugars (up to 200 g/L) and other many compounds, direct immersion mode leads to a rapid degradation of the fiber surface. To avoid this effect, all the studies were performed in headspace mode.
In order to establish optimal extraction parameters, the six fibers named above were studied. Experiences were made varying extraction time from 15 min to 90 min using a spiked synthetic wine.
Figure 2 shows normalized peak areas (absolute peak area/analyte concentration) for different analytes as a function of extraction time for the fibers studied.
Figure 2.

Extraction profiles of the different analytes versus extraction time for the different fibers.
CAR/PDMS fiber was immediately discarded because it offered very wide and low peaks shapes resulting in a poor peak resolution with lactone peaks overlapping between them.
As can be seen, γ-butyrolactone shows the lower peak area for all fibers studied. On the contrary, γ-undecalactone is used to show the higher peak area.
Almost all fibers show a fast initial increase in extraction during the first 15 min, and then it slows down until 45 min. After these time differences among compounds appear, some compounds like γ-heptalactone reach a saturation state shown as a horizontal graph line. On the contrary, some others like γ-octalactone, whiskey-lactone II, or δ-decalactone continue increasing with extraction time without reaching a saturation state. This is a general pattern for DVB/CAR/PDMS fiber and most compounds except γ-butyrolactone and γ-heptalactone. Finally, some analytes decrease in peak area at high extraction time probably due to competition for fiber active points with other compounds.
The extraction performance increases all over homologous series of n-γ-lactones. This is a general behaviour in all studied fibers.
PDMS fiber presents the lower extraction ability for γ-hexalactone, γ-heptalactone, γ-decalactone, and δ-decalactone; so this fiber was discarded from further optimization.
3.2. Extraction Reproducibility
A reproducibility study was made injecting five times a wine synthetic solution spiked with lactones (Table 3).
Table 3.
RSD (%) (n = 5) of the relative areas of studied compounds obtained with studied fibers.
| Compound | PA | DVB/CAR/PDMS | PDMS/DVB | CW/DVB |
|---|---|---|---|---|
| γ-Butyrolactone | 8.14 | 36.43 | 14.41 | 8.47 |
| γ-Hexalactone | 8.24 | 11.76 | 6.56 | 8.94 |
| Whiskey-lactone I | 1.26 | 31.44 | 1.47 | 4.33 |
| γ-Octalactone | 1.72 | 26.08 | 2.45 | 2.56 |
| Whiskey-lactone II | 2.19 | 27.04 | 3.39 | 4.27 |
| γ-Nonalactone | 1.40 | 46.96 | 1.80 | 0.56 |
| γ-Decalactone | 2.45 | 58.36 | 4.40 | 3.24 |
| δ-Decalactone* | 2.91 | 16.77 | 5.23 | 3.71 |
| γ-Undecalactone | 2.78 | 62.05 | 3.96 | 3.37 |
*3,4-Dimethylphenol as IS.
As can be observed, DVB/CAR/PDMS shows the worst RSD values for most compounds; so it was discarded for the rest of the studies.
Focusing on the rest of the three fibers, all of them present good overall reproducibility. Nevertheless, the value of 14.41 for γ-butyrolactone with PDMS/DVB fiber is bad enough for discarding this fiber.
Finally, PA and CW/DVB present a similar behaviour in terms of extraction and reproducibility; so any of them would be adequate. As CW/DVB has been discontinued by the manufacturer, PA fiber was selected as the best fiber for extracting lactones in wine samples. So, the rest of this study was done using PA fiber.
3.3. Extraction Temperature
Extraction temperature plays an important role in extraction but in two opposite ways. Increasing temperature produces desorption of molecules on the fiber decreasing sensibility. Simultaneously increasing temperature modifies liquid-gas equilibrium enriching gas phase with analytes [31, 32].
An extraction temperature, study temperature, was done using a synthetic wine spiked with lactones. Temperature was set to 60°C, 42°C, and 25°C using 45 min extraction time. Results of normalized peak area (peak area/concentration) versus temperatures are shown in Figure 3.
Figure 3.
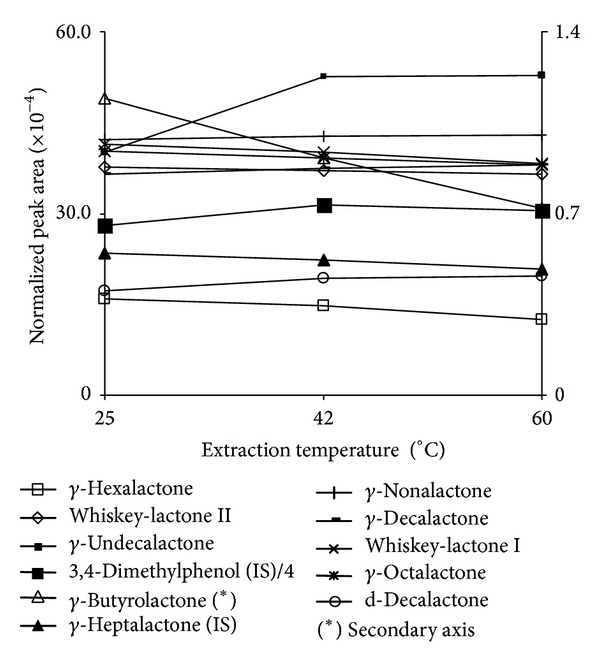
Temperature influence on the extraction of the different analytes on a PA fiber.
Figure 3 shows that increasing temperature leads to a decreasing extraction of both whiskey lactones and γ-lactones from γ-butyrolactone to γ-octalactone. The rest of the compounds show a moderate increase in extraction specially at 42°C. As lower temperatures enlarge fiber life and increasing temperature does not have a great effect, 25°C was selected as extraction temperature.
3.4. Desorption Time and Temperature
Desorption time and temperature were also tested within the range recommended by manufacturer. Injections were made at 250°C and 300°C desorption temperatures and 2 min, 5 min, and 10 min desorption time using the rest of selected parameters. Results are shown in Table 4.
Table 4.
Normalized peak areas at different temperatures and desorption time.
| Compound | 250°C | 300°C | ||||
|---|---|---|---|---|---|---|
| 2 min | 5 min | 10 min | 2 min | 5 min | 10 min | |
| γ-Butyrolactone | 2033 | 7644 | 10782 | 3820 | 9353 | 11068 |
| γ-Hexalactone | 20681 | 129160 | 155069 | 53359 | 143513 | 155513 |
| γ-Heptalactone (IS) | 38894 | 185530 | 230138 | 81918 | 209111 | 230259 |
| Whiskey-lactone I | 110605 | 364837 | 413042 | 199319 | 387667 | 413463 |
| γ-Octalactone | 111825 | 360337 | 407674 | 197209 | 384203 | 408474 |
| Whiskey-lactone II | 90756 | 320900 | 380063 | 183103 | 351145 | 380190 |
| γ-Nonalactone | 119260 | 374890 | 417670 | 212936 | 389701 | 418269 |
| γ-Decalactone | 106828 | 327823 | 363834 | 186756 | 339222 | 364786 |
| 3,4-Dimethylphenol (IS) | 444709 | 999600 | 1090969 | 490860 | 1032004 | 1092175 |
| δ-Decalactone | 16550 | 130347 | 175598 | 49251 | 138751 | 175672 |
| γ-Undecalactone | 135335 | 367100 | 412196 | 217489 | 381118 | 411959 |
Values of normalized peak area show increasing values with time for all analytes indicating that short desorption time leads to incomplete desorption.
On the other hand, higher temperature shows higher areas until 10 min desorption time. Taking into account that fiber life is longer at lower desorption temperatures, we selected 250°C as desorption temperature and 10 min as desorption time. Blank injections showed no memory effect in desorption for any analyte.
3.5. Ionic Strength
Ionic strength affects analyte extraction, particularly those of polar character. In order to study this effect, increasing quantities of solid NaCl were added to spiked synthetic wine. Quantities of 0 mL, 80 mL, 160 mL, 200 mL, and 240 mg in 0.77 mL of sample were added to reach 0%, 10.3%, 20.7%, 25.9%, and 31.1% NaCl solutions, respectively. Results are shown in Figure 4.
Figure 4.
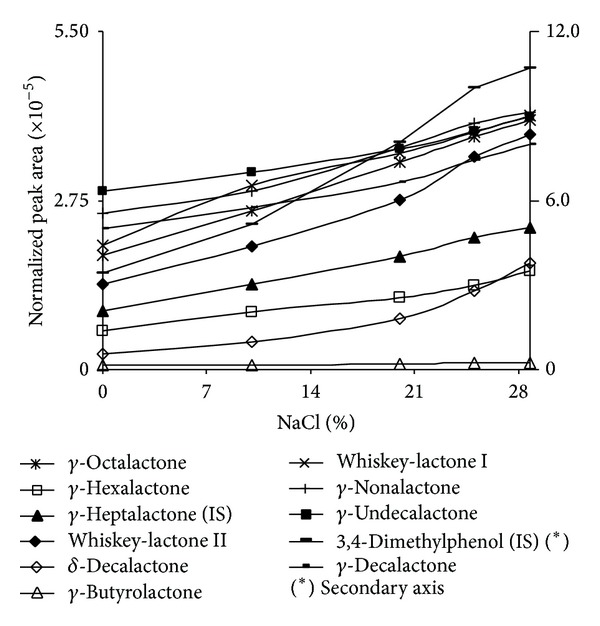
Influence of ionic strength on the extraction of the different analytes on a PA fiber.
Increasing ionic strength produces an increase in extraction. The best values are those obtained by saturation of NaCl and this condition is selected for further studies.
3.6. Phenolics, Sugar, pH, and Sulphur Dioxide Effect
Wine sample matrix has a wide variety of compounds that can affect extraction process. So it is necessary to study the effect of pH, phenolic compounds, sugar, sulphur dioxide, and ethanol content as influencing extraction process.
Polyphenol content presents wide variations in wines especially from white to red wine. An extraction study was made in order to test its influence in the process.
Synthetic wine spiked solution was prepared with tannins concentrations ranging from 0 g/L to 1 g/L and anthocyanins from 0 g/L to 5 g/L. Obtained results do not show tannins or anthocyanins influence in the extraction.
In the same way, sugar content varies widely from dry to sweet wine reaching even values higher than 200 g/L. Spiking synthetic wine with concentrations up to 200 g/L showed no influence. This is in coincidence with results reported for other compounds [18]. Wine pH usually ranges between 3 and 4 depending on grape variety and kind of wine; it is higher in red than in white wines. Different pH implies variation in dominant chemical species when acid-base properties are present. Solid-phase microextraction only extracts molecular components so ionized acids or bases remain unextracted. An extraction study was made varying pH from 3 to 4. Results showed no variation in extraction process for analytes studied.
Sulphur dioxide is a commonly used additive in wine making due to its antiseptic, antioxidant, and antioxidasic properties. Sulphur dioxide added to wine reacts with carbonyl compounds forming the so-called “combined sulphur,” especially with acetaldehyde, changing the expected concentration of free carbonyl compounds. Added sulphur dioxide quantities also change from red to white wine. To study this effect, synthetic wine solutions spiked with lactones and sodium metabisulphite ranging up to 200 mg/L were extracted. All lactones showed no influence in extraction in the range studied.
3.7. Ethanol Effect
Behind water, ethanol is the major component in wines. Obviously, ethanol is extracted in fiber and effectively competes with analytes by active positions. This effect has been previously described by several authors [2, 19, 33–38]. Alcoholic degree usually ranges from 9% to 15%, but most of wines vary between 11% and 14%. So the ethanol influence was studied over synthetic wine spiked solutions with alcoholic degree in this range. Results are shown in Figure 5.
Figure 5.
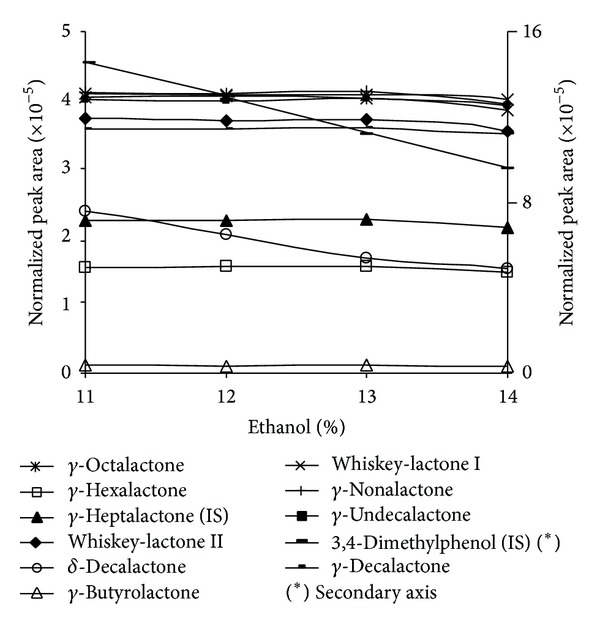
Normalized peak areas of the different analytes versus alcoholic degree on a PA fiber.
Figure 5 shows that δ-decalactone presented a strong decrease with increasing alcoholic degree. A similar pattern is presented by the internal standard 3,4-dimethylphenol.
The rest of lactones including the internal standard γ-heptalactone show no variation with ethanol increase until 13%. Higher ethanol concentrations produce a small decrease in extraction. According to these patterns, 3,4-dimethylphenol was chosen as internal standard for quantifying δ-decalactone and γ-heptalactone for the rest of analytes [16].
Figure 6 shows the relative peak areas. Relative peak areas were calculated, dividing individual standard peak area between internal standard peak areas in each chromatogram. As can be seen, relative areas appear now independent from alcoholic degree. So, internal standard quantification was selected using internal standards named above.
Figure 6.
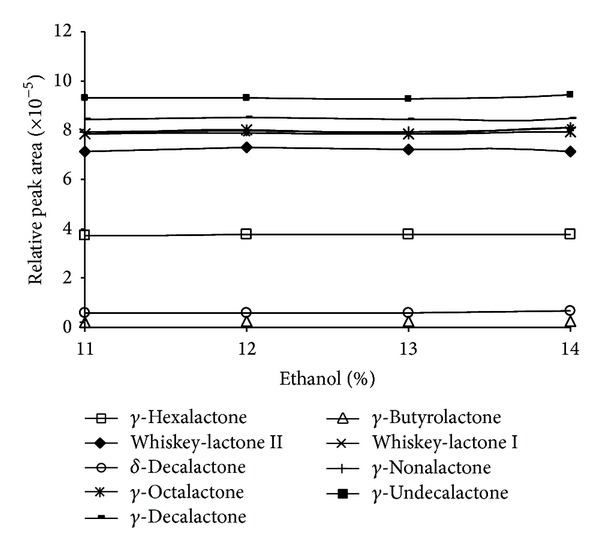
Relative peak areas versus alcoholic degree.
3.8. Validation
Method validation was developed in terms of linearity, detection and quantification limits, precision, and matrix effect influence.
Calibration curves were elaborated using eight synthetic wine solutions spiked with lactones, internal standards, and using the parameters selected above. Table 5 summarizes the results. All correlation coefficients show an excellent linearity.
Table 5.
O.O, slope, R 2, and linear range of lactones (n = 8).
| Compound | Intercept | Slope | R 2 | Linear range (μg/L) |
|---|---|---|---|---|
| γ-Butyrolactone | 0.001 ± 0.001 | 0.110 ± 0.001 | 0.999 | 0.17–60.26* |
| γ-Hexalactone | 0.003 ± 0.002 | 1.445 ± 0.009 | 0.999 | 11–609 |
| Whiskey-lactone I | 0.003 ± 0.002 | 3.425 ± 0.003 | 0.999 | 1–401 |
| γ-Octalactone | 0.003 ± 0.001 | 3.418 ± 0.003 | 0.999 | 1–21 |
| Whiskey-lactone II | 0.005 ± 0.002 | 3.272 ± 0.002 | 0.999 | 1–401 |
| γ-Nonalactone | 0.004 ± 0.001 | 3.665 ± 0.003 | 0.997 | 2–206 |
| γ-Decalactone | 0.001 ± 0.001 | 3.294 ± 0.002 | 0.997 | 1–20 |
| δ-Decalactone | 0.001 ± 0.001 | 0.311 ± 0.001 | 0.997 | 4–517 |
| γ-Undecalactone | 0.004 ± 0.001 | 3.724 ± 0.003 | 0.999 | 1–15 |
*mg/L.
Detection and quantification limits were calculated as the concentration corresponding to 3 and 10 times signal/noise, respectively. Values are shown in Table 5. Most of analytes present low detection and quantification limits. The highest value is presented by γ-butyrolactone; but it is much lower than concentrations found in wines. In every case, detection limit is lower than odor thresholds reported.
Method repeatability and reproducibility were obtained analyzing 5 replicates of synthetic wine spiked with lactones intermediate concentration of calibration. The 5 replicates were repeated during 3 different days. Results are shown in Table 6. Both repeatability and reproducibility fall below 5% for all analytes.
Table 6.
| Compound | LOD (μg/L) |
LOQ (μg/L) |
Odor threshold (μg/L) |
Repeatability RSD (%) |
Reproducibility RSD (%) |
|---|---|---|---|---|---|
| γ-Butyrolactone | 170.97 | 569.93 | 35000 | 0.63 | 1.93 |
| γ-Hexalactone | 10.51 | 35.04 | 359000 | 2.98 | 2.57 |
| Whiskey-lactone I | 0.97 | 3.24 | 790 | 3.54 | 4.41 |
| γ-Octalactone | 1.19 | 3.98 | 7 | 4.31 | 3.96 |
| Whiskey-lactone II | 0.60 | 2.02 | 67 | 4.13 | 5.25 |
| γ-Nonalactone | 2.11 | 7.05 | 30 | 3.24 | 2.73 |
| γ-Decalactone | 0.86 | 2.89 | 88 | 2.50 | 2.78 |
| δ-Decalactone | 4.17 | 13.92 | 386 | 3.19 | 4.56 |
| γ-Undecalactone | 0.63 | 2.10 | 60 | 2.88 | 4.45 |
Wine is a complex matrix that includes hundreds of different compounds besides those studied here. So it is necessary to perform a matrix effect study to evidence the existence of extraction interferences [7, 31–33]. In order to establish these interferences, a recovery study was realized.
Three samples of white and red wine were spiked with lactones at two different concentration levels shown in Experimental section. Results are shown in Table 7.
Table 7.
Mean (%) and RSD (%) of recoveries.
| Compound | Low level | High Level | ||||||
|---|---|---|---|---|---|---|---|---|
| Mean | Mean | RSD | RSD | Mean | Mean | RSD | RSD | |
| White | Red | White | Red | White | Red | White | Red | |
| γ-Butyrolactone | 189.1 | 191.9 | 1.45 | 2.56 | 189.4 | 198.6 | 4.23 | 0.14 |
| γ-Hexalactone | 88.1 | 86.7 | 3.05 | 5.62 | 88.6 | 88.5 | 1.80 | 0.77 |
| Whiskey-lactone I | 144.3 | 145.2 | 9.11 | 1.98 | 144.2 | 143.1 | 1.05 | 1.46 |
| γ-Octalactone | 76.8 | 81.3 | 2.06 | 3.21 | 78.1 | 80.1 | 0.65 | 2.10 |
| Whiskey-lactone II | 76.8 | 80.1 | 1.36 | 8.15 | 74.7 | 76.9 | 1.26 | 1.65 |
| γ-Nonalactone | 93.2 | 101.6 | 3.73 | 1.79 | 94.2 | 107.0 | 1.26 | 1.05 |
| γ-Decalactone | 136.5 | 142.8 | 0.96 | 3.45 | 137.5 | 147.4 | 2.33 | 0.38 |
| δ-Decalactone | 109.3 | 108.2 | 1.79 | 4.58 | 108.5 | 109.1 | 1.27 | 2.01 |
| γ-Undecalactone | 112.0 | 116.5 | 0.99 | 2.10 | 111.0 | 112.6 | 1.31 | 1.17 |
γ-Nonalactone and δ-decalactone are free from matrix effect showing recoveries in the range of 100 ± 10%. The most affected compounds are γ-butyrolactone, whiskey-lactone I, and γ-decalactone. Matrix effect was revealed to be similar for white and red wines.
Table 8 presents average recovery for all wines and all concentration levels. These values were used as a correction factor to obtain real concentration for analytes in real samples.
Table 8.
Mean of recoveries and RSD.
| Compound | Mean (%) | RSD (%) |
|---|---|---|
| γ-Butyrolactone | 192.3 | 3.01 |
| γ-Hexalactone | 88.0 | 2.98 |
| Whiskey-lactone I | 144.2 | 4.09 |
| γ-Octalactone | 79.1 | 2.95 |
| Whiskey-lactone II | 77.1 | 4.52 |
| γ-Nonalactone | 99.0 | 6.23 |
| γ-Decalactone | 141.1 | 3.72 |
| δ-Decalactone | 108.8 | 2.37 |
| γ-Undecalactone | 113.0 | 2.27 |
3.9. Analysis of Wine Samples
Optimized method was applied to 72 wine samples including white, red, and rosé wines. Table 9 shows average concentration values (μg/L) and standard deviations.
Table 9.
Concentration mean and SD (μg/L).
| Compound |
White wines1
(n = 35) |
Rosé wines2
(n = 8) |
Red wines3
(n = 29) |
Significative differences (P < 0.05) |
|||
|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | ||
| γ-Butyrolactone | 26287 | 8478 | 25940 | 6553 | 32652 | 6403 | 1–3, 2-3 |
| γ-Hexalactone | 200 | 89 | 202 | 72 | 211 | 67 | — |
| Whiskey-lactone I | 10.60 | 14.38 | 5.44 | 2.20 | 45.73 | 34.42 | 1–3, 2-3 |
| γ-Octalactone | 5.41 | 2.64 | 5.63 | 2.20 | 6.95 | 3.13 | — |
| Whiskey-lactone II | 20.31 | 50.46 | d-nq | — | 138.44 | 111.13 | 1–3, 2-3 |
| γ-Nonalactone | 14.77 | 7.39 | 19.98 | 5.64 | 42.09 | 27.45 | 1–3, 2-3 |
| γ-Decalactone | d-nq | — | 4.16 | 2.91 | 3.85 | 2.65 | 1-2, 1–3 |
| δ-Decalactone | 157 | 72 | 149 | 37 | 228 | 96 | 1–3, 2-3 |
| γ-Undecalactone | nd | — | d-nq | — | d-nq | — | — |
nd: not detected; d-nq: detected not quantified.
As expected, γ-butyrolactone is the most abundant compound for the three kinds of wine. For the rest of analytes, γ-hexalactone and δ-decalactone present the higher medium values and γ-octalactone, γ-decalactone, and γ-undecalactone the lower.
3.10. Statistical Analysis
ANOVA for these samples revealed that γ-hexalactone, γ-octalactone, and γ-undecalactone presented no statistically significant differences among the three kinds of wines but red wine and rosé wines presented statistically significant differences values for γ-butyrolactone, whiskey-lactones I and II, γ-nonalactone, and δ-decalactone. Finally, red wines had contents in γ-decalactone significantly higher than white wines.
4. Conclusions
Solid-phase microextraction is a suitable technique for determining concentrations of different lactones in wine matrix. The proposed methodology covers the range of concentrations usually found in wines with an acceptable uncertainty. The use of two internal standards corrects the influence of ethanol content. Matrix effect exists but can be corrected using both standard addition calibration and experimental correction factors, allowing the quantification of all the compounds studied using gas chromatography, mass spectrometry detection, and electronic impact.
Acknowledgments
This work has been funded by the Spanish CICYT (Comisión Interministerial de Ciencia y Tecnología), Project AGL 2003-04911/ALI. The authors acknowledge Bodegas Viñatigo (Tenerife) for sample supply.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Câmara JS, Alves MA, Marques JC. Changes in volatile composition of Madeira wines during their oxidative ageing. Analytica Chimica Acta. 2006;563(1-2):188–197. [Google Scholar]
- 2.Campo E, Ferreira V, Escudero A, Marqués JC, Cacho J. Quantitative gas chromatography-olfactometry and chemical quantitative study of the aroma of four Madeira wines. Analytica Chimica Acta. 2006;563(1-2):180–187. [Google Scholar]
- 3.Perestrelo R, Barros AS, Câmara JS, Rocha SM. In-depth search focused on furans, lactones, volatile phenols, and acetals as potential age markers of Madeira wines by comprehensive two-dimensional gas chromatography with time-of-flight mass spectrometry combined with solid phase microextraction. Journal of Agricultural and Food Chemistry. 2011;59(7):3186–3204. doi: 10.1021/jf104219t. [DOI] [PubMed] [Google Scholar]
- 4.Câmara JS, Alves MA, Marques JC. Multivariate analysis for the classification and differentiation of Madeira wines according to main grape varieties. Talanta. 2006;68(5):1512–1521. doi: 10.1016/j.talanta.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Fernández de Simón B, Cadahía E, Hernández T, Estrella I. Evolution of oak-related volatile compounds in a Spanish red wine during 2 years bottled, after aging in barrels made of Spanish, French and American oak wood. Analytica Chimica Acta. 2006;563(1-2):198–203. [Google Scholar]
- 6.Schneider R, Baumes R, Bayonove C, Razungles A. Volatile compounds involved in the aroma of sweet fortified wines (vins doux naturels) from grenache noir. Journal of Agricultural and Food Chemistry. 1998;46(8):3230–3237. [Google Scholar]
- 7.Guth H. Identification of character impact odorants of different white wine varieties. Journal of Agricultural and Food Chemistry. 1997;45(8):3022–3026. [Google Scholar]
- 8.Rocha SM, Rodrigues F, Coutinho P, Delgadillo I, Coimbra MA. Volatile composition of Baga red wine: assessment of the identification of the would-be impact odourants. Analytica Chimica Acta. 2004;513(1):257–262. [Google Scholar]
- 9.Nakamura S, Crowell EA, Ough CS, Totsuka A. Quantitative analysis of γ-nonalactone in wines and its threshold determination. Journal of Food Science. 1988;53(4):1243–1244. [Google Scholar]
- 10.Pollnitz AP, Jones GP, Sefton MA. Determination of oak lactones in barrel-aged wines and in oak extracts by stable isotope dilution analysis. Journal of Chromatography A. 1999;857(1-2):239–246. doi: 10.1016/s0021-9673(99)00785-2. [DOI] [PubMed] [Google Scholar]
- 11.Tateo F, Bononi M. Determination of gamma-butyrolactone (GBL) in foods by SBSE-TD/GC/MS. Journal of Food Composition and Analysis. 2003;16(6):721–727. [Google Scholar]
- 12.Guth H. Quantitation and sensory studies of character impact odorants of different white wine varieties. Journal of Agricultural and Food Chemistry. 1997;45(8):3027–3032. [Google Scholar]
- 13.Pollnitz AP, Pardon KH, Sykes M, Sefton MA. The effects of sample preparation and gas chromatograph injection techniques on the accuracy of measuring guaiacol, 4-methylguaiacol and other volatile oak compounds in oak extracts by stable isotope dilution analyses. Journal of Agricultural and Food Chemistry. 2004;52(11):3244–3252. doi: 10.1021/jf035380x. [DOI] [PubMed] [Google Scholar]
- 14.Hayasaka Y, Bartowsky EJ. Analysis of diacetyl in wine using solid-phase microextraction combined with gas chromatography-mass spectrometry. Journal of Agricultural and Food Chemistry. 1999;47(2):612–617. doi: 10.1021/jf9807667. [DOI] [PubMed] [Google Scholar]
- 15.Câmara JS, Marques JC, Alves MA, Silva Ferreira AC. 3-Hydroxy-4,5-dimethyl-2(5H)-furanone levels in fortified Madeira wines: relationship to sugar content. Journal of Agricultural and Food Chemistry. 2004;52(22):6765–6769. doi: 10.1021/jf049547d. [DOI] [PubMed] [Google Scholar]
- 16.Mestres M, Sala C, Martí MP, Busto O, Guasch J. Headspace solid-phase microextraction of sulphides and disulphides using Carboxen-polydimethylsiloxane fibers in the analysis of wine aroma. Journal of Chromatography A. 1999;835(1-2):137–144. doi: 10.1016/s0021-9673(98)01050-4. [DOI] [PubMed] [Google Scholar]
- 17.Mestres M, Martí MP, Busto O, Guasch J. Simultaneous analysis of thiols, sulphides and disulphides in wine aroma by headspace solid-phase microextraction-gas chromatography. Journal of Chromatography A. 1999;849(1):293–297. doi: 10.1016/s0021-9673(99)00538-5. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q, O’Reilly J, Pawliszyn J. Determination of low-molecular mass aldehydes by automated headspace solid-phase microextraction with in-fibre derivatisation. Journal of Chromatography A. 2005;1071(1-2):147–154. doi: 10.1016/j.chroma.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 19.Bueno M, Culleré L, Cacho J, Ferreira V. Chemical and sensory characterization of oxidative behavior in different wines. Food Research International. 2010;43(5):1423–1428. [Google Scholar]
- 20.Armada L, Fernández E, Falqué E. Influence of several enzymatic treatments on aromatic composition of white wines. LWT—Food Science and Technology. 2010;43(10):1517–1525. [Google Scholar]
- 21.Rodríguez-Bencomo JJ, Conde JE, Rodríguez-Delgado MA, García-Montelongo F, Pérez-Trujillo JP. Determination of esters in dry and sweet white wines by headspace solid-phase microextraction and gas chromatography. Journal of Chromatography A. 2002;963(1-2):213–223. doi: 10.1016/s0021-9673(02)00551-4. [DOI] [PubMed] [Google Scholar]
- 22.Rodríguez-Bencomo JJ, Conde JE, García-Montelongo F, Pérez-Trujillo JP. Determination of major compounds in sweet wines by headspace solid-phase microextraction and gas chromatography. Journal of Chromatography A. 2003;991(1):13–22. doi: 10.1016/s0021-9673(03)00211-5. [DOI] [PubMed] [Google Scholar]
- 23.Azenha M, Vasconcelos MT. Headspace solid-phase micro-extraction gas chromatography-mass detection method for the determination of butyltin compounds in wines. Analytica Chimica Acta. 2002;458(1):231–239. [Google Scholar]
- 24.de la Calle García D, Reichenbächer M, Danzer K, Hurlbeck C, Bartzsch C, Feller K-H. Use of solid-phase microextraction-capillary-gas chromatography (SPME-CGC) for the varietal characterization of wines by means of chemometrical methods. Fresenius’ Journal of Analytical Chemistry. 1998;360(7-8):784–787. [Google Scholar]
- 25.Câmara JS, Herbert P, Marques JC, Alves MA. Varietal flavour compounds of four grape varieties producing Madeira wines. Analytica Chimica Acta. 2004;513(1):203–207. [Google Scholar]
- 26.Câmara JS, Marques JC, Alves A, Silva Ferreira AC. Heterocyclic acetals in Madeira wines. Analytical and Bioanalytical Chemistry. 2003;375(8):1221–1224. doi: 10.1007/s00216-003-1818-0. [DOI] [PubMed] [Google Scholar]
- 27.Zeng Z, Zhang H, Zhang T, Tamogami S, Chen JY. Analysis of flavor volatiles of glutinous rice during cooking by combined gas chromatography-mass spectrometry with modified headspace solid-phase microextraction method. Journal of Food Composition and Analysis. 2009;22(4):347–353. [Google Scholar]
- 28.Alanis P, Ashkan S, Krauter C, Campbell S, Hasson AS. Emissions of volatile fatty acids from feed at dairy facilities. Atmospheric Environment. 2010;44(39):5084–5092. [Google Scholar]
- 29.Demyttenaere JCR, Dagher C, Sandra P, Kallithraka S, Verhé R, de Kimpe N. Flavour analysis of Greek white wine by solid-phase microextraction-capillary gas chromatography-mass spectrometry. Journal of Chromatography A. 2003;985(1-2):233–246. doi: 10.1016/s0021-9673(02)01467-x. [DOI] [PubMed] [Google Scholar]
- 30.Francioli S, Guerra M, López-Tamames E, Guadayoi JM, Caixach J. Aroma of sparkling wines by headspace/solid phase microextraction and gas chromatography/mass spectrometry. American Journal of Enology and Viticulture. 1999;50(4):404–408. [Google Scholar]
- 31.Emel’yanenko VN, Kozlova SA, Verevkin SP, Roganov GN. Vapour pressures and enthalpies of vapourization of a series of the γ-lactones. Journal of Chemical Thermodynamics. 2008;40(6):911–916. [Google Scholar]
- 32.Emel’yanenko VN, Kozlova SA, Verevkin SP, Roganov GN. Vapour pressures and enthalpies of vaporization of a series of δ-lactones. Journal of Chemical Thermodynamics. 2007;39(1):10–15. [Google Scholar]
- 33.Zhang Z, Pawliszyn J. Analysis of organic-compounds in environmental-samples by headspace solid-phase microextraction. HRC: Journal of High Resolution Chromatography. 1993;16(12):689–692. [Google Scholar]
- 34.Urruty L, Montury M. Influence of ethanol on pesticide extraction in aqueous solutions by solid-phase microextraction. Journal of Agricultural and Food Chemistry. 1996;44(12):3871–3877. [Google Scholar]
- 35.Fischer C, Fischer U. Analysis of cork taint in wine and cork material at olfactory subthreshold levels by solid phase microextraction. Journal of Agricultural and Food Chemistry. 1997;45(6):1995–1997. [Google Scholar]
- 36.de la Calle D, Reichenbächer M, Danzer K, Hurlbeck C, Bartzsch C, Feller K. Analysis of wine bouquet components using headspace solid-phase microextraction-capillary gas chromatography. HRC: Journal of High Resolution Chromatography. 1998;21(7):373–377. [Google Scholar]
- 37.Liu M, Zeng Z, Tian Y. Elimination of matrix effects for headspace solid-phase microextraction of important volatile compounds in red wine using a novel coating. Analytica Chimica Acta. 2005;540(2):341–353. [Google Scholar]
- 38.de la Calle García D, Reichenbächer M, Danzer K, Hurlbeck C, Bartzsch C, Feller K-H. Investigations on Wine Bouquet Components by Solid-Phase Microextraction-Capillary Gas Chromatography (SPME-CGC) using Different Fibers. HRC: Journal of High Resolution Chromatography. 1997;20(12):665–668. [Google Scholar]