

Abstract
T helper (TH) cells orchestrate appropriate cellular and humoral immune responses to a wide variety of pathogens and are central to the success of vaccines. However, their dysregulation can cause allergies and autoimmune diseases. The TH cell universe is characterized by a diversity of distinct cell types, such as TH1, TH2, TH17 cells, regulatory T cells, and T follicular helper cells, each featuring specific functions and gene expression programs, but also by plasticity among the different TH cell subsets. Here, we review recent advances and emerging concepts on how microRNAs, small endogenously expressed oligonucleotides that modulate gene expression, fit into the regulatory networks that govern T helper cell fate decisions and regulate their effector functions.
Introduction
Following their activation by antigen and costimulatory signals, CD4+ T cells can differentiate into several distinct types of effector T helper (TH) cells. Since the discovery of TH1 and TH2 cells approximately 25 years ago1, an ever-increasing number of TH cell subsets have been described. Historically, TH cell subsets were distinguished based on the cytokines they secrete following restimulation with antigen. For instance, TH1 cells produce interferon-γ (IFNγ which is required for clearance of intracellular pathogens, whereas TH2 cells produce interleukin-4 (IL-4), IL5 and IL-13, which mediate immune responses against helminths. However, as the diversity of subsets increased and distinct subsets were found to express overlapping sets of cytokines, ‘lineage-defining’ or ‘master’ transcription factors have become important classifiers of Th cell subsets. For a long time, TH1 and TH2 cells have been widely referred to as stably differentiated lineages. However, the recent emergence of additional subsets, such as peripherally derived regulatory T (TReg) cells, T follicular helper (TFH) cells, TH17, TH9 and TH22 cells, forced some reconsideration in the field and focused attention on the plasticity of TH cells2–5. It has become clear that a complex network of transcription factors, epigenetic changes, and post-transcriptional regulators is responsible for the development and maintenance of the different T helper cell subsets and their characteristic gene expression programs6–10.
MicroRNAs (miRNAs) are small (~21 nucleotide) endogenously expressed RNAs that regulate gene expression. They are sequentially processed from longer transcripts by the RNase III enzymes DROSHA and DICER and exert their function by guiding the Argonaute (AGO) protein-containing miRNA-induced silencing complex (miRISC) [G] to specific target mRNAs by complementary base pairing (Box 1). The miRISC destabilizes target mRNAs and reduces their translation into protein11, 12. Whether an mRNA is targeted by miRISC depends on several factors, including alternative splicing and poly-A site usage, and interplay with RNA binding proteins. Moreover, the expression of miRNAs is regulated at several stages during their biogenesis, often involving feedback from their target gene products13. Each miRNA has many targets, and several mRNAs are subject to regulation by more than one miRNA (Box 2). Thus, similarly to transcription factors, miRNAs are integral parts of gene expression networks that determine cell identity and function. Conventional methods for the study of coding genes have been complemented by a large number of miRNA-specific technologies that improve our ability to measure miRNA expression, determine their biological functions, and empirically identify their mRNA targets (Box 3).
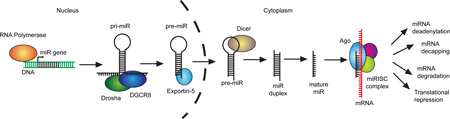
Box 1 | miRNA biogenesis and function.
MicroRNA genes are transcribed into primary miRNAs (pri-miRNAs) by RNA polymerase II. Pri-miRNAs are bound by Dgcr8 and processed by the RNase III activity of Drosha into hairpin structures called pre-miRNAs. Exportin-5 shuttles pre-miRNAs from the nucleus into the cytoplasm where the RNase III Dicer cleaves off the pre-miRNA’s hairpin loop. The resulting duplex segregates and the mature single-stranded miRNA associates with Argonaute and other accessory proteins to form the miRNA-induced silencing complex (miRISC), which mediates translational repression and increased degradation of its mRNA targets. A mature miRNA bound to an Argonaute (Ago) protein forms the core of the miRISC. Ago recruits other protein complexes that antagonize translation and deadenylate the targeted mRNA129. This ultimately leads to mRNA decapping and degradation, so the effect of miRNA repression can be observed at both the protein and mRNA level. The miRNA provides specificity through complementary base pairing with target mRNAs11. Nucleotides in positions 2–8 from the 5′ end of a miRNA, termed the seed sequence, are a major determinant of target recognition. However, complementarity in the 3′ half of the miRNA does contribute to binding, and ‘seedless’ targets that rely on non-seed sequences for binding also exist. Most functional miRNA binding sites occur in the 3′ UTR of target mRNAs, and many of these are deeply conserved, indicating co-evolution of miRNAs and their targets. These principles have been exploited to develop algorithms for bioinformatic prediction of miRNA targets. Though useful for hypothesis generation, these programs remain imperfect. Predicted targets must be confirmed experimentally, and many true targets are missed.
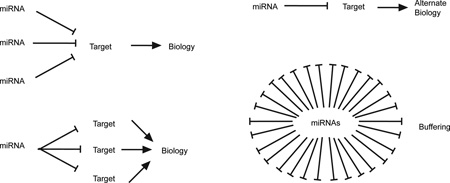
Box 2 | Regulation of gene expression networks by miRNAs.
The magnitude of repression of direct miRNA targets is relatively low – less than 50% at both the protein and mRNA level in the vast majority of cases. Nevertheless, miRNAs can have big effects on biological processes. Target genes are frequently inhibited by a combination of several miRNAs. Similarly, each miRNA targets many genes at the same time. In some cases, miRNA effects are amplified through coordinated repression of several target mRNAs in a common pathway. Small changes in individual targets can also have big effects in systems governed by feedback mechanisms and activation thresholds. In this context, miRNAs can be important to maintain effective gene silencing in the face of “leaky” transcription that produces a functionally significant amount of mRNA in an inappropriate cell type or biological condition. Finally, the combined effect of miRNAs acting on many target genes can buffer noise associated with transcriptional processes and effectively set thresholds for signals that induce programmed changes in gene expression, such as those that mediate TH cell differentiation and plasticity.
Box 3 | Methods for studying miRNAs and their function.
Several methods exist to profile and quantify miRNA expression in cells, tissues and body fluids. These range from measuring single miRNAs by conventional or multiplexed qPCR in purified T cell subsets to genome-wide analysis of miRNA expression by microarrays and RNA-sequencing 74, 130. Online databases catalog miRNA sequences and annotation131, and both bioinformatically predicted132 and experimentally validated133 miRNA targets.
MicroRNA function can be studied by a variety of gain- and loss-of-function technologies. Conditional deletion of key molecules of the miRNA biogenesis pathway showed that miRNAs as a whole regulate many aspects of CD4+ T cell behavior, but the current challenge is to understand how each miRNA contributes to these effects. Two consortia have generated freely available miRNA-deficient ES cells134 and conditional knock-out mice135 that will be very helpful in advancing our understanding of miRNA function in vivo. Specific miRNA inhibition can also be achieved using a variety of transfectable or self-deliverable synthetic antagonists (often called ‘antagomirs’), or by overexpressing target mRNAs that act as ‘sponges’ of miRNAs, decreasing their activity against endogenous targets136. Overexpression of specific miRNAs can be achieved with transfectable synthetic miRNA mimics, expression constructs, viral vectors, or transgenesis in mice.
Understanding the mechanism of miRNA function requires methods to identify its targets137. Genome-wide transcriptional profiling of specific miRNA-deficient or overexpressing cells and controls can be used to assess global target gene expression changes, though it is often difficult to distinguish direct and indirect targets with this approach. Direct targeting can be validated with 3´ UTR luciferase reporter assays, but validating functional relevance requires genetic rescue experiments. Recently, biochemical techniques such as high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP)138 and photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP)139, have been developed to identify RNA sequences that bind to RNA-binding proteins such as Argonaute proteins.
Because of their evolutionary conserved abundance and wide range of functions, miRNAs are emerging as an integral part of the cellular machinery that confers overall robustness to biological processes14. There is also growing evidence that miRNAs are critical modulators of development and function in the immune system and that miRNAs regulate important aspects of TH cell differentiation15–21.
In this review, we first discuss how miRNAs are important for modulating naive CD4+ T cell maintenance, activation and expansion and how miRNA expression itself is regulated during these processes. We then examine how miRNAs regulate the differentiation of distinct effector TH cell subsets and their lineage-defining effector functions by focusing on TH1, TH2, TH17 and TFH cells. Finally, we review the literature that is available on the miRNA-mediated regulation of TReg cell differentiation and function, before concluding with an outlook on future challenges and the clinical relevance of miRNA biology.
MicroRNA regulation of T cell activation
Effector TH cell differentiation is highly influenced by the strength of T cell antigen receptor (TCR) stimulation, the nature of the co-stimulatory molecules expressed on antigen presenting cells, and by the cytokine environment 9. Early systematic miRNA profiling studies in cells of the hematopoietic system identified cell type-specific patterns of miRNA expression that suggested an important role for miRNAs in cell lineage specification and effector functions22. These findings were corroborated and extended in subsequent miRNA profiling studies that used small RNA sequencing or high throughput quantitative PCR to profile CD4+ and CD8+ T cells23, naive and in vitro activated CD4+ T cells24, several human TH cell subsets25 and a broad range of hematopoietic cell types26.
Alterations in miRNA-mediated control following T cell activation
Not surprisingly, T-cell specific deletion of essential factors in the miRNA biogenesis pathway, such as DICER27–29, DROSHA30 and DGCR831, reduced the survival and proliferation of T cells following activation (Figure 1a). However, miRNA-deficient CD4+ T cells also exhibit a surprising increase in effector TH cell differentiation and cytokine production27, 30, 32. The enhanced differentiation of miRNA-deficient T cells indicates that miRNAs are critically involved in the maintenance of the naïve T cell state27, 30, 32. This raises the question of how T cells overcome the miRNA barrier when they receive signals that induce differentiation.
Figure 1. miRNA regulation of T helper cell activation.

a | MicroRNAs are important regulators of effector T cell differentiation, including T cell activation, acquisition of effector functions such as cytokine production, and T cell proliferation (top panel). Genetic ablation of key molecules of the miRNA biogenesis pathway in CD4+ T cells underlines the importance of miRNAs for these processes (lower panel). Activation of microRNA-deficient CD4+ T cells results in increased and aberrant cytokine production, reduced cell proliferation. b | Mechanistic overview illustrating miRNA participation in regulatory networks that control T cell activation, expansion, and effector cell differentiation. Note that specific miRNAs are both regulated targets and upstream regulators of signaling pathways that govern T cell behavior. Selected miRNA target genes are indicated. TEFF, effector T cell; Tnaive, naïve T cell.
One possibility is that key mRNAs that are held in check by miRNA control in naïve T cells are transcriptionally induced to such a large degree upon activation that their repression by miRNAs is rendered relatively insignificant. However, at least two other contributing mechanisms have been discovered. Proliferating T cells utilize alternative polyadenylation sites to produce mRNAs with shortened 3′ UTRs and thus fewer miRNA binding sites33. In addition, T cell activation induces the ubiquitylation of AGO2 and its proteasome-dependent turnover, reducing its half-life to less than 2 hours32. This results in a global reduction in miRNA abundance, and also rapidly remodels the miRNA repertoire to favor expression of a few miRNAs that are transcriptionally upregulated upon T cell activation.
Among the miRNAs that are substantially downregulated upon T cell activation are some, such as miR-29, that inhibit the differentiation of particular TH cell subsets31, 34, 35. Others, such as miR-125, appear to inhibit effector T cell differentiation in general. miR-125b is highly expressed in human naïve CD4+ T cells as compared with various memory T cell populations, and it inhibits several genes involved in T cell differentiation, including IFNG, IL2RB, IL10RA, and PRDM1 (encoding BLIMP1)25 (Figure 1b). Interestingly, miR-125b is not regulated in this way in mouse CD4+ T cells25. Instead, its close relative miR-125a is highly expressed in naïve mouse T cells and rapidly downregulated upon activation32. In addition, among the miR-125b target genes identified in human T cells, only Prdm1 has retained the predicted miR-125 binding site during the evolution of its respective murine gene25. Attention to species differences like these is important, since the miRNomes of human and murine lymphocytes and their targets are apparently under evolutionary pressure and are not strictly conserved.
miRNA-mediated regulation of TCR signaling
During their development in the thymus, T cells undergo the processes of positive selection [G] and negative selection [G]. In the periphery, activation of mature naïve T cells that encounter foreign antigen leads to clonal selection and differentiation into effector cells. In all of these processes, the sensitivity of a TCR to recognize cognate peptides has important implications for the fate of the T cell. It is not surprising that the signaling cascade downstream of TCR activation is highly regulated at multiple levels, and several miRNAs have been shown to critically participate in this regulation (Figure 1b).
The miR-181 miRNA family [G] is one such regulator of TCR signaling with clear effects on T cell development and homeostasis. Inhibition of miR-181 in thymocytes reduces TCR sensitivity and impairs positive and negative selection in the thymus36. Conversely, overexpression of miR-181a in mature T cells augments sensitivity to peptide antigen in the periphery36. miR-181 targets multiple phosphatases that dampen TCR signals, including SH2 domain-containing protein tyrosine phosphatase 2 (SHP2), protein tyrosine phosphatase, non-receptor type 22 (PTPN22), dual-specificity protein phosphatase 5 (DUSP5) and DUSP6, thereby reducing the TCR engagement threshold for cellular responses36 (Figure 1b). As a consequence, an endogenous peptide that positively selects T cells in the thymus but fails to activate specific T cell responses in the periphery can become an autoantigen when T cells develop in the absence of miR-181 activity37.
The miR-181 family consists of 4 unique mature miRNAs encoded in three miRNA clusters [G], but one of these, miR-181a1b1, is particularly important in T cells. Three independent studies identified defects in thymocyte development in mir181a1b1-deficient mice38–40. Natural killer T (NKT) cell development is particularly dependent on miR-18138, 40. Providing an agonistic TCR ligand40 or eliminating the miR-181 target phosphatase and tensin homologue (PTEN)38 rescued the defective NKT cell development of mir181a1b1-deficient mice, indicating an important role for miR-181 in regulating PI3K signaling. In addition to its effect on TCR signals, miR-181 targets Nrarp (Notch-regulated ankyrin repeat protein) and dampens Notch signals that are critical for thymocyte development39.
miR-181a1b1-deficiency also compromises peripheral T cell homeostasis and TCR signaling38, 39. In addition, naturally occurring changes in miR-181 expression have been implicated in the age-associated decline in CD4+ T cell immunity. Decreased miR-181a expression with age is associated with increased DUSP6 expression and reduced TCR sensitivity41. Neonatal CD4+ T cells expressed more miR-181a than did adult peripheral blood CD4+ T cells, concomitant with increased activation-induced calcium flux and extracellular signal-regulated kinase (ERK) phosphorylation. However, neonatal cells display an independent defect in AP1-dependent transcription, ultimately resulting in impaired T cell activation42. Taken together, these data suggest that regulation of miR-181a expression may influence age-dependent changes in T cell responsiveness, but additional mechanisms have a dominant influence in special contexts, as in the case of human neonates.
miR-146a is an important feedback regulator of nuclear factor-κB (NF-κB) signaling (Figure 1b). It is abundantly expressed in human memory T cells, and further induced by NF-κB p50 activation in response to TCR engagement43–45. In turn, miR-146a targets TRAF6 and IRAK1, forming a negative feedback loop that controls the intensity and duration of NF-κB signaling46. Absence of this feedback loop causes spontaneous myeloproliferation and myeloid cell tumorigenesis in miR-146a-deficient mice45. In T cells, it causes hyperresponsiveness to TCR signals and failure to resolve T cell-mediated inflammation46. Together with its role in TReg cells (discussed below), these effects make miR-146a a powerful inhibitor of autoimmunity.
miRNA-mediated control of T cell proliferation and survival
The miR-17~92 cluster comprises six miRNAs from four miRNA families that regulate several aspects of T cell activation and activated T cell fate. This cluster and the related miR-106a~363 and miR-106b~25 clusters are all expressed in CD4+ and CD8+ T cells32, 47. Overexpression of miR-17~92 in T cells causes lymphoproliferation and an autoimmune-like disease characterized by elevated serum antibody levels and production of autoantibodies48. Both miR-17 and miR-92 family miRNAs can enhance T cell proliferation in the absence of other miRNAs31. Another miR-17~92 cluster member, miR-19, positively regulates activated T cell survival. These miRNAs mediate their effects through many direct target genes, including PTEN and the pro-apoptotic protein BIM48, 49 (Figure 1b).
PI3K signaling and downstream regulators of metabolic activity are subject to intense miRNA control in T cells. PTEN is a common target of several miRNA families that are differentially expressed during T cell development and activation, including miR-17, miR-19, miR-181 and miR-2138, 48, 50. Another phosphatidylinositol phosphatase, SHIP-1, is a validated direct target of miR-15549, 51, the miRNA most highly induced by TCR stimulation32, 52, 53. miR-155 deficiency reduces T cell-dependent humoral and cellular immune responses, likely through its activity on many mRNA targets53–56. Transcriptome-wide biochemical analysis of Ago2 binding to mRNAs in activated primary mouse T cells identified over 300 sites containing miR-155 seed recognition sequences, and almost 200 miR-155-dependent sites, including a large fraction (~40%) that lack canonical miR-155 seed recognition sequences49. Thus, the regulation of T cell behavior by miR-155, and by extension miRNAs in general, involves a large network of co-regulated target mRNAs.
IL-2 is another important regulator of CD4+ T cell metabolism and proliferation57. It is produced by activated T cells and can act in an autocrine fashion, and it is also frequently added to in vitro T helper cell cultures as a growth factor. miR-146a has been implicated as a negative regulator of IL-2 production in activated human CD4+ T cells44, and miR-9 has been described as a positive regulator of IL-2 production through a mechanism involving direct targeting of PRDM1, which encodes the transcriptional repressor B lymphocyte-induced maturation protein 1 (BLIMP1)58, 59. In addition, miR-182 enhances clonal selection downstream of IL-2 by inhibiting forkhead box O1 (FOXO1), a transcription factor that is highly expressed in naïve T cells and limits T cell proliferation60. At early time points during T cell stimulation, FOXO1 is inactivated by phosphorylation downstream of TCR signals. Later in the response, however, IL-2 produced by activated T cells induces miR-182, which represses Foxo1 post-transcriptionally (Figure 1b).
In summary, miRNAs can have powerful effects on biological processes by regulating genes that control signaling thresholds. Several miRNAs operate in this capacity to regulate T cell development, homeostasis, and activation. These effects have important consequences for T cell immune function, and they are complemented by miRNA regulation of particular T cell differentiation pathways and effector mechanisms.
MicroRNA regulation of effector T helper cells
MicroRNAs are increasingly recognized as important modulators of TH cell fate decisions and effector functions. Their ‘fine-tuning’ activity is particularly well suited to act early in the response when small changes in the expression of key genes can have a big effect on cell fate. In addition, miRNAs can regulate the plasticity and effector functions of differentiated TH cell subsets.
TH1 cell differentiation and function
TH1 cells, orchestrate immune responses against viruses, intracellular pathogens and tumors. The first evidence for miRNA regulation of TH cell differentiation came from studies with DICER-deficient CD4+ T cells, which exhibit increased differentiation into effector cells expressing the TH1 cell lineage-defining transcription factor T-bet and its transcriptional target, the TH1 cell hallmark cytokine IFNγ27. T cells lacking DROSHA or DGCR8 display a very similar phenotype of aberrant TH1 cell differentiation30, 31.
A functional screen in DGCR8-deficient CD4+ T cells revealed that among CD4+ T cell-expressed miRNAs, miR-29a and miR-29b are the most potent inhibitors of TH1 cell differentiation and IFNγ expression31. miR-29 is highly expressed in naïve T cells, and it inhibits TH1 cell differentiation through multiple direct targets (Figure 2). T-bet is one of the direct miR-29 targets in CD4+ T cells, but miR-29 also targets the closely related transcription factor eomesodermin (EOMES), which is usually not expressed in CD4+ T cells31. EOMES regulates IFNγ production in natural killer (NK) cells and CD8+ T cells, and in the absence of miRNAs it is also inappropriately expressed in CD4+ T cells and contributes to the aberrant IFNγ31. miR-29 may also directly target the Ifng mRNA35. Transgenic mice expressing a miRNA sponge [G] that reduces miR-29a-mediated control exhibited higher serum IFN-γ levels and more effective clearance of the intracellular bacterium Listeria monocytogenes35. A recent study confirmed the importance of miR-29 in restraining IFN-γ expression by T cells using mir-29ab1 knock-out mice34.
Figure 2. miRNA regulation of IFNγ production.

IFN-γ production and signaling in Th1 cells is regulated by several miRNAs at distinct levels. The lineage determining transcription factor T-bet induces expression of the Th1 hallmark cytokine IFN-γ. T-bet is induced by TCR signaling, by IL-12 via STAT4 signaling, and by IFN-γ itself in a positive feedback loop via STAT1. miR-146a directly targets STAT1 and the NFκB signaling molecules TRAF6 and IRAK1. TCR activation induces miR-155, which in turn downregulates negative regulators of cytokine signaling SHIP1 and SOCS1. However, miR-155 has also been proposed to induce downregulation of IFNγR1. miR-29 limits Th1 cell differentiation and IFN-γ production by targeting the mRNAs encoding T-bet, Eomes, and IFN-γ itself.
Several other miRNAs regulate TH1 cell differentiation and function (Figure 2). miR-146a-deficient CD4+ T cells produce more IFNγ in vitro and in vivo46, 51, 61. As discussed above, miR-146a inhibits T cell responses in general by targeting TRAF6 and IRAK146. However, it may also specifically regulate TH1 differentiation by targeting signal transducer and activator of transcription 1 (STAT1)61. By contrast, miR-155 enhances both TH1 and TH17 cell–dependent tissue inflammation55. Among its many identified targets in T cells49, miR-155 inhibits the negative regulators of cytokine signaling SOCS1 and SHIP151, 62. In addition, miR-155 targets Ifngr1 mRNA and may be responsible for the downregulation of IFNγ receptor in activated CD4+ T cells that differentiate into TH1 cells52. Epistasis experiments showed that miR-155 deficiency is dominant over miR-146 deficiency, as cells lacking both miRNAs are as defective in IFN-γ expression and anti-tumor immunity as those lacking miR-155 only51.
The miR-17~92 cluster also impacts TH1 cell differentiation. CD4+ T cells freshly isolated from peripheral lymph nodes of 10- to 12-month-old mice that expressed a transgene encoding the human miR-17~92 cluster showed a dramatic increase in the percentage of IFNγ-expressing cells48. Similarly, naïve miR-17~92 transgenic CD4+ T cells produced higher amounts of IFNγ under TH1 polarizing conditions in vitro48. Conversely, T cell-specific deletion of the miR-17~92 cluster in mice resulted in a gene dose-dependent reduction of IFNγ-secreting CD4+ T cells63. Reconstitution of miR-17~92-deficient CD4+ T cells with individual miRNA mimics [G] identified miR-19b as sufficient to restore IFNγ production63.
miRNAs can influence TH1 cell differentiation indirectly by targeting genes in immune cells other than CD4+ T cells. For example, miR-21 regulates TH1 cell differentiation by modulating IL-12 production by dendritic cells (DCs). IL-12 is an important cytokine that induces T-bet and IFNγ expression and supports the growth and survival of TH1 cells64. miR-21 is highly expressed in DCs, and directly targets the mRNA that encodes IL-12p3565. miR-21 deficiency increases IL-12 expression in DCs, and enhances TH1 cell development and delayed type hypersensitivity responses in vivo66. It is possible that other miRNAs that modulate DC activity and cytokine production also have important indirect effects on TH cell differentiation and immune function.
Studies of TH1 cell differentiation and function have led the way to insights into the T-cell intrinsic and extrinsic roles for miRNAs in T cell differentiation, and set paradigms for the logic of miRNA-mediated regulation of cell differentiation in general. Some of the miRNAs that regulate TH1 cells also play identical or related roles in other IFN-γ producing lymphocytes, such as cytotoxic CD8+ T cells and NK cells67, 68. Lessons learned through the study of TH1 cells will likely continue to inform our understanding of miRNA regulation of the immune system.
TH2 cell differentiation
TH2 cells orchestrate innate immune responses against helminths69, and dysregulated TH2 responses cause allergies and asthma70. Over the past decades, much progress has been made in our understanding of the molecular requirements for the generation of TH2 cells. In vitro differentiation systems and more recently elegant in vivo approaches have established the powerful influence of a positive feedback loop involving the hallmark TH2 cytokine IL-4 and the transcription factors STAT6 and GATA-binding protein 3 (GATA3) in amplifying TH2 responses, though the initial signals leading to TH2 cell differentiation have been harder to define71. In addition to IL-4, TH2 cells express the genetically linked cytokines IL-5 and IL-13, which drive type 2 inflammation through their effects on myeloid and non-hematopoietic cells7.
Compared to TH1 cells, miRNA regulation of TH2 cell differentiation is poorly understood. Overexpression of miR-21 in T cells increases TH2 cell differentiation in vitro72, whereas overexpression of miR-27 or miR-128 decreases the secretion of IL-4 and IL-5 by activated CD4+ T cells73. Further work is needed to characterize the importance of these miRNAs in TH2 cell responses. In addition, miR-155-deficient T cells display a mild bias towards TH2 differentiation in vitro. This is accompanied by dysregulation of many target genes, including MAF (also known as c-MAF), a transcription factor that regulates IL-4 production53, 54. Approximately 50% of ageing miR-155 knockout mice develop spontaneous lung inflammation, which displays some but not all of the hallmarks of type 2 inflammation that have been associated with asthma54. This pathology may be caused by cell-intrinsic defects in TH2 cell differentiation, but it may also reflect other changes in T cell responses or requirements for miR-155 in other cells.
The lung’s accessibility for local drug delivery makes asthma an attractive target for miRNA-directed therapies. MicroRNA expression patterns associated with asthma have been detected in both lung epithelial cells and purified T cells from human asthmatic subjects74, 75. In addition, several proof-of-concept studies have shown that miRNA inhibitors can be protective in mouse models of allergic asthma. House dust mite allergen (HDM) challenge increases miR-126 expression in the airway wall, and intranasal administration of a miR-126-specific antagomir [G] (cholesterol-linked single stranded RNA complementary to miR-126) reduces airway hyperresponsiveness and eosinophil recruitment to the lung76. Treatment with the miR-126-specific antagomir also reduced lung eosinophilia in a mouse model of chronic asthma, but it did not have sustained effects on inflammation or airway remodeling77. As miR-126 is strongly expressed in the airway wall76, 77, but is present in T cells and other hematopoietic cells at only very low abundance26, 31, it seems likely that miR-126-specific antagomir acts on epithelial cells or other non-hematopoietic cells in the lung rather than directly on infiltrating TH2 cells in these models.
A miR-145-specific antagomir also inhibited allergic airway disease in the HDM model, possibly through effects on smooth muscle cells, which express a large amount of miR-14578. Members of the let-7 family of miRNAs are highly expressed in most cells. Let-7 directly targets Il13 mRNA and reduces IL-13 production by T cells in vitro79, 80. However, intravenous administration of locked nucleic acid [G] (LNA) inhibitors of let-7 ameliorated lung inflammation and airway hyperresponsiveness in a mouse model of asthma, rather than exacerbating the disease as may have been expected if the dominant effect of let-7 inhibitors was to derepress IL-13 expression in TH2 cells. Let-7 also targets Il10 mRNA, and increased IL-10 levels in HIV patients correlate with decreased let-7 expression in CD4+ T cells81.
Taken together, these studies show that individual miRNAs can have a strong impact on type 2 immunity, but also underscore the complexity of miRNA biology in vivo. In particular, they indicate that targeted delivery of miRNAs or miRNA inhibitors specifically to T cells will be important for manipulating miRNA-mediated control of TH2 cell gene expression, differentiation and effector functions, be it for scientific or therapeutic purposes.
TH17 cell differentiation
TH17 cells are defined by expression of the transcription factor retinoic acid receptor-related orphan receptor-γt (RORγt) and cytokines of the IL-17 family82 and have an important role in the defense against extracellular bacteria and fungi. In addition, they have been implicated in the etiology and pathology of several autoimmune diseases including psoriasis, multiple sclerosis, colitis, arthritis, and even asthma83.
MiRNA profiling in T cells from patients with multiple sclerosis, as well as studies in mice and rats with experimental autoimmune encephalomyelitis (EAE) [G], a model system for multiple sclerosis, have revealed distinct contributions for miRNAs in the regulation of TH cell subsets involved in this disease84. Expression of miR-326 in TH cells correlated with disease severity in mice with EAE and also in patients with multiple sclerosis85. miR-326 targets ETS1, a negative regulator of TH17 cell differentiation and increases TH17 differentiation in vitro85.
Conversely, expression of miR-10a in CD4+ T cells can limit TH17 cell differentiation86. This effect depends on the presence of retinoic acid, which induces both miR-10a and T-bet. Suppression of the miR-10a targets Bcl6 or Ncor by RNA interference limited TH17 cell differentiation in wild-type, but not T-bet-deficient T cells. As BCL-6 and NCOR modulate the expression and transcriptional activity of T-bet87–89, and T-bet in turn inhibits the expression of RORγt90, miR-10a and T-bet may cooperate to balance TH1 and TH17 differentiation in retinoic acid-rich microenvironments. In line with these observations, miR-10a overexpression in myelin oligodendrocyte glycoprotein (MOG)-specific CD4+ T cells delays neurological disease in EAE86.
miR-155 knockout mice are strongly resistant to EAE. Detailed analysis revealed both a T cell-intrinsic role for miR-155 in TH17 cell differentiation, as well as an indirect role through the regulation of production of TH17 cell-polarizing cytokines by DCs55, 91. Impaired TH17 cell responses of miR-155-deficient T cells were also observed in a mouse model of Helicobacter pylori infection and in a mouse model of TH17-driven chronic colitis56. Thus, miR-155 promotes T cell–mediated tissue inflammation through the regulation of both TH1 and TH17 cell responses. Finally, miR-301a enhances TH17 cell differentiation, possibly through targeting PIAS3, which inhibits STAT3 signaling and TH17 cell differentiation92.
Although the last few years have seen substantial progress in defining the transcriptional requirements for TH17 cells93, studies on miRNA function in TH17 cells remain sparse. Given the high plasticity associated with this TH cell subset, it is anticipated that miRNAs will play an important role in defining and maintaining the gene expression program of differentiated TH17 cells.
TFH cell differentiation
T follicular helper (TFH) cells preferentially localize in close proximity to B cells in the follicles and germinal centers (GCs) of secondary lymphoid organs94–96 and regulate humoral immunity97. TFH cells are necessary for effective T cell-dependent antibody responses such as those induced by common vaccines, and TFH cell dysregulation is involved in several autoimmune diseases98. TFH cells express the chemokine receptor CXC chemokine receptor 5 (CXCR5), the co-inhibitory molecule programmed cell death 1 (PD-1) and the co-stimulatory molecule inducible T cell co-stimulator (ICOS), and they shape the humoral immune response by providing cytokines that influence immunoglobulin production by B cells97. The transcriptional repressor BCL-6 is necessary and sufficient to induce TFH cell differentiation88, 89, 99. An initial BCL6+CXCR5+ TFH cell population develops directly from naïve CD4+ T cells after priming by DCs100, 101. Subsequent interactions with antigen-specific B cells further polarize TFH cells, induce GCs, and contribute to the maintenance of the TFH cell phenotype99, 102–104.
Several miRNAs are differentially expressed in TFH cells as compared to other TH cell subsets26. miRNAs are essential for an early step in TFH cell differentiation, as activated DGCR8-deficient CD4+ T cells fail to upregulate CXCR5 and downregulate CCR7, thus preventing their migration to the T-B zone border and into B cell follicles105. This defect stands in sharp contrast to the role of miRNAs in other TH cell subsets, such as TH1 and TH2, as these can still be generated from naïve miRNA-deficient CD4+ T cells, even with aberrantly high efficiency in some cases27, 31. The fact that TFH cell differentiation strictly requires a transcriptional repressor (BCL-6) and post-transcriptional repressors (miRNAs) indicates that silencing of alternative differentiation pathways has a crucial role in specifying or maintaining the TFH cell gene expression program.
To date, only a few studies have provided insight into the roles of individual miRNAs or miRNA families in TFH cells. In sanroque mice, a mutation of the RNA-binding protein Roquin-1 leads to excessive TFH cell accumulation, which ultimately causes a lupus-like autoimmune disease106. Initially, it was suggested that Roquin limits TFH cell responses by repressing the 3’ UTR of Icos mRNA through a process involving miR-101 binding within processing bodies (P-bodies) [G]107. However, later studies clarified that Roquin represses Icos by direct binding to its 3’ UTR in a miRNA-independent manner108.
The miR-17~92 cluster promotes TFH cell differentiation and function independent of its effect on T cell proliferation105, 109. miR-17~92 targets PTEN in CD4+ T cells48, and this leads to a modest reduction in the number of developing BCL6+CXCR5+ TFH cells very early following protein immunization105, 109. However, other derepressed miR-17~92 target genes likely contribute to the significantly reduced frequency of TFH cells that accrues later in the response. In this regard, derepression of the miR-17~92 target gene Phlpp22, an Akt phosphatase involved in the ICOS signaling pathway, has been linked to the defective migration of activated miR-17~92-deficient T cells into B cell follicles109. A feedback mechanism may limit the expression of miR-17~92 in TFH cells, as BCL-6 transduction reduced miR-17~92 expression in primary T cells, and miR-17~92 overexpression reduced CXCR5 expression in B cells89. However, the degrees to which the reported miRNA expression differences reflected the T cell activation status versus direct effects of BCL6 remain unclear.
miR-17~92 also prevents the expression of TFH cell-inappropriate genes characteristic of Th17 or Th22 cells, including CC chemokine receptor 6 (Ccr6), Il22, and Rora, which encodes the transcription factor RORα105. All four miRNA families represented in the miR-17~92 cluster target the Rora mRNA, and aberrant RORα expression is at least partly responsible for the increased expression of CCR6 and IL-22 in miR-17~92-deficient TFH cells. These findings highlight the ability of miRNAs to ensure the fidelity of TH subset-specific gene expression programs. Similarly, miR-10a inhibits the conversion of peripherally derived TReg cells into TFH cells under lymphopenic conditions by directly targeting BCL6 and its co-repressor NCOR286. Transient BCL-6 expression occurs during the development of other TH cell subsets, including central memory T (TCM) cells and TH1 cells101, 110–112. Although TCM and effector memory T (TEM) cells are important for protective immunity against reinfection with certain pathogens and memory T cells contribute to the pathogenesis of chronic autoimmune diseases113, not much is known about the involvement of miRNAs in the generation and maintenance of TCM and TEM cells.
Further work is needed to comprehensively profile miRNA expression in developing TFH cells and to dissect how these miRNAs contribute to the balance between TFH versus effector cell differentiation and to the maintenance of the TFH cell identity.
MiRNA-mediated control of TReg cells
CD4+ regulatory T (TReg) cells are essential mediators of peripheral tolerance114. They counterbalance the actions of effector T cells, preventing inappropriate or exaggerated immune responses that cause autoimmunity and immunopathology. Thymus-derived TReg cells are formed during positive selection in the thymus, but naïve CD4+ T cells can also differentiate into peripherally derived TReg cells 115. The gene expression program and functional identity of both TReg cell types depend on the lineage-defining transcription factor forkhead box P3 (FOXP3). Research on the requirements for miRNA expression in TReg cells, as well as on the specific contributions of individual miRNAs in TReg cell development, function, and plasticity has generally outpaced research on conventional T cells, though much remains to be learned.
MiRNAs support TReg cell development and homeostasis, and they also are important for the maintenance of their immunosuppressive function. Early studies suggested that miRNAs have a predominantly tolerogenic role in T cells, as CD4+ T cells lacking miRNAs failed to develop into TReg cells in the thymus and showed reduced differentiation into in vitro-induced TReg cells28. Moreover, a substantial percentage of mice with miRNA-deficient CD4+ T cells developed spontaneous autoimmunity as they aged28. Notably, it was recently confirmed that miRNA deficiency in conventional CD4+ T cells is in fact protective against the induction of autoimmunity29. By contrast, conditional deletion of DICER or DROSHA only in FOXP3+ TReg cells causes early onset of severe spontaneous autoimmunity30, 116, 117. Thus, miRNA expression in TReg cells is important for the maintenance of self tolerance (Figure 3a).
Figure 3. miRNA regulation of TReg function and plasticity.

a | Treg-specific miRNA expression is required to restrain effector T cell responses. In the absence of miRNA expression in TReg cells, for example due to deletion of essential components of the miRNA biogenesis pathway, such as DICER or DROSHA, TReg cells fail to maintain tolerance, which might result in autoimmunity. b | Examples of miRNA pathways that contribute to the regulation of TReg cell function and plasticity. TEFF, effector T cell; TReg, regulatory T cell.
Several miRNAs have been identified to impact TReg cell development and function (Figure 3b). Although TReg cells and conventional (non-TReg) effector CD4+ T cells express a very similar set of miRNAs, expression profiling has uncovered several miRNAs that are differentially expressed in TReg cells25, 26, 28. miR-155 is highly expressed in TReg cells, and miR-155-deficient mice exhibit reduced numbers of thymus-derived and peripherally derived TReg cells62. FOXP3 controls this high level of miR-155 expression in TReg cells, and binds to an intronic region of Bic, the gene that encodes the primary transcript of miR-15562, 118, 119. miR-155 ensures TReg cell homeostasis by targeting the suppressor of cytokine signaling 1 (SOCS1)62, a negative regulator of the IL-2 signaling pathway, which has a crucial role in TReg cell development. miR-155-deficiency also reduces IL-2 production by conventional CD4+ T cells54, suggesting that this miRNA may regulate IL-2-directed Treg cell homeostasis through both cell intrinsic and extrinsic mechanisms.
miR-146a also impacts TReg cell function substantially. miR-146a-deficiency limited mainly to FOXP3+ TReg cells using a mixed bone marrow chimera approach resulted in an IFNγ- and TH1 cell-mediated pathology61, similar to the disease observed in mice with TReg cell-specific DICER or DROSHA deficiency30, 116, 117. This pathology could result from increased expression of the miR-146a target STAT1 in TReg cells61. However, dysregulated miR-146a-deficient myeloid cells45 may also contribute in this system, so confirmation of a TReg cell-intrinsic role for miR-146a in the development of autoimmunity remains to be confirmed with conditional miR-146a-deficient mice.
The miR-17~92 cluster represses TReg cell formation in vitro63. miR-17 directly targets TGF-β receptor II (Tgfbr2) and cAMP-responsive element binding protein 1 (Creb1), both of which are implicated in TReg cell differentiation in vitro63. In thymus-derived TReg cells, miR-17~92 seems to be particularly important during antigenic responses.miR-17~92 is dispensable for the development of thymus-derived TReg cells in vivo63, 120, but miR-17~92-deficiency does reduce the number of MOG-specific TReg cells that are peripherally induced during EAE120. Similarly, transgenic overexpression of miR-17~92 does not affect total FOXP3+ TReg cell numbers in a protein immunization model, but does increase the frequency of follicular TReg cells that emerge from thymus-derived TReg cells105. TReg cell-specific miR-17~92-deficiency also reduces the frequency of IL-10 producing TReg cells, which are particularly important for dampening immune responses120. Further studies are needed to identify the molecular targets and mechanisms of miR-17~92 in the regulation of TReg cell activity.
miR-10a is selectively expressed in TReg cells26, and it regulates the stability of peripherally derived and in vitro-induced TReg cells. Retinoic acid synergizes with TGFβ to induce miR-10a expression in conventional CD4+ T cells, and these conditions also promote the differentiation of peripherally derived TReg cells86, 121. miR-10a is not required for FOXP3 induction in in vitro-induced TReg cells, but it contributes to TReg cell stability by maintaining high FOXP3 expression121. miR-10a also inhibited the conversion of peripherally derived TReg cells into TFH cells under lymphopenic conditions86, a phenomenon that was originally described for FOXP3+ TReg cells that had been adoptively transferred in the Peyer’s patches of T cell-deficient mice122. miR-10a expression in TReg cells correlates inversely with susceptibility to autoimmune disease: TReg cells from the autoimmunity-resistant C57Bl/6 mouse strain have high miR-10a expression, whereas TReg cells from the autoimmunity-prone NOD mouse strain have low miR-10a expression levels121. miR-10a may prevent inappropriate activation of a TFH cell gene expression program in TReg cells by directly targeting the mRNAs encoding the TFH cell lineage-defining transcriptional repressor BCL-6 and its co-repressor NCOR286. As discussed above, miR-10a also limits TH17 cell differentiation86, adding another layer to the complex regulatory network that constrains TReg cell plasticity.
The availability of genetic tools that facilitate the study of TReg cells in vivo has hastened the pace of research on miRNA function in TReg cells. miRNAs clearly perform critical functions in regulating the development and function of TReg cells in maintaining immune tolerance. Still, further studies are needed to dissect the impact of individual miRNAs and their targets in TReg cells, and to elucidate their role in the plasticity that is associated with this TH cell subset.
Concluding remarks and future perspectives
MiRNAs are now generally recognized as important regulators of gene expression in most, if not all, vertebrate cells. The last few years have seen much progress in the field of miRNA regulation of lymphocyte development and immune function123, and particularly in T helper cells67, 124, 125. T helper cell differentiation is particularly sensitive to small changes in transcriptional regulation, which is also reflected in the variety of known TH cell subsets and the observed plasticity among some of these apparently differentiated cell types. Since miRNAs have evolved as a mechanism to fine tune biological processes14, it is very likely that miRNAs represent an equally plastic and adjustable system for the regulation of TH cell differentiation and plasticity.
The complexity and redundancy of miRNA networks underscores the importance of precise gene regulation in the programming of the great variety of cells that comprise a human body and their appropriate responses to developmental and environmental cues. This is also reflected in the observation that defects in single miRNAs within the same cell type can have diverse effects in different experimental settings, as discussed in this review for the different TH cell subsets. Most of the progress on miRNA function in TH cells has so far been based on mouse in vitro and in vivo models, and relatively little is known about miRNA function in human TH cells. As one of the means to compensate for this lack of knowledge, it will be interesting to use systems biology approaches such as exome sequencing to discover mutations in patients that might correlate with altered miRNA expression. Such tools might also allow screening for alterations in miRNA target sequences that might contribute to the etiology of immune diseases.
Elucidating miRNA function in TH cells can be leveraged to promote our understanding of the complex molecular pathways that govern the behavior and cell fate decisions of these cells. Future studies will not only provide insights into the role of miRNAs themselves, but most likely will also reveal novel mechanisms of immune regulation through the identification and functional analysis of miRNA target genes. Ideally, these studies will identify novel targets for the treatment of conditions in which T helper cell functions are impaired or exaggerated. Recent years have seen considerable progress in the field of miRNA-based therapies126. The first miRNA-targeted therapy, miraversen, blocks miR-122 in hepatocytes and reduced hepatitis C virus RNA in patients enrolled in a phase 2a trial127. Serious challenges remain though before miRNA-targeted therapies can be used in immunological diseases. It will be particularly important to develop enhanced methods for the delivery of these modalities specifically to TH cells in patients128. One advantage of the immune system is that many of its effector cells recirculate between sites of inflammation and secondary lymphoid organs via the blood and lymph. Thus, systemic delivery of agents that target certain immune cell types might be easier accomplished than delivery to solid organs. Local administration at sites of inflammation may also be sufficient to safely alter immune activation while averting the need for cell type specific targeting. In any case, our expanding knowledge of these powerful but tiny regulators of TH cell function promises new avenues for harnessing and shaping immune responses.
Online summary.
T helper (TH) cells represent a central component of the adaptive immune system. They coordinate cellular and humoral responses by producing cytokines and growth factors. Several TH cell subsets have been described, including TH1, TH2, TH17, TH9, TH22 cells, regulatory T (TReg) cells, and T follicular helper (TFH) cells.
MicroRNAs are small evolutionarily conserved nucleotide sequences that regulate gene expression by interfering with mRNA translation and stability.
MicroRNA-deficient CD4+ T cells display impaired survival and proliferation, but also exhibit increased sensitivity to signals that induce effector TH cell differentiation and cytokine production.
An expanding list of individual microRNAs and co-expressed microRNA clusters have been shown to have significant effects on TH cell fate decisions and immune functions.
MicroRNAs are critical for the proper regulation of Treg development, homeostasis, plasticity, and the maintenance of immune tolerance.
Research on microRNA function can be used as a tool for the discovery of novel pathways that regulate TH cell biology, and may identify novel targets for the treatment of conditions in which TH cell functions are impaired or exaggerated.
Acknowledgements
This work was supported by the NIH (HL109102, HL107202), and a Scholar Award from The Leukemia & Lymphoma Society (K.M.A.), and by the Swiss Foundation for Grants in Biology and Medicine (D.B.).
Glossary terms
- T helper cell
Effector T cell that develops from naïve CD4+ T cells. Upon activation in the periphery, Th cells produce cytokines that orchestrate cellular and humoral immunity.
- secondary lymphoid organs
Organized lymphoid structures in which adaptive immune responses are elicited, e.g. spleen and lymph nodes.
- positive selection
Immature T cells are selected in the thymus for expression of a functional TCR.
- negative selection
Immature T cells that harbor high affinity for self-antigens are deleted in the thymus to prevent egress of auto-reactive T cells into the circulation.
- germinal center
Specialized anatomic structure in secondary lymphoid organs in which TFH cells provide help to B cells to generate high affinity antibodies, memory B cells, and long-lived plasma cells.
- processing bodies
Molecular structures within the cytoplasm that are major sites for mRNA turnover.
- miRISC
The miRNA-induced silencing complex consists of a miRNA bound to an Argonaute protein. The miRNA provides sequence specificity for the complex’s function in translational repression and decreasing target mRNA stability.
- miRNA cluster
miRNAs are often found within clusters in the genome, and these clusters are typically transcribed together to form primary miRNA transcripts that are processed to yield multiple mature miRNAs.
- miRNA family
miRNAs are classified into ‘families’ that share the same seed sequence, and are therefore predicted to share many of the same target mRNAs. For example, the polycistronic miR-17~92 cluster comprises six miRNAs representing four separate miRNA families: the miR-17 family (miR-17, miR-20a), the miR-18 family (miR-18a), the miR-19 family (miR-19a, miR-19b), and the miR-25 family (miR-92a).
- miRNA sponge
A genetically engineered construct containing several miRNA binding sites that compete with endogenous miRNA binding sites, reducing specific miRNA availability and function.
- miRNA mimics
Small transfectable synthetic RNAs that mimic endogenous miRNAs used to study the effects of miRNA overexpression.
- antagomir
Also known as anti-miR, miRNA inhibitor, and miRNA antagonist. Small synthetic nucleic acid oligonucleotides that bind to endogenous miRNAs and inhibit their function. Antagomirs are often chemically modified to promote their stability and/or entry into target cells.
- locked nucleic acids (LNA)
RNA oligonucleotides bearing a modification of the ribose moiety in their backbone that “locks” them in a favorable conformation for base pairing, increasing their binding affinity. LNAs are used in various applications for miRNA detection and in miRNA inhibitors for experimental and therapeutic use.
- experimental autoimmune encephalitis (EAE)
An animal model of multiple sclerosis — a chronic demyelinating disease in humans. In animals, EAE is induced by the injection of several different antigens that are derived from the myelin sheath, including myelin basic protein, proteolipid protein or myelin oligodendrocyte glycoprotein, together with a potent adjuvant.
Biographies
Dirk Baumjohann is currently a postdoctoral fellow in the laboratory of Dr. K. Mark Ansel at the University of California, San Francisco. He received his Ph.D. in Cell Biology/Immunology from the University of Bern, Switzerland, for work with Dr. Federica Sallusto at the Institute for Research in Biomedicine in Bellinzona, Switzerland. His recent studies focus on understanding how miRNAs and transcription factors regulate T helper cell differentiation.
K. Mark Ansel is an Assistant Professor of Microbiology & Immunology in the Sandler Asthma Basic Research Center at the University of California, San Francisco (UCSF). He completed his Ph.D. in Biomedical Sciences in Dr. Jason Cyster’s laboratory at UCSF, and conducted postdoctoral research with Dr. Anjana Rao at Harvard Medical School. His laboratory studies miRNA regulation in the immune system.
Footnotes
Further information
Web site1 title: “Ansel Lab”, Web site1 URL: http://ansel.ucsf.edu/
Web site2 title: “miRBase”, Web site2 URL: http://www.mirbase.org
Web site3 title: “TargetScan”, Web site3 URL: http://www.targetscan.org
Web site4 title: “miRTarBase”, Web site4 URL: http://mirtarbase.mbc.nctu.edu.tw
References
- 1.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 2.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Locksley RM. Nine lives: plasticity among T helper cell subsets. J Exp Med. 2009;206:1643–1646. doi: 10.1084/jem.20091442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. 2010;11:674–680. doi: 10.1038/ni.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 7.Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol. 2006;24:607–656. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 8.Kanno Y, Vahedi G, Hirahara K, Singleton K, O'Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol. 2012;30:707–731. doi: 10.1146/annurev-immunol-020711-075058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ansel KM, Lee DU, Rao A. An epigenetic view of helper T cell differentiation. Nat Immunol. 2003;4:616–623. doi: 10.1038/ni0703-616. [DOI] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 13.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 14.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoefig KP, Heissmeyer V. MicroRNAs grow up in the immune system. Curr Opin Immunol. 2008;20:281–287. doi: 10.1016/j.coi.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Ceribelli A, Satoh M, Chan EK. MicroRNAs and autoimmunity. Curr Opin Immunol. 2012;24:686–691. doi: 10.1016/j.coi.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 18.Nakayamada S, Takahashi H, Kanno Y, O'Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol. 2012;24:297–302. doi: 10.1016/j.coi.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136:26–36. doi: 10.1016/j.cell.2008.12.027. [DOI] [PubMed] [Google Scholar]
- 20.Belver L, Papavasiliou FN, Ramiro AR. MicroRNA control of lymphocyte differentiation and function. Curr Opin Immunol. 2011;23:368–373. doi: 10.1016/j.coi.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeker LT, Bluestone JA. Small RNA regulators of T cell-mediated autoimmunity. J Clin Immunol. 2010;30:347–357. doi: 10.1007/s10875-010-9392-7. [DOI] [PubMed] [Google Scholar]
- 22. Monticelli S, et al. MicroRNA profiling of the murine hematopoietic system. Genome Biol. 2005;6:R71. doi: 10.1186/gb-2005-6-8-r71. This study was the first to systematically profile miRNA expression in the murine hematopoietic system.
- 23.Landgraf P, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barski A, et al. Chromatin poises miRNA- and protein-coding genes for expression. Genome Res. 2009;19:1742–1751. doi: 10.1101/gr.090951.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rossi RL, et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat Immunol. 2011;12:796–803. doi: 10.1038/ni.2057. Elegant study that used high-throughput miRNA profiling to establish miRNA expression in human lymphocyte subsets.
- 26. Kuchen S, et al. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity. 2010;32:828–839. doi: 10.1016/j.immuni.2010.05.009. High-throughput sequencing technology was used to comprehensively characterize the microRNome in many different immune cell types.
- 27. Muljo SA, et al. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202:261–269. doi: 10.1084/jem.20050678. This study provided the first evidence that miRNAs are important regulators of T helper cell differentiation.
- 28.Cobb BS, et al. A role for Dicer in immune regulation. J Exp Med. 2006;203:2519–2527. doi: 10.1084/jem.20061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian L, et al. Loss of T cell microRNA provides systemic protection against autoimmune pathology in mice. J Autoimmun. 2012;38:39–48. doi: 10.1016/j.jaut.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 30. Chong MM, Rasmussen JP, Rudensky AY, Littman DR. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J Exp Med. 2008;205:2005–2017. doi: 10.1084/jem.20081219. These three papers described that miRNA expression in Treg cells is required to prevent autoimmunity.
- 31. Steiner DF, et al. MicroRNA-29 Regulates T-Box Transcription Factors and Interferon-gamma Production in Helper T Cells. Immunity. 2011;35:169–181. doi: 10.1016/j.immuni.2011.07.009. These papers showed that miR-29 is an important regulator of IFN-γ production in Th1 cells.
- 32.Bronevetsky Y, et al. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. J Exp Med. 2013;210:417–432. doi: 10.1084/jem.20111717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3' untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma F, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol. 2011;12:861–869. doi: 10.1038/ni.2073. These papers showed that miR-29 is an important regulator of IFN-γ production in Th1 cells.
- 35. Smith KM, et al. miR-29ab1 Deficiency Identifies a Negative Feedback Loop Controlling Th1 Bias That Is Dysregulated in Multiple Sclerosis. J Immunol. 2012 doi: 10.4049/jimmunol.1103171. These papers showed that miR-29 is an important regulator of IFN-γ production in Th1 cells.
- 36.Li QJ, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Ebert PJ, Jiang S, Xie J, Li QJ, Davis MM. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nat Immunol. 2009;10:1162–1169. doi: 10.1038/ni.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henao-Mejia J, et al. The MicroRNA miR-181 Is a Critical Cellular Metabolic Rheostat Essential for NKT Cell Ontogenesis and Lymphocyte Development and Homeostasis. Immunity. 2013;38:984–997. doi: 10.1016/j.immuni.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fragoso R, et al. Modulating the strength and threshold of NOTCH oncogenic signals by mir-181a-1/b-1. PLoS Genet. 2012;8:e1002855. doi: 10.1371/journal.pgen.1002855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zietara N, et al. Critical role for miR-181a/b-1 in agonist selection of invariant natural killer T cells. Proc Natl Acad Sci U S A. 2013;110:7407–7412. doi: 10.1073/pnas.1221984110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li G, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med. 2012;18:1518–1524. doi: 10.1038/nm.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palin AC, Ramachandran V, Acharya S, Lewis DB. Human Neonatal Naive CD4+ T Cells Have Enhanced Activation-Dependent Signaling Regulated by the MicroRNA miR-181a. J Immunol. 2013 doi: 10.4049/jimmunol.1202534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rusca N, et al. miR-146a and NF-kappaB1 Regulate Mast Cell Survival and T Lymphocyte Differentiation. Mol Cell Biol. 2012;32:4432–4444. doi: 10.1128/MCB.00824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Curtale G, et al. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood. 2010;115:265–273. doi: 10.1182/blood-2009-06-225987. [DOI] [PubMed] [Google Scholar]
- 45.Zhao JL, et al. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A. 2011;108:9184–9189. doi: 10.1073/pnas.1105398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang L, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med. 2012;209:1655–1670. doi: 10.1084/jem.20112218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu T, et al. Temporal expression of microRNA cluster miR-17-92 regulates effector and memory CD8+ T-cell differentiation. Proc Natl Acad Sci U S A. 2012;109:9965–9970. doi: 10.1073/pnas.1207327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xiao C, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. Elegant study showing that overexpression of the miR-17~92 cluster in lymphocytes leads to lymphoproliferative disease and autoimmunity.
- 49. Loeb GB, et al. Transcriptome-wide miR-155 Binding Map Reveals Widespread Noncanonical MicroRNA Targeting. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.10.002. Transcripome-wide analysis of miRNA binding sites that revealed an unexpectedly high frequency of functional non-canonical target sites.
- 50.Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39:493–506. doi: 10.1016/j.molcel.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huffaker TB, et al. Epistasis between MicroRNAs 155 and 146a during T Cell-Mediated Antitumor Immunity. Cell Rep. 2012;2:1697–1709. doi: 10.1016/j.celrep.2012.10.025. This paper revealed epistasis between miR-155 and miR-146a in T cell-mediated anti-tumour immunity.
- 52.Banerjee A, Schambach F, DeJong CS, Hammond SM, Reiner SL. Micro-RNA-155 inhibits IFN-gamma signaling in CD4+ T cells. Eur J Immunol. 2010;40:225–231. doi: 10.1002/eji.200939381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thai TH, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. These two studies were the first to show that genetic disruption of a single miRNA in vivo can have adverse effects on immune cell homeostasis and function.
- 54. Rodriguez A, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. These two studies were the first to show that genetic disruption of a single miRNA in vivo can have adverse effects on immune cell homeostasis and function.
- 55.O'Connell RM, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607–619. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oertli M, et al. MicroRNA-155 is essential for the T cell-mediated control of Helicobacter pylori infection and for the induction of chronic Gastritis and Colitis. J Immunol. 2011;187:3578–3586. doi: 10.4049/jimmunol.1101772. [DOI] [PubMed] [Google Scholar]
- 57.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38:13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thiele S, Wittmann J, Jack HM, Pahl A. miR-9 enhances IL-2 production in activated human CD4(+) T cells by repressing Blimp-1. Eur J Immunol. 2012;42:2100–2108. doi: 10.1002/eji.201142203. [DOI] [PubMed] [Google Scholar]
- 59.Seddiki N, et al. The microRNA-9/B-lymphocyte-induced maturation protein-1/IL-2 axis is differentially regulated in progressive HIV infection. Eur J Immunol. 2013;43:510–520. doi: 10.1002/eji.201242695. [DOI] [PubMed] [Google Scholar]
- 60.Stittrich AB, et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol. 2010;11:1057–1062. doi: 10.1038/ni.1945. [DOI] [PubMed] [Google Scholar]
- 61.Lu LF, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu LF, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang S, et al. Molecular dissection of the miR-17-92 cluster's critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood. 2011;118:5487–5497. doi: 10.1182/blood-2011-05-355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lazarevic V, Glimcher LH. T-bet in disease. Nat Immunol. 2011;12:597–606. doi: 10.1038/ni.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu TX, Munitz A, Rothenberg ME. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol. 2009;182:4994–5002. doi: 10.4049/jimmunol.0803560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu TX, et al. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J Immunol. 2011;187:3362–3373. doi: 10.4049/jimmunol.1101235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jeker LT, Bluestone JA. MicroRNA regulation of T-cell differentiation and function. Immunol Rev. 2013;253:65–81. doi: 10.1111/imr.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beaulieu AM, et al. MicroRNA function in NK-cell biology. Immunol Rev. 2013;253:40–52. doi: 10.1111/imr.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allen JE, Maizels RM. Diversity and dialogue in immunity to helminths. Nat Rev Immunol. 2011;11:375–388. doi: 10.1038/nri2992. [DOI] [PubMed] [Google Scholar]
- 70.Locksley RM. Asthma and allergic inflammation. Cell. 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sawant DV, Wu H, Kaplan MH, Dent AL. The Bcl6 target gene microRNA-21 promotes Th2 differentiation by a T cell intrinsic pathway. Mol Immunol. 2013;54:435–442. doi: 10.1016/j.molimm.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guerau-de-Arellano M, et al. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain. 2011;134:3578–3589. doi: 10.1093/brain/awr262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seumois G, et al. An integrated nano-scale approach to profile miRNAs in limited clinical samples. Am J Clin Exp Immunol. 2012;1:70–89. [PMC free article] [PubMed] [Google Scholar]
- 75.Solberg OD, et al. Airway epithelial miRNA expression is altered in asthma. Am J Respir Crit Care Med. 2012;186:965–974. doi: 10.1164/rccm.201201-0027OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mattes J, Collison A, Plank M, Phipps S, Foster PS. Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc Natl Acad Sci U S A. 2009;106:18704–18709. doi: 10.1073/pnas.0905063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collison A, et al. Altered expression of microRNA in the airway wall in chronic asthma: miR-126 as a potential therapeutic target. BMC Pulm Med. 2011;11:29. doi: 10.1186/1471-2466-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Collison A, Mattes J, Plank M, Foster PS. Inhibition of house dust mite-induced allergic airways disease by antagonism of microRNA-145 is comparable to glucocorticoid treatment. J Allergy Clin Immunol. 2011;128:160–167. e4. doi: 10.1016/j.jaci.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 79.Polikepahad S, et al. Proinflammatory role for let-7 microRNAS in experimental asthma. J Biol Chem. 2010;285:30139–30149. doi: 10.1074/jbc.M110.145698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar M, et al. Let-7 microRNA-mediated regulation of IL-13 and allergic airway inflammation. J Allergy Clin Immunol. 2011;128:1077–1085. e1–e10. doi: 10.1016/j.jaci.2011.04.034. [DOI] [PubMed] [Google Scholar]
- 81.Swaminathan S, et al. Differential regulation of the Let-7 family of microRNAs in CD4+ T cells alters IL-10 expression. J Immunol. 2012;188:6238–6246. doi: 10.4049/jimmunol.1101196. [DOI] [PubMed] [Google Scholar]
- 82.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 83.Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol. 2013;8:477–512. doi: 10.1146/annurev-pathol-011110-130318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thamilarasan M, Koczan D, Hecker M, Paap B, Zettl UK. MicroRNAs in multiple sclerosis and experimental autoimmune encephalomyelitis. Autoimmun Rev. 2012;11:174–179. doi: 10.1016/j.autrev.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 85.Du C, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10:1252–1259. doi: 10.1038/ni.1798. [DOI] [PubMed] [Google Scholar]
- 86.Takahashi H, et al. TGF-beta and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol. 2012;13:587–595. doi: 10.1038/ni.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oestreich KJ, Huang AC, Weinmann AS. The lineage-defining factors T-bet and Bcl-6 collaborate to regulate Th1 gene expression patterns. J Exp Med. 2011;208:1001–1013. doi: 10.1084/jem.20102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nurieva RI, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu D, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 90.Lazarevic V, et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat Immunol. 2011;12:96–104. doi: 10.1038/ni.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2011;187:2213–2221. doi: 10.4049/jimmunol.1003952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mycko MP, et al. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci U S A. 2012;109:E1248–E1257. doi: 10.1073/pnas.1114325109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Basu R, Hatton RD, Weaver CT. The Th17 family: flexibility follows function. Immunol Rev. 2013;252:89–103. doi: 10.1111/imr.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med. 1999;190:1123–1134. doi: 10.1084/jem.190.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Breitfeld D, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192:1545–1552. doi: 10.1084/jem.192.11.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schaerli P, et al. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:1553–1562. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Crotty S. Follicular Helper CD4 T Cells (T(FH)) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 98.Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. 2012;8:337–347. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Choi YS, et al. ICOS Receptor Instructs T Follicular Helper Cell versus Effector Cell Differentiation via Induction of the Transcriptional Repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baumjohann D, Okada T, Ansel KM. Cutting Edge: Distinct Waves of BCL6 Expression during T Follicular Helper Cell Development. J Immunol. 2011;187:2089–2092. doi: 10.4049/jimmunol.1101393. [DOI] [PubMed] [Google Scholar]
- 102.Haynes NM, et al. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007;179:5099–5108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 103.Deenick EK, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity. 2010;33:241–253. doi: 10.1016/j.immuni.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baumjohann D, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 105.Baumjohann D, et al. The microRNA cluster miR-17~92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat Immunol. 2013 doi: 10.1038/ni.2642. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vinuesa CG, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 107.Yu D, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature. 2007;450:299–303. doi: 10.1038/nature06253. [DOI] [PubMed] [Google Scholar]
- 108.Glasmacher E, et al. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol. 2010;11:725–733. doi: 10.1038/ni.1902. [DOI] [PubMed] [Google Scholar]
- 109.Kang, et al. Nat Immunol. 2013 in press. [Google Scholar]
- 110.Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35:583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakayamada S, et al. Early Th1 Cell Differentiation Is Marked by a Tfh Cell-like Transition. Immunity. 2011;35:919–931. doi: 10.1016/j.immuni.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. 2012;13:405–411. doi: 10.1038/ni.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 114.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 115.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhou X, et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008;205:1983–1991. doi: 10.1084/jem.20080707. These three papers described that miRNA expression in Treg cells is required to prevent autoimmunity.
- 117. Liston A, Lu LF, O'Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205:1993–2004. doi: 10.1084/jem.20081062. These three papers described that miRNA expression in Treg cells is required to prevent autoimmunity.
- 118.Zheng Y, et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- 119.Marson A, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–935. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.de Kouchkovsky D, et al. miR-17~92 regulates interleukin-10 production by Tregs and control of experimental autoimmune encephalomyelitis. J Immunol. 2013 doi: 10.4049/jimmunol.1203567. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jeker LT, et al. MicroRNA 10a Marks Regulatory T Cells. PLoS One. 2012;7:e36684. doi: 10.1371/journal.pone.0036684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tsuji M, et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science. 2009;323:1488–1492. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- 123.Ansel KM. RNA regulation of the immune system. Immunol Rev. 2013;253:5–11. doi: 10.1111/imr.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pagani M, et al. Role of microRNAs and long-non-coding RNAs in CD4(+) T-cell differentiation. Immunol Rev. 2013;253:82–96. doi: 10.1111/imr.12055. [DOI] [PubMed] [Google Scholar]
- 125.Dooley J, Linterman MA, Liston A. MicroRNA regulation of T-cell development. Immunol Rev. 2013;253:53–64. doi: 10.1111/imr.12049. [DOI] [PubMed] [Google Scholar]
- 126.Petrocca F, Lieberman J. Promise and challenge of RNA interference-based therapy for cancer. J Clin Oncol. 2011;29:747–754. doi: 10.1200/JCO.2009.27.6287. [DOI] [PubMed] [Google Scholar]
- 127.Janssen HL, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 128.Peer D. A daunting task: manipulating leukocyte function with RNAi. Immunol Rev. 2013;253:185–197. doi: 10.1111/imr.12044. [DOI] [PubMed] [Google Scholar]
- 129.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 130.Leshkowitz D, Horn-Saban S, Parmet Y, Feldmesser E. Differences in microRNA detection levels are technology and sequence dependent. RNA. 2013;19:527–538. doi: 10.1261/rna.036475.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hsu SD, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39:D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Prosser HM, Koike-Yusa H, Cooper JD, Law FC, Bradley A. A resource of vectors and ES cells for targeted deletion of microRNAs in mice. Nat Biotechnol. 2011;29:840–845. doi: 10.1038/nbt.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Park CY, et al. A resource for the conditional ablation of microRNAs in the mouse. Cell Rep. 2012;1:385–391. doi: 10.1016/j.celrep.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Brown BD, Naldini L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat Rev Genet. 2009;10:578–585. doi: 10.1038/nrg2628. [DOI] [PubMed] [Google Scholar]
- 137.Thomas M, Lieberman J, Lal A. Desperately seeking microRNA targets. Nat Struct Mol Biol. 2010;17:1169–1174. doi: 10.1038/nsmb.1921. [DOI] [PubMed] [Google Scholar]
- 138.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]