

SUMMARY
The endothelial layer of cells lining the intimal surface of blood vessels is essential for proper vascular function. The endothelium releases multiple vasodilator and protective factors including nitric oxide (NO), prostacyclin (PGI2) and endothelium- derived hyperpolarizing factor (EDHF); an imbalance in these factors predisposes to vascular diseases such as stroke. These factors are differentially regulated by vessel size, sex hormones and disease state; therefore, playing differential roles in different tissues following vascular injury. In particular, the EDHF candidate called epoxyeicosatrienoic acid (EETs), plays a prominent role in microvessel function, especially after ischemia, thereby making this signalling pathway an attractive target for therapy in vascular disease, including stroke.
Keywords: EDHF, EET, cerebrovascular, endothelial, vasodilation, ischemia
Endothelial dysfunction
The endothelium was once thought of as a simple layer of cells providing a barrier between the bloodstream and surrounding tissue. We now know that this monolayer is of critical importance to vascular function. It not only maintains tone, it regulates angiogenesis, blood coagulation as well as inflammatory responses. A delicate balance of these factors is maintained in healthy physiology; disrupting this balance causes endothelial dysfunction, which is a contributing factor to cardiovascular disease and stroke. In humans endothelial dysfunction is characterized by an imbalance in the release of vasoactive factors, which leads to an alteration in vascular tone. The endothelium releases both relaxant and constricting substances that act together to regulate vascular tone. In 1980, the importance of the endothelial cells lining vessel walls to acetylcholine (ACh)- induced relaxation of the blood vessel was revealed (1). Since then, three key players of vasodilation have been identified and studied; nitric oxide (NO), prostacylin (PGI2) and endothelium- derived hyperpolarizing factor (EDHF), with the identity of this last factor still being a subject of debate. These three factors will be discussed here in the context of the cerebrovasculature, with particular emphasis on EDHF (2, 3, 4).
The contribution of endothelial- derived factors to vasodilation: the effects of vessel size, injury and gender
1. Nitric Oxide
Before being identified as nitric oxide by Ignarro and colleagues (5), the vasodilating substance released by endothelial cells was referred to as endothelium- derived relaxing factor (EDRF). NO is generated from L-arginine by NO synthase (NOS), and diffuses to the vascular smooth muscle from the endothelial cell causing relaxation through activation of soluble guanylate cyclase, and increasing cyclic guanosine monophospate (cGMP) (6).
It has been shown that the amount of NO released from the endothelium varies with vessel size, resulting in varying relaxation of smooth muscle in vessels of different sizes, with NO- mediated dilation being most important in large vessels (7). Inhibition of NOS by NG-nitro-L-arginine (L-NNA) is more effective at attenuating ACh- induced relaxations of rat peripheral vessels in the aorta compared to smaller mesenteric vessels indicating that NO plays a more prominent role in larger than smaller vessel relaxation (8). This is consistent with higher endothelial NOS (eNOS) expression in the rat aorta than mesenteric arteries (9). Studying purinoceptor- induced vasodilation in isolated rat middle cerebral arteries (MCA), its branches and penetrating arterioles, it has also been demonstrated that the importance of NO- mediated vasodilation decreases along the cerebrovascular tree, with its effects being most pronounced in the MCA and decreasing with vessel size (10). In contrast to these studies where agonist-induced relaxations were studied, experiments have also been carried out aimed at investigating the effect of NO on basal tone by treatment of rat cerebral vessels with the NOS inhibitor, NG-monomethyl-L-arginine (L-NMMA) and measuring vessel constriction. In this paradigm, diameter- dependent vessel constrictions were observed, with large vessels exhibiting larger contractory responses than arterioles, showing that NO has a larger influence on basal tone of larger arteries than on the microvasculature in the brain (11). The possibility of arterioles having inherent impaired constrictor capacity has been discounted; these effects were shown to be due to decreased nitric oxide (11). Thus, both in the peripheral and cerebral circulations, the contribution of NO- induced relaxation is dependent on vessel size.
The vasodilatory responses of vascular smooth muscle to nitric oxide have been investigated following ischemic brain injury. Ex vivo using cerebral arterioles, the contractory effect of inhibition of NOS has been shown be diminished following ischemia/reperfusion, indicating that the regulation of blood flow is altered following stroke. It has been suggested, that this is not a result of an attenuated responsiveness to NO by the vascular smooth muscle but that production of NO is decreased following ischemia (12,13). Following MCA occlusion in vivo, enhancing levels of NO, either using its precursor L-arginine or the NO donor sodium nitroprusside, has been shown to be protective, increasing cerebral blood flow (CBF) to the ischemic region thus reducing tissue damage in rats (14,15). Conversely, reducing NO levels has deleterious effects, eNOS null mice display reduced CBF and larger infarct sizes following MCA occlusion compared to wild-type controls (16). Inhibition of NOS during reperfusion in rat stroke models results in a lack of CBF recovery, whereas vehicle treated rats recover CBF to pre-occlusion levels, highlighting the importance of endogenous NO in restoring blood flow following ischemia to large cerebral vessels (14,17). While this evidence suggests that NO donors may be therapeutically useful following stroke, it should be stressed that while endothelial NO has beneficial effects following stroke due to its effects on the cerebral vasculature and CBF, there is evidence that neuronal NO has deleterious effects on neurons (16,18). Also, while enhancing NO levels in the vasculature of mechanical stroke models in the rat is beneficial, stroke paradigms modelling arterial thrombosis- induced stroke showed no beneficial effects to L-arginine on CBF or infarct volume (19).
Sex differences have been shown to exist in stroke incidence, and estrogen is known to be neuroprotective in experimental models of cerebral ischemia (20, 21); this protection, in part, has been attributed to estrogen’s vasodilatory ability (22). It has been shown that estrogen receptor activation in cerebrovascular tissue leads to increased protein levels and activity of endothelial nitric oxide synthase (eNOS), indicating that increased NO production might play a role in the neuroprotective effects of estrogen (23). Taken together, it appears that, although it is down- regulated during ischemia, NO is protective in large cerebral vessels in cycling females compared to males.
2. Prostacyclin
Prostacyclin (PGI2), a product of arachidonic acid metabolism by cyclooxygenase (COX) enzymes, is produced by endothelial cells and has vasodilating properties. Consistent with this, exogenous PGI2 has been shown to increase CBF in baboons (24). The role of prostacyclin as an endothelial- derived vasodilator is of particular importance in infants. In human cerebral arteries, endothelium- dependent relaxations to ACh have been shown to be larger in infants than in adults; these relaxations in infants are suppressed by the PGI2 inhibitor indomethacin, but not L-NMMA, indicating that the primary relaxing factor in infants is PGI2. The reverse was true in adult vessels where NO was the primary factor responsible for vasodilation and PGI2 inhibition had no effect (25). It appears that the sensitivity to PGI2 decreases with age, with cerebrovascular dilation being prostacyclin- dependent and NO- independent in the infant, dependent on both in the juvenile and NO- dependent in the adult (25, 26).
Studies are scarce on whether PGI2- dependent vasodilation is influenced by vessel diameter. A study using different sizes of rat peripheral vessels showed that indomethacin did not affect the relaxant responses to ACh in any of the vessels, regardless of size (8). Given that PGI2- mediated vasodilation is age- dependent, and these experiments were not carried out in infants, a diameter- dependent response may have been masked.
In the cerebrovasculature, prostacyclin levels are regulated following ischemic injury; protein and mRNA levels of prostacyclin synthase, the enzyme involved in the last step of the metabolism of arachidonic acid in generating PGI2, are increased following cerebral ischemia in the rat. It is co-localized with PECAM-1 to the endothelial cells of both large and small vessels in the brain. Increased PGI2 levels have been shown to be protective against ischemic injury, as forced expression of prostacyclin synthase, as well as administration of PGI2 analogues decreases infarct size (27–29). This effect, unlike the protective effect of NO, may be attributed to both endothelial and neuronal actions as PGI2 has been shown to protect cultured cortical neurons from hypoxia/reperfusion injury in vitro, while in vivo PGI2 increases CBF after MCA occlusion in cats and rats (29–31).
The expression of PGI2 appears to be sexually dimorphic. Endothelial cells isolated from human vein umbilical cord from male and female babies, show that male cells synthesize more PGI2 than female cells (32). Although PGI2 does not play a major role in vasodilation in the adult, this sex difference does continue into adulthood where ACh- dependent relaxation of eNOS null mesenteric arteries is unaffected by indomethacin in females but greatly reduced in males. It is thought that COX activity, to an extent, compensates for lack of NO in eNOS knockout mice in males as plasma PGI2 levels are increased in eNOS null mice in males but not in females (33).
3. Endothelium- derived hyperpolarizing factor
In the early 1980s a factor released from the endothelium that causes hyperpolarization in smooth muscle cells via opening of potassium channels was suggested, this relaxant factor has since been termed endothelium- derived hyperpolarizing factor (EDHF) (34–36). It was shown that acetylcholine, in addition to relaxation, was able to cause endothelium- dependent transient hyperpolarizations of vascular smooth muscle, and that these two phenomena were independent of each other. This was attributed to an endothelium- derived factor, one that was neither prostacyclin nor EDRF (NO) (36–38). The non- NO and non- PGI2 relaxations were shown to be mediated by potassium, as they were blocked by potassium channel inhibitors, while extracellular potassium ions mimicked the effects of EDHF. EDHF hyperpolarizes endothelial cells, increasing potassium ions in the myoendothelial space which cause smooth muscle relaxation. Potassium ions are also another candidate for EDHF (39). Accordingly, there are 4 main criteria for a vasodilatory response to be defined as being EDHF- mediated. First, as with NO and PGI2, the dilation must be endothelial dependent; removal of the endothelium would abolish the smooth muscle relaxation. Second, dilations must be independent of NO and PGI2, therefore inhibition of NOS or COX enzymes would not eliminate the vasodilatory response. Third, EDHF- mediated dilations must be accompanied by hyperpolarization of the vascular smooth muscle. Fourth, dilations are sensitive to potassium channel inhibition, in particular the calcium- activated potassium channel (K+Ca) located on the endothelial cells (10, 35,40). In the periphery both small (SKCa) and intermediate (IKCa) conductance channels must be inhibited to prevent EDHF- mediated vasodilation, while in the cerebral circulation IKCa are important in mediating hyperpolarization in the absence of NO, with SKCa contributing additionaly only when NOS is activated (41–43) The identity of EDHF is to this day a much-debated topic, it has been suggested that there are multiple EDHFs depending on the vascular bed. As discussed later, we believe that epoxyeicosatrienoic acids contribute to the EDHF response in the cerebral microvasculature.
The importance of EDHF- mediated vasodilation has, like nitric oxide, been shown to be influenced by vessel size. Much of the work regarding vessel size has been carried out on the aorta and various branches of mesenteric artery in the rat. Using isolated blood vessels from these vascular beds, it was shown that the smaller vessels were more sensitive to agents modulating the potassium channel, than larger vessels, but insensitive to NOS inhibition, demonstrating that the contribution of NO is more prominent in the aorta, while that of EDHF is more prominent in the smaller vessels of the mesenteric arteries. This therefore indicates that the EDHF contribution to endothelium- dependent relaxations and hyperpolarizations increases as the vessel size decreases (7–9,44). Similarly studies on human gastric arteries and rat conduit arteries have shown that the contribution of EDHF is greater in smaller than in larger vessels (45, 46). The same holds true in the cerebrovasculature. It has been observed in the rat brain, when studying purinoceptor- mediated vasodilation, that the role of EDHF becomes more prominent as vessel sizes decrease along the cerebrovascular tree (10), and a significant portion of the vasodilatory response to ADP in brain microvessels has been attributed to EDHF (47). It therefore appears that in both peripheral and cerebral vascular beds, the significance of EDHF to endothelium- dependent vasodilation increases as the vessel diameter decreases, which is the opposite for NO. This differing contribution of EDHF to vasodilation, as well as that of NO and PGI2, depending on vessel size is summarized in figure 1.
Figure 1.
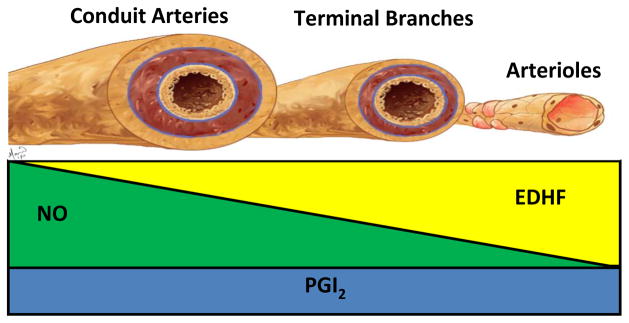
Schematic representation of the relative contribution of the 3 major endothelial- derived vasodilating factors. The vasodilatory contribution of nitric oxide (green) is largest in large calibre vessels such as conduit arteries, decreasing with vessel size. The opposite is observed for EDHF (yellow). Its vasodilatory role is most pronounced in small calibre vessels such as arterioles, this contribution to vessel relaxation decreases as vessel size increases. Whether the influence of prostacyclin (blue) is dependent on vessel size is less established, though it is expressed in both large and small vessels. The influence of contribution of prostacyclin in the adult is much smaller than that of NO and EDHF.
As with nitric oxide and prostacyclin, vasodilatory responses of EDHF are altered following ischemic injury. EDHF- mediated dilations seem to be potentiated following middle cerebral artery (MCA) occlusion and similar observations have been made using other cerebral injury models (48–50). The vast majority of studies into EDHF- mediated vasodilation following cerebral ischemia have been carried out in direct comparison to the altered responses to NO. Following ischemia/reperfusion (I/R), the potentiated EDHF-mediated dilations of the rat MCA were in contrast to diminished NO- mediated dilations; it has been suggested that the up-regulation of the EDHF mechanism compensates for a decline in NO- mediated vasodilation (48). This suggestion, together with findings in the peripheral circulation, lead to the belief that NO dampens EDHF- mediated responses in the brain, however it has been shown that NO does not suppress EDHF- mediated vasodilation in the MCA, and as mentioned previously both SKCa and IKCa are involved in mediating EDHF responses in the presence of NO, indicating that EDHF is also important in the uninjured state in the presence of NO (43, 51, 52). It has been reported that, following I/R, parenchymal arterioles were able to maintain EDHF responsiveness, however this was lost in the MCA (12). This resistance of the arterioles compared to the arteries is consistent with EDHF- mediated relaxations being more significant in small diameter vessels, and EDHF responses being more resistant to I/R than NO. The concept of compensation by EDHF for the diminished NO- mediated responses has also been demonstrated in the periphery, where EDHF- mediated vasodilation was upregulated in eNOS- null mice (53, 54). In both the brain and periphery it appears that, while in the normal physiological state both NO and EDHF contribute to vasodilation, EDHF plays a more pronounced role when NO responses are dampened following injury (12,48,51,52,55,56).
The actions of EDHF have, as for nitric oxide, been shown to be sexually dimorphic. EDHF has been shown to be the principal endothelium- derived vasodilator in female mice as mean arterial blood pressure was unaffected in eNOS and COX-1 double knock-out mice (the “EDHF” mouse); however it was elevated in male mice. This indicates that either the compensatory mechanism of EDHF for the loss of NO is functionally more important in females than in males or that the EDHF component is physiologically more important in regulating blood pressure in females than males (33,57). This latter explanation appears to be the case in mesenteric arteries where EDHF release is increased in females compared to males. This sex difference has been shown to be estrogen- dependent, as reduced EDHF- mediated relaxation is observed in ovariectomized females, but can be reversed by addition of exogenous estrogen (57–59). A greater EDHF component has also been observed in the femoral artery, where female mice exhibited greater EDHF- mediated relaxation compared to males in response to ACh (60). In the cerebrovasculature, the sex differences in EDHF- mediated vasodilation are of stark contrast to those just described in the periphery. It has been shown that smooth muscle cell hyperpolarization of the MCA is decreased in females compared to males (61). Furthermore, compared to males, ATP- induced EDHF-mediated dilations are negligible in the MCA of female rats, though dilations can be increased by ovariectomy, indicating that estrogen also plays a role in mediating this sex difference (62). These contrasting gender differences in the brain and periphery highlight that the nature of EDHF- mediated responses are very much dependent on the particular vascular bed. A possible explanation for these seemingly conflicting phenomena may be that these gender differences were studied in the MCA, which is a large vessel. As discussed earlier, the EDHF contribution to vasodilation increases as vessel size decreases; further studies are needed to shed light on whether the effects of estrogen observed in the periphery may hold true in the cerebral microvasculature (10).
Different identity of EDHF in these different vascular beds. Also, as will be discussed later, EDHF responses vary in large versus small diameter vessels in the brain. These studies showing a very small effect in the female have been carried out on the MCA. The results may be very different in the cerebral microvessels.
Is epoxyeicosatrienoic acid the EDHF in the brain?
The identity of EDHF has remained elusive and hotly debated over recent years. Candidates include epoxyeicosatrienoic acids, potassium ions, hydrogen peroxide and myoendothelial gap junctions among others (39, 63–66). The contribution of each of these has been the subject of many reviews (55, 67); this review will focus on epoxyeicosatrienoic acids as EDHF.
Epoxyeicosatrienoic acids (EETs) are endothelium- derived vasodilating eicosanoids. They are metabolites of arachidonic acid, formed by cytochrome P450 epoxygenase (CYP450). There are 4 EET regioisomers formed from arachidonic acid: 5,6-, 8,9-, 11,12-, 14–15-EET. In the brain, EETs are produced by both endothelial cells and by astrocytes indicating that the cerebrovasculature has a dual supply of EETs; for the purposes of this review, endothelium- derived EETs only will be discussed (68–70, unpublished results).
Endothelial EETs are produced in response to chemical and physical stimuli including muscarinic agonists such as acetylcholine and methacholine as well as bradykinin and shear stress (68, 70). They have vasodilatory actions in the brain and heart, as well as in other vascular beds. In bovine coronary arteries, exogenous arachidonic acid has been shown to induce endothelium- dependent relaxations. Inhibition of CYP450, using SKF 525a, reduces this relaxation suggesting that the product of arachidonic acid metabolism, EET, is involved in this relaxation of the coronary artery. In fact, all 4 EET regioisomers have been shown to be synthesized in bovine coronary artery endothelial cells from arachidonic acid and all produce concentration- dependent relaxation of coronary smooth muscle (71–73). Circulating EETs are known to be taken back up by endothelial cells and incorporated into phospholipids; it has been suggested that this stored supply of EETs is able to produce chronic vasoactive effects. The addition of exogenous EETs to coronary arterial ring preparations is able to increase the relaxation response to bradykinin, however on inhibition of EET incorporation into endothelial phospholipids, this potentiation is lost. This suggests that, in the coronary circulation at least, the phospholipase activity of bradykinin is able to release stored EETs, which act to enhance vasodilation (74, 75). Vasodilatory EETs are therefore synthesized by, and stored in vascular endothelial cells. If EETs are indeed an EDHF, it is possible that this store is important for the compensatory actions of EDHF when NO is compromised.
In the brain, EETs play an important role in the regulation of blood flow. Studies using laser-Doppler flowmetry have shown that, in the rat, inhibition of CYP450 results in reduced blood flow in the cerebral microcirculation, this is independent of any effect of CYP450 inhibition on NOS and therefore attributed to a decrease in formation of EETs globally in the brain (76). Experiments aimed at altering EETs levels by manipulating their metabolism rather than inhibiting their formation have shown similar results. Mice lacking the enzyme responsible for degrading EETs, soluble epoxide hydrolase (sEH), exhibit increases in cortical blood flow, while over-expression of sEH conditionally in the endothelium decreases cerebral blood flow (77); unpublished results). These effects are presumably due to the vasodilatory actions of increased and decreased EET levels, respectively, in the brain. Using ischemic models of cerebral injury, our laboratory has shown that sEH- null mice display increased cerebral blood flow during vessel occlusion and reduced infarct size following the insult, suggesting that this protection is linked to the vasodilatory effects of increased levels of EETs in the brain microvasculature during ischemia, resulting in increased cerebral perfusion and thus reduced injury. Such observations highlight the importance of EETs in regulating vascular function in the brain during stroke (77).
While EETs are established vasodilatory agents, since it was first suggested in the 1980s, speculation still remains on the matter of whether EETs constitute EDHF (78). There is much experimental evidence supporting this notion in certain vascular beds. Indeed, the work carried out in the cardiac circulation has led to the acceptance that EETs are the EDHF in the heart. Using isolated preparations of pre-contracted bovine coronary artery it has been shown that EDHF- mediated relaxations (i.e. nitric oxide- independent dilations sensitive to K+Ca channel inhibitors) are sensitive to CYP450 inhibition indicating that EETs are involved in this response. 14,15- and 11,12- EET are known to increase the open-state probability of K+Ca channels; it therefore follows that dilations induced by exogenous 11,12-EET are abolished on K+Ca channel inhibition. Furthermore, agonist- induced hyperpolarization of bovine and porcine coronary smooth muscle is abolished by CYP450 inhibition but enhanced by CYP450 induction in the pig. This suggests that, in the cardiac circulation, endothelial- derived EETs, synthesized by CYP450, are a vasodilating, hyperpolarizing factor that act by opening K+Ca channels, consistent with the criteria discussed earlier for EDHF (64, 65, 79).
Evidence for EETs being an EDHF are not confined to the cardiac circulation. Studies on skeletal muscle arterioles in eNOS null mice have shown that EDHF- mediated dilations in response to acetylcholine are attenuated by inhibitors of cytochrome P450, indicating that EDHF in these vessels is, as in the heart, synthesized by cytochrome P450. Similar results have been observed in the rat aorta and renal circulation. In humans a CYP450- mediated EDHF response is seen in mammary and forearm circulations as well as cardiac and skeletal circulations (54, 55, 80, 81).
There is also evidence in the brain that the actions of EETs are consistent with that of EDHF. NO- and prostacyclin- independent vasodilations have been shown to be mediated by a cytochrome P450 product, inhibition of this enzyme in the guinea pig cerebral artery attenuated this relaxation indicating that a decrease in the production of EETs reduces vasodilation (82). Mechanisticaly, the relaxation to exogenous 8,9- and 11,12- EET have been shown to be mediated by an increase in activation of smooth muscle cell potassium channels in feline cerebral arteries (83). Recent evidence also suggests that, via TRPV4 channels on cerebral artery smooth muscle cells, 11,12-EET is able to cause membrane hyperpolarization, which in turn causes activation of calcium- activated potassium channels (84). It is important to note that other components of the arachidonic acid pathway, upstream of EETs have also been shown to act in a similar manner. Phospholipase A2, which releases arachidonic acid from phospholipids, has been shown in the MCA to be involved in UTP- induced increases in endothelial calcium concentrations and vasodilation by regulating TRPV4 channels (85).
In the cerebral vasculature, evidence suggests that there is not a single factor operating as EDHF, and that the identity of EDHF may change along the vascular tree. For instance, there is evidence indicating that EETs do not contribute to the EDHF response in large calibre vessels (86). The majority of studies investigating the identity of EDHF in the brain have been carried out on the rat MCA where potassium ions and gap junctions appear to mediate the EDHF response. It has been shown that UTP- induced dilations, which are accompanied by endothelial cell hyperpolarization, require stimulation of the IKCa channel (42), and it has been suggested that potassium ions act as an EDHF in the MCA (87). It has been demonstrated that the manner in which the endothelial cell hyperpolarization becomes transmitted to the vascular smooth muscle, allowing for vasodilation to occur, is via gap junctions. Blockade of these junctions, while having no effect on endothelial hyperpolarization, reduced smooth muscle hyperpolarization and subsequent dilation. (88). It therefore appears that in the MCA, EDHF- mediated vasodilation can be explained by activation of IKCa channels, potassium ions and myoendothelial gap junctions which allow for communication between the endothelial cells and vascular smooth muscle.
In the large calibre MCA You and colleagues demonstrated that EETs are not an EDHF in this vessel. Using epoxygenase inhibitors, thus blocking EET production, they showed that UTP- induced EDHF- mediated dilations were only slightly decreased. Also, use of the EET antagonist 14,15-EEZE had no effect (86). However, in small calibre vessels in the brain there is evidence that EETs may in fact mediate the EDHF response. Using isolated cerebral penetrating arterioles, it has been shown that, ATP- induced vasodilation is dependent on EETs. Inhibition of EET production by MS-PPOH reduced vessel dilation in response to ATP but had no effect on basal tone. These studies were not carried out in the presence of NO or prostacyclin inhibitors. However, it was observed that the ATP- induced dilations were sensitive to IKCa inhibition, but not SKCa, consistent with a small contribution of NO to vasodilation in microvessels (10, 43, 89). Taken together, this data indicates that while EETs may not be an EDHF in large calibre vessels in the brain, they may contribute to EDHF- mediated vasodilation in the cerebral microvasculature.
Consistent with the known mediators of endothelial- mediated vasodilation NO and PGI2, and also EDHF, the actions of EETs have been also been shown to be sexually dimorphic in the periphery. Using gracilis arterioles, experiments have shown that vasodilation in females is mediated by EETs, however in males NO is the predominant vasodilatory mechanism, this is consistent with EETs being EDHF in the periphery. Vasodilation is decreased in OVX females and increased by estrogen replacement indicating that this gender difference is dependent on estrogen (90, 91). In the brain, estradiol has been shown to decrease sEH levels in OVX rats, however there have been no studies into gender differences in the levels of EETs in the brain as a whole or in the cerebral endothelium (92). Investigations into whether EETs levels in the brain, particularly in microvascular endothelial cells, differ between males and females, and their influence on vasodilation will be pertinent in assessing EETs as EDHF in the brain.
It is important to note that in the periphery, there have been lines of argument against a role for EETs as EDHF, these however, are largely confined to gastric and mesenteric circulations. In the human gastric vascular bed, while there is evidence for the actions of an EDHF in microvessels, potassium channel- mediated relaxations of smooth muscle are not inhibited by CYP450 inhibition, indicating that EETs are not involved in the EDHF response (45). In the mouse mesentery, the EDHF response was shown to be mediated by hydrogen peroxide and not P450 metabolites. In one study P450 eicosanoids were vasodilatory, however this was brought about by their ability to activate eNOS, while another study found that inhibiting P450 epoxygenases had no effect on vasodilation in either male or female rats; both of these studies indicate that EETs are not EDHF in the mesentery (93, 94). Also, in both mouse hindlimb and small femoral arteries, inhibition of P450 epoxygenase has been shown not to effect EDHF- mediated relaxation, where gap junctions were implicated instead (60). Even in the coronary circulation, where it is established that EETs constitute the EDHF response, it has been suggested that in the human microcirculation, hydrogen peroxide is more important as an EDHF than EETs as hydrogen peroxide inhibits CYP450 enzymes, and that EETs compensate when there is lack of hydrogen peroxide (95).
The issue of whether EETs are EDHF has therefore been the subject of many investigations in many species and vascular beds. It has been suggested that the identity of EDHF may be different between vascular beds (82), this is consistent with contradicting evidence on whether EETs are EDHF in different vessels.
In the cerebrovasculature, EETs do appear to fulfil the 4 criteria described earlier that have to be met by an EDHF. (1) EETs are produced by cerebral endothelial cells and cause vasodilation, which is (2) independent of NO and PGI2. (3) This relaxation is brought about via vascular smooth muscle cell hyperpolarization and (4) activation of calcium- activated potassium channels. There is also evidence that the physiological actions of EETs may be consistent with EDHF in the brain. While it has been shown that EETs may not be an EDHF in the MCA, the vasodilatory effects of EDHF in the brain are known to be more pronounced in small rather than large diameter vessels and evidence does indicate that EETs may contribute to EDHF- mediated vasodilation in small calibre cerebral vessels (10, 86, 89). EDHF responses in the brain have been shown to be regulated by the sex steroid estrogen, with EDHF- mediated dilations being diminished in the female MCA compared to males; whether such a gender difference is observed for the actions of EETs remains to be determined.
Whether the physiological actions of EETs are compatible with those of EDHF are less clear.. To date, there have not been any studies directly comparing the effect of EETs on vasodilation between cerebral vessels of different diameters. EDHF responses in the brain have been shown to be regulated by the sex steroid estrogen, with EDHF- mediated dilations being diminished in females compared to males.
The role of EETs in the brain following stroke and influence of sex
As already mentioned, EETs have been shown to be neuroprotective following stroke. Experimentally, EETs levels are commonly manipulated indirectly by altering the levels of the soluble epoxide hydrolase (sEH) enzyme responsible for the hydrolysis of EETs into their biologically less active metabolites. Enhancement of EET signaling in the brain is protective by both vascular and non-vascular means.
We have previously shown that reducing sEH levels in the brain is protective to mice following middle cerebral artery occlusion (MCAO). Using a pharmacological inhibitor of sEH, 12-(3-adamantan-1-yl-ureido)- dodecanoic acid butyl ester (AUDA-BE), infarct volume was reduced; an effect that was attributed to a non- vascular mechanism, as cerebral blood flow was unaltered on sEH inhibition (96)]. This protection was abolished on co-administration of N-methylsulfonyl-6-(2-propargyloxyphenyl) hexanamide (MS-PPOH), an inhibitor of P450 expoygenase, indicating that these results are due to increased EETs in the brain. Similarly, sEH- null mice exhibit smaller infarct sizes following MCAO (77)]. However in this paradigm and in contrast to the inhibitor study, the protective effects were attributed to increased cerebral blood flow.
Different polymorphisms in the EPHX2 gene, which encodes sEH, are associated with different susceptibilities to stroke. In humans, certain populations carrying an EPHX2 polymorphism (R287Q) resulting in decreased sEH activity (and thus presumably increased EET levels) have a reduced risk of ischemic stroke (97)]. When this same polymorphism is introduced into cultured cortical neurons, they are protected from an in-vitro model of ischemia, oxygen- glucose deprivation (OGD). While in contrast, neurons expressing a human sEH polymorphism resulting in increased sEH enzymatic activity displayed increased cell death following OGD (98).
It therefore appears that sEH inhibition is protective against ischemic stroke by a variety of methods, not only by vasodilation and increased cerebral blood flow, but also by direct actions on neurons.
In the brain, the expression and activity of sEH are lower in females than males. This corresponds with increased protection in females against cerebral ischemia, who display both increased blood flow and a smaller infarct size compared to males. In rodents, these sex differences have been demonstrated to be due to estrogen. Estrogen decreases sEH protein in the brain, while ovariectomy increases levels, and functionally ovariectomy of females increases infarct size to levels similar to those seen in males. This sex difference is also abolished in sEH- null mice, where male infarct sizes are reduced to those seen in females (92, 99).
FUTURE PERSPECTIVE
With further investigation into the influence of gender differences on endothelial EETs in the brain, and in particular of estrogen, manipulation of epoxyeicosanoid levels may provide an attractive avenue for gender- specific medical intervention for devastating cerebrovascular diseases such as stroke. Although manipulating sEH/EET levels in humans poses its own problems, a positive aspect of this route is that EETs are not deleterious to neurons, which is in contrast to other endothelial- derived substances such as nitric oxide. Furthermore if, true to being an EDHF in the brain, the vasodilator influence of EETs are dependent on vessel size, EETs may not just represent a gender- specific therapy, but may also be used differentially in small vessel disease, such as lacunar stroke and white matter lesion (WML)-associated vascular cognitive disorder (VCD).
EXECUTIVE SUMMARY.
Endothelial dysfunction
An imbalance in vasoactive substances released from the endothelium is a factor contributing to vascular disease including stroke.
The contribution of endothelial- derived factors to vasodilation; the effects of vessel size, injury and gender
-
Nitric Oxide
The vasodilator effects of NO are most important in large calibre blood vessels
NO levels are decreased following ischemic insult.
Increasing endothelial NO levels is protective against ischemic injury in the brain by increasing cerebral blood flow, however this may not be therapeutically useful as NO has deleterious effects on neurons.
Estrogen may be protective against ischemia by increasing NO levels, affording protection to large diameter vessels in females.
-
Prostacyclin
The vasodilatory effects of PGI2 do not appear to be dependent on vessel size.
Vasodilatory properties are most pronounced in infants.
Prostacyclin synthase levels are increased following ischemic insult, and increased PGI2 levels are protective against ischemia.
Prostacyclin levels are increased in males compared to females, indicating that it plays a compensatory role for a lack of NO in males but not females.
-
EDHF
The vasodilatory effects of EDHF are most important in small calibre vessels.
Compensates for lack of NO after injury, but is also important under normal physiological conditions.
In the periphery, EDHF plays are more prominent role in females than males; in the cerebral vasculature the reverse appears to occur.
Is epoxyeicosatrienoic acid the EDHF in the brain?
EETs in the brain meet the 4 criteria of the mechanism of action of EDHF.
EETs are vasodilatory; increasing their levels during ischemic insult is protective by increasing cerebral blood flow.
Gender differences in EETs levels in brain endothelial cells have not been studied; this will be important in assessing whether the physiological actions of EETs in the brain hold true to being part of the EDHF mechanism.
The effects of EETs on vasodialtion are dependent on vessel size; they are more prominent in small rather than large calibre vessels.
The role of EETs in the brain following stroke and influence of sex
-
sEH inhibition is protective against cerebral ischemia:
Pharmacological inhibition and genetic ablation of sEH decreases infarct size in vivo, by both vascular and non- vascular actions.
Human polymorphisms decreasing sEH levels decrease neuronal death following ischemia in vitro. The reverse is true for polymorphisms increasing sEH.
sEH expression is lower in the female brain, affording them protection against ischemia. sEH levels are regulated by estrogen.
Conclusions
EETs contribute to EDHF- mediated vasodilation in the cerebral vasculature.
It is possible that endothelial EETs are preferentially protective against ischemic brain insult in females than males. Further studies are needed to determine this.
EETs appear to have a greater vasodilatory role in small versus large calibre vessels, however further studies are needed to investigate a direct comparison between cerebral vessels of different diameters.
EETs, by increasing cerebral blood flow, may be protective to brain microvessels following stroke.
Contributor Information
Catherine M Davis, Email: davis@ohsu.edu.
Dominic A Siler, Email: silerd@ohsu.edu.
Nabil J Alkayed, Email: alkayedn@ohsu.edu.
References
- 1.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980 Nov 27;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 2.Flammer AJ, Luscher TF. Human endothelial dysfunction: EDRFs. Pflugers Arch. May;459:1005–1013. doi: 10.1007/s00424-010-0822-4. [DOI] [PubMed] [Google Scholar]
- 3.Feletou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond) 2009 Aug;117:139–155. doi: 10.1042/CS20090096. [DOI] [PubMed] [Google Scholar]
- 4.Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 2009 Jun;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 5.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987 Dec;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toda N, Ayajiki K, Okamura T. Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev. 2009 Mar;61:62–97. doi: 10.1124/pr.108.000547. [DOI] [PubMed] [Google Scholar]
- 7.Hwa JJ, Ghibaudi L, Williams P, Chatterjee M. Comparison of acetylcholine-dependent relaxation in large and small arteries of rat mesenteric vascular bed. Am J Physiol. 1994 Mar;266:H952–958. doi: 10.1152/ajpheart.1994.266.3.H952. [DOI] [PubMed] [Google Scholar]
- 8.Tomioka H, Hattori Y, Fukao M, et al. Relaxation in different-sized rat blood vessels mediated by endothelium-derived hyperpolarizing factor: importance of processes mediating precontractions. J Vasc Res. 1999 Jul-Aug;36:311–320. doi: 10.1159/000025659. [DOI] [PubMed] [Google Scholar]
- 9.Shimokawa H, Yasutake H, Fujii K, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996 Nov;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- 10.You J, Johnson TD, Marrelli SP, Bryan RM., Jr Functional heterogeneity of endothelial P2 purinoceptors in the cerebrovascular tree of the rat. Am J Physiol. 1999 Sep;277:H893–900. doi: 10.1152/ajpheart.1999.277.3.H893. [DOI] [PubMed] [Google Scholar]
- 11.Faraci FM. Role of endothelium-derived relaxing factor in cerebral circulation: large arteries vs microcirculation. Am J Physiol. 1991 Oct;261:H1038–1042. doi: 10.1152/ajpheart.1991.261.4.H1038. [DOI] [PubMed] [Google Scholar]
- 12.Cipolla MJ, Smith J, Kohlmeyer MM, Godfrey JA. SKCa and IKCa Channels, myogenic tone, and vasodilator responses in middle cerebral arteries and parenchymal arterioles: effect of ischemia and reperfusion. Stroke. 2009 Apr;40:1451–1457. doi: 10.1161/STROKEAHA.108.535435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cipolla MJ, Bullinger LV. Reactivity of brain parenchymal arterioles after ischemia and reperfusion. Microcirculation. 2008 Aug;15:495–501. doi: 10.1080/10739680801986742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang F, Iadecola C. Nitroprusside improves blood flow and reduces brain damage after focal ischemia. Neuroreport. 1993 May;4:559–562. doi: 10.1097/00001756-199305000-00024. [DOI] [PubMed] [Google Scholar]
- 15.He Z, Ibayashi S, Nagao T, Fujii K, Sadoshima S, Fujishima M. L-arginine ameliorates cerebral blood flow and metabolism and decreases infarct volume in rats with cerebral ischemia. Brain Res. 1995 Nov 20;699:208–213. doi: 10.1016/0006-8993(95)00907-8. [DOI] [PubMed] [Google Scholar]
- 16.Dawson VL, Dawson TM. Nitric oxide neurotoxicity. J Chem Neuroanat. 1996 Jun;10:179–190. doi: 10.1016/0891-0618(96)00148-2. [DOI] [PubMed] [Google Scholar]
- 17.Prado R, Watson BD, Wester P. Effects of nitric oxide synthase inhibition on cerebral blood flow following bilateral carotid artery occlusion and recirculation in the rat. J Cereb Blood Flow Metab. 1993 Jul;13:720–723. doi: 10.1038/jcbfm.1993.91. [DOI] [PubMed] [Google Scholar]
- 18.Dalkara T, Yoshida T, Irikura K, Moskowitz MA. Dual role of nitric oxide in focal cerebral ischemia. Neuropharmacology. 1994 Nov;33:1447–1452. doi: 10.1016/0028-3908(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 19.Prado R, Watson BD, Zhao W, et al. L-arginine does not improve cortical perfusion or histopathological outcome in spontaneously hypertensive rats subjected to distal middle cerebral artery photothrombotic occlusion. J Cereb Blood Flow Metab. 1996 Jul;16:612–622. doi: 10.1097/00004647-199607000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998 Jan;29:159–165. doi: 10.1161/01.str.29.1.159. discussion 166. [DOI] [PubMed] [Google Scholar]
- 21.Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab. 2000 Apr;20:631–652. doi: 10.1097/00004647-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe Y, Littleton-Kearney MT, Traystman RJ, Hurn PD. Estrogen restores postischemic pial microvascular dilation. Am J Physiol Heart Circ Physiol. 2001 Jul;281:H155–160. doi: 10.1152/ajpheart.2001.281.1.H155. [DOI] [PubMed] [Google Scholar]
- 23.Mcneill AM, Zhang C, Stanczyk FZ, Duckles SP, Krause DN. Estrogen increases endothelial nitric oxide synthase via estrogen receptors in rat cerebral blood vessels: effect preserved after concurrent treatment with medroxyprogesterone acetate or progesterone. Stroke. 2002 Jun;33:1685–1691. doi: 10.1161/01.str.0000016325.54374.93. [DOI] [PubMed] [Google Scholar]
- 24.Pickard J, Tamura A, Stewart M, Mcgeorge A, Fitch W. Prostacyclin, indomethacin and the cerebral circulation. Brain Res. 1980 Sep 22;197:425–431. doi: 10.1016/0006-8993(80)91127-0. [DOI] [PubMed] [Google Scholar]
- 25.Charpie JR, Schreur KD, Papadopoulos SM, Webb RC. Endothelium dependency of contractile activity differs in infant and adult vertebral arteries. J Clin Invest. 1994 Mar;93:1339–1343. doi: 10.1172/JCI117093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willis AP, Leffler CW. Endothelial NO and prostanoid involvement in newborn and juvenile pig pial arteriolar vasomotor responses. Am J Physiol Heart Circ Physiol. 2001 Dec;281:H2366–2377. doi: 10.1152/ajpheart.2001.281.6.H2366. [DOI] [PubMed] [Google Scholar]
- 27.Fang YC, Wu JS, Chen JJ, et al. Induction of prostacyclin/PGI2 synthase expression after cerebral ischemia-reperfusion. J Cereb Blood Flow Metab. 2006 Apr;26:491–501. doi: 10.1038/sj.jcbfm.9600205. [DOI] [PubMed] [Google Scholar]
- 28.Dogan A, Temiz C, Turker RK, Egemen N, Baskaya MK. Effect of the prostacyclin analogue, iloprost, on infarct size after permanent focal cerebral ischemia. Gen Pharmacol. 1996 Oct;27:1163–1166. doi: 10.1016/s0306-3623(96)00051-1. [DOI] [PubMed] [Google Scholar]
- 29.Shima K, Umezawa H, Chigasaki H, Okuyama S, Araki H. Stable prostacyclin improves postischaemic microcirculatory changes in hypertensive rats. Acta Neurochir (Wien) 1995;137:89–95. doi: 10.1007/BF02188788. [DOI] [PubMed] [Google Scholar]
- 30.Cazevieille C, Muller A, Bonne C. Prostacyclin (PGI2) protects rat cortical neurons in culture against hypoxia/reoxygenation and glutamate-induced injury. Neurosci Lett. 1993 Sep 17;160:106–108. doi: 10.1016/0304-3940(93)90924-a. [DOI] [PubMed] [Google Scholar]
- 31.De La Torre JC. Synergic activity of combined prostacyclin: dimethyl sulfoxide in experimental brain ischemia. Can J Physiol Pharmacol. 1991 Feb;69:191–198. doi: 10.1139/y91-028. [DOI] [PubMed] [Google Scholar]
- 32.Batres RO, Dupont J. Gender differences in prostacyclin and prostaglandin E2 synthesis by human endothelial cells. Prostaglandins Leukot Med. 1986 May;22:159–171. doi: 10.1016/0262-1746(86)90085-5. [DOI] [PubMed] [Google Scholar]
- 33.Scotland RS, Madhani M, Chauhan S, et al. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005 Feb 15;111:796–803. doi: 10.1161/01.CIR.0000155238.70797.4E. [DOI] [PubMed] [Google Scholar]
- 34.Bolton TB, Lang RJ, Takewaki T. Mechanisms of action of noradrenaline and carbachol on smooth muscle of guinea-pig anterior mesenteric artery. J Physiol. 1984 Jun;351:549–572. doi: 10.1113/jphysiol.1984.sp015262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kauser K, Stekiel WJ, Rubanyi G, Harder DR. Mechanism of action of EDRF on pressurized arteries: effect on K+ conductance. Circ Res. 1989 Jul;65:199–204. doi: 10.1161/01.res.65.1.199. [DOI] [PubMed] [Google Scholar]
- 36.Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br J Pharmacol. 1988 Dec;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br J Pharmacol. 1988 Mar;93:515–524. doi: 10.1111/j.1476-5381.1988.tb10306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komori K, Lorenz RR, Vanhoutte PM. Nitric oxide, ACh, and electrical and mechanical properties of canine arterial smooth muscle. Am J Physiol. 1988 Jul;255:H207–212. doi: 10.1152/ajpheart.1988.255.1.H207. [DOI] [PubMed] [Google Scholar]
- 39.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998 Nov 19;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 40.Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor. Clin Exp Pharmacol Physiol. 1996 Dec;23:1082–1090. doi: 10.1111/j.1440-1681.1996.tb01174.x. [DOI] [PubMed] [Google Scholar]
- 41.Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002 Aug;23:374–380. doi: 10.1016/s0165-6147(02)02050-3. [DOI] [PubMed] [Google Scholar]
- 42.Marrelli SP, Eckmann MS, Hunte MS. Role of endothelial intermediate conductance KCa channels in cerebral EDHF-mediated dilations. Am J Physiol Heart Circ Physiol. 2003 Oct;285:H1590–1599. doi: 10.1152/ajpheart.00376.2003. [DOI] [PubMed] [Google Scholar]
- 43.Mcneish AJ, Sandow SL, Neylon CB, Chen MX, Dora KA, Garland CJ. Evidence for involvement of both IKCa and SKCa channels in hyperpolarizing responses of the rat middle cerebral artery. Stroke. 2006 May;37:1277–1282. doi: 10.1161/01.STR.0000217307.71231.43. [DOI] [PubMed] [Google Scholar]
- 44.Adeagbo AS, Triggle CR. Varying extracellular [K+]: a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J Cardiovasc Pharmacol. 1993 Mar;21:423–429. [PubMed] [Google Scholar]
- 45.Urakami-Harasawa L, Shimokawa H, Nakashima M, Egashira K, Takeshita A. Importance of endothelium-derived hyperpolarizing factor in human arteries. J Clin Invest. 1997 Dec 1;100:2793–2799. doi: 10.1172/JCI119826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagao T, Illiano S, Vanhoutte PM. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-L-arginine in rats. Am J Physiol. 1992 Oct;263:H1090–1094. doi: 10.1152/ajpheart.1992.263.4.H1090. [DOI] [PubMed] [Google Scholar]
- 47.Faraci FM, Lynch C, Lamping KG. Responses of cerebral arterioles to ADP: eNOS-dependent and eNOS-independent mechanisms. Am J Physiol Heart Circ Physiol. 2004 Dec;287:H2871–2876. doi: 10.1152/ajpheart.00392.2004. [DOI] [PubMed] [Google Scholar]
- 48.Marrelli SP, Khorovets A, Johnson TD, Childres WF, Bryan RM., Jr P2 purinoceptor-mediated dilations in the rat middle cerebral artery after ischemia-reperfusion. Am J Physiol. 1999 Jan;276:H33–41. doi: 10.1152/ajpheart.1999.276.1.H33. [DOI] [PubMed] [Google Scholar]
- 49.Marrelli SP. Altered endothelial Ca2+ regulation after ischemia/reperfusion produces potentiated endothelium-derived hyperpolarizing factor-mediated dilations. Stroke. 2002 Sep;33:2285–2291. doi: 10.1161/01.str.0000027439.61501.39. [DOI] [PubMed] [Google Scholar]
- 50.Golding EM, You J, Robertson CS, Bryan RM., Jr Potentiated endothelium-derived hyperpolarizing factor-mediated dilations in cerebral arteries following mild head injury. J Neurotrauma. 2001 Jul;18:691–697. doi: 10.1089/089771501750357636. [DOI] [PubMed] [Google Scholar]
- 51.Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996 Dec 15;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- 52.Schildmeyer LA, Bryan RM., Jr Effect of NO on EDHF response in rat middle cerebral arteries. Am J Physiol Heart Circ Physiol. 2002 Feb;282:H734–738. doi: 10.1152/ajpheart.00583.2001. [DOI] [PubMed] [Google Scholar]
- 53.Waldron GJ, Ding H, Lovren F, Kubes P, Triggle CR. Acetylcholine-induced relaxation of peripheral arteries isolated from mice lacking endothelial nitric oxide synthase. Br J Pharmacol. 1999 Oct;128:653–658. doi: 10.1038/sj.bjp.0702858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang A, Sun D, Smith CJ, et al. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am J Physiol Heart Circ Physiol. 2000 Mar;278:H762–768. doi: 10.1152/ajpheart.2000.278.3.H762. [DOI] [PubMed] [Google Scholar]
- 55.Luksha L, Agewall S, Kublickiene K. Endothelium-derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis. 2009 Feb;202:330–344. doi: 10.1016/j.atherosclerosis.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 56.Zygmunt PM, Plane F, Paulsson M, Garland CJ, Hogestatt ED. Interactions between endothelium-derived relaxing factors in the rat hepatic artery: focus on regulation of EDHF. Br J Pharmacol. 1998 Jul;124:992–1000. doi: 10.1038/sj.bjp.0701893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCulloch AI, Randall MD. Sex differences in the relative contributions of nitric oxide and EDHF to agonist-stimulated endothelium-dependent relaxations in the rat isolated mesenteric arterial bed. Br J Pharmacol. 1998 Apr;123:1700–1706. doi: 10.1038/sj.bjp.0701781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White RM, Rivera CO, Davison CA. Nitric oxide-dependent and -independent mechanisms account for gender differences in vasodilation to acetylcholine. J Pharmacol Exp Ther. 2000 Jan;292:375–380. [PubMed] [Google Scholar]
- 59.Liu MY, Hattori Y, Fukao M, Sato A, Sakuma I, Kanno M. Alterations in EDHF-mediated hyperpolarization and relaxation in mesenteric arteries of female rats in long-term deficiency of oestrogen and during oestrus cycle. Br J Pharmacol. 2001 Mar;132:1035–1046. doi: 10.1038/sj.bjp.0703899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luksha L, Poston L, Gustafsson JA, Hultenby K, Kublickiene K. The oestrogen receptor beta contributes to sex related differences in endothelial function of murine small arteries via EDHF. J Physiol. 2006 Dec 15;577:945–955. doi: 10.1113/jphysiol.2006.121939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sokoya EM, Burns AR, Marrelli SP, Chen J. Myoendothelial gap junction frequency does not account for sex differences in EDHF responses in rat MCA. Microvasc Res. 2007 Jul;74:39–44. doi: 10.1016/j.mvr.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Golding EM, Kepler TE. Role of estrogen in modulating EDHF-mediated dilations in the female rat middle cerebral artery. Am J Physiol Heart Circ Physiol. 2001 Jun;280:H2417–2423. doi: 10.1152/ajpheart.2001.280.6.H2417. [DOI] [PubMed] [Google Scholar]
- 63.Edwards G, Thollon C, Gardener MJ, et al. Role of gap junctions and EETs in endothelium-dependent hyperpolarization of porcine coronary artery. Br J Pharmacol. 2000 Mar;129:1145–1154. doi: 10.1038/sj.bjp.0703188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996 Mar;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 65.Fisslthaler B, Popp R, Kiss L, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999 Sep 30;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 66.Matoba T, Shimokawa H, Nakashima M, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000 Dec;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Feletou M, Vanhoutte PM. Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol. 2006 Jun;26:1215–1225. doi: 10.1161/01.ATV.0000217611.81085.c5. [DOI] [PubMed] [Google Scholar]
- 68.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 2010 May;459:881–895. doi: 10.1007/s00424-010-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman Rj, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996 May;27:971–979. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- 70.Medhora M, Narayanan J, Harder D. Dual regulation of the cerebral microvasculature by epoxyeicosatrienoic acids. Trends Cardiovasc Med. 2001 Jan;11:38–42. doi: 10.1016/s1050-1738(01)00082-2. [DOI] [PubMed] [Google Scholar]
- 71.Amruthesh SC, Falck JR, Ellis EF. Brain synthesis and cerebrovascular action of epoxygenase metabolites of arachidonic acid. J Neurochem. 1992 Feb;58:503–510. doi: 10.1111/j.1471-4159.1992.tb09749.x. [DOI] [PubMed] [Google Scholar]
- 72.Rosolowsky M, Campbell WB. Role of PGI2 and epoxyeicosatrienoic acids in relaxation of bovine coronary arteries to arachidonic acid. Am J Physiol. 1993 Feb;264:H327–335. doi: 10.1152/ajpheart.1993.264.2.H327. [DOI] [PubMed] [Google Scholar]
- 73.Rosolowsky M, Campbell WB. Synthesis of hydroxyeicosatetraenoic (HETEs) and epoxyeicosatrienoic acids (EETs) by cultured bovine coronary artery endothelial cells. Biochim Biophys Acta. 1996 Jan 19;1299:267–277. doi: 10.1016/0005-2760(95)00216-2. [DOI] [PubMed] [Google Scholar]
- 74.Vanrollins M, Kaduce TL, Fang X, Knapp HR, Spector AA. Arachidonic acid diols produced by cytochrome P-450 monooxygenases are incorporated into phospholipids of vascular endothelial cells. J Biol Chem. 1996 Jun 14;271:14001–14009. doi: 10.1074/jbc.271.24.14001. [DOI] [PubMed] [Google Scholar]
- 75.Weintraub NL, Fang X, Kaduce TL, Vanrollins M, Chatterjee P, Spector AA. Potentiation of endothelium-dependent relaxation by epoxyeicosatrienoic acids. Circ Res. 1997 Aug;81:258–267. doi: 10.1161/01.res.81.2.258. [DOI] [PubMed] [Google Scholar]
- 76.Alkayed NJ, Birks EK, Hudetz AG, Roman RJ, Henderson L, Harder DR. Inhibition of brain P-450 arachidonic acid epoxygenase decreases baseline cerebral blood flow. Am J Physiol. 1996 Oct;271:H1541–1546. doi: 10.1152/ajpheart.1996.271.4.H1541. [DOI] [PubMed] [Google Scholar]
- 77.Zhang W, Otsuka T, Sugo N, et al. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008 Jul;39:2073–2078. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rubanyi GM, Vanhoutte PM. Nature of endothelium-derived relaxing factor: are there two relaxing mediators? Circ Res. 1987 Nov;61:II61–67. [PubMed] [Google Scholar]
- 79.Gebremedhin D, Harder DR, Pratt PF, Campbell WB. Bioassay of an endothelium-derived hyperpolarizing factor from bovine coronary arteries: role of a cytochrome P450 metabolite. J Vasc Res. 1998 Jul-Aug;35:274–284. doi: 10.1159/000025594. [DOI] [PubMed] [Google Scholar]
- 80.Woodman OL, Boujaoude M. Chronic treatment of male rats with daidzein and 17 beta-oestradiol induces the contribution of EDHF to endothelium-dependent relaxation. Br J Pharmacol. 2004 Jan;141:322–328. doi: 10.1038/sj.bjp.0705603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Imig JD, Falck JR, Wei S, Capdevila JH. Epoxygenase metabolites contribute to nitric oxide-independent afferent arteriolar vasodilation in response to bradykinin. J Vasc Res. 2001 May-Jun;38:247–255. doi: 10.1159/000051053. [DOI] [PubMed] [Google Scholar]
- 82.Dong H, Jiang Y, Cole WC, Triggle CR. Comparison of the pharmacological properties of EDHF-mediated vasorelaxation in guinea-pig cerebral and mesenteric resistance vessels. Br J Pharmacol. 2000 Aug;130:1983–1991. doi: 10.1038/sj.bjp.0703474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gebremedhin D, Ma YH, Falck JR, Roman RJ, Vanrollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol. 1992 Aug;263:H519–525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- 84.Earley S. Endothelium-Dependent Cerebral Artery Dilation Mediated by Transient Receptor Potential and Ca2+-Activated K+ Channels. J Cardiovasc Pharmacol. 2010 Aug; doi: 10.1097/FJC.0b013e3181f580d9. [DOI] [PubMed] [Google Scholar]
- 85.Marrelli SP, O’Neil RG, Brown RC, Bryan RM., Jr PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol. 2007 Mar;292:H1390–1397. doi: 10.1152/ajpheart.01006.2006. [DOI] [PubMed] [Google Scholar]
- 86.You J, Golding EM, Bryan RM., Jr Arachidonic acid metabolites, hydrogen peroxide, and EDHF in cerebral arteries. Am J Physiol Heart Circ Physiol. 2005 Sep;289:H1077–1083. doi: 10.1152/ajpheart.01046.2004. [DOI] [PubMed] [Google Scholar]
- 87.Mcneish AJ, Dora KA, Garland CJ. Possible role for K+ in endothelium-derived hyperpolarizing factor-linked dilatation in rat middle cerebral artery. Stroke. 2005 Jul;36:1526–1532. doi: 10.1161/01.STR.0000169929.66497.73. [DOI] [PubMed] [Google Scholar]
- 88.Sokoya EM, Burns AR, Setiawan CT, Coleman HA, Parkington HC, Tare M. Evidence for the involvement of myoendothelial gap junctions in EDHF-mediated relaxation in the rat middle cerebral artery. Am J Physiol Heart Circ Physiol. 2006 Jul;291:H385–393. doi: 10.1152/ajpheart.01047.2005. [DOI] [PubMed] [Google Scholar]
- 89.Dietrich HH, Horiuchi T, Xiang C, Hongo K, Falck JR, Dacey RG., Jr Mechanism of ATP-induced local and conducted vasomotor responses in isolated rat cerebral penetrating arterioles. J Vasc Res. 2009;46:253–264. doi: 10.1159/000167273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu Y, Huang A, Sun D, Falck JR, Koller A, Kaley G. Gender-specific compensation for the lack of NO in the mediation of flow-induced arteriolar dilation. Am J Physiol Heart Circ Physiol. 2001 Jun;280:H2456–2461. doi: 10.1152/ajpheart.2001.280.6.H2456. [DOI] [PubMed] [Google Scholar]
- 91.Huang A, Sun D, Jacobson A, Carroll MA, Falck JR, Kaley G. Epoxyeicosatrienoic acids are released to mediate shear stress-dependent hyperpolarization of arteriolar smooth muscle. Circ Res. 2005 Feb 18;96:376–383. doi: 10.1161/01.RES.0000155332.17783.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koerner IP, Zhang W, Cheng J, Parker S, Hurn PD, Alkayed NJ. Soluble epoxide hydrolase: regulation by estrogen and role in the inflammatory response to cerebral ischemia. Front Biosci. 2008;13:2833–2841. doi: 10.2741/2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hercule HC, Schunck WH, Gross V, et al. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol. 2009 Jan;29:54–60. doi: 10.1161/ATVBAHA.108.171298. [DOI] [PubMed] [Google Scholar]
- 94.Morton JS, Rueda-Clausen CF, Davidge ST. Mechanisms of endothelium-dependent vasodilation in male and female, young and aged offspring born growth restricted. Am J Physiol Regul Integr Comp Physiol. 2010 Apr;298:R930–938. doi: 10.1152/ajpregu.00641.2009. [DOI] [PubMed] [Google Scholar]
- 95.Larsen BT, Gutterman DD, Sato A, et al. Hydrogen peroxide inhibits cytochrome p450 epoxygenases: interaction between two endothelium-derived hyperpolarizing factors. Circ Res. 2008 Jan 4;102:59–67. doi: 10.1161/CIRCRESAHA.107.159129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang W, Koerner IP, Noppens R, et al. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007 Dec;27:1931–1940. doi: 10.1038/sj.jcbfm.9600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang L, Ding H, Yan J, et al. Genetic variation in cytochrome P450 2J2 and soluble epoxide hydrolase and risk of ischemic stroke in a Chinese population. Pharmacogenet Genomics. 2008 Jan;18:45–51. doi: 10.1097/FPC.0b013e3282f313e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koerner IP, Jacks R, Debarber AE, et al. Polymorphisms in the human soluble epoxide hydrolase gene EPHX2 linked to neuronal survival after ischemic injury. J Neurosci. 2007 Apr 25;27:4642–4649. doi: 10.1523/JNEUROSCI.0056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang W, Iliff JJ, Campbell CJ, Wang RK, Hurn PD, Alkayed NJ. Role of soluble epoxide hydrolase in the sex-specific vascular response to cerebral ischemia. J Cereb Blood Flow Metab. 2009 Aug;29:1475–1481. doi: 10.1038/jcbfm.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]