

Abstract
Transforming growth factor β1 (TGF-β1) is a pleiotropic cytokine expressed throughout the CNS. Previous studies demonstrated that TGF-β1 contributes to maintain neuronal survival, but mechanistically this effect is not well understood. We generated a CNS-specific TGF-β1-deficient mouse model to investigate the functional consequences of TGF-β1-deficiency in the adult mouse brain. We found that depletion of TGF-β1 in the CNS resulted in a loss of the astrocyte glutamate transporter (GluTs) proteins GLT-1 (EAAT2) and GLAST (EAAT1) and decreased glutamate uptake in the mouse hippocampus. Treatment with TGF-β1 induced the expression of GLAST and GLT-1 in cultured astrocytes and enhanced astroglial glutamate uptake. Similar to GLT-1-deficient mice, CNS-TGF-β1-deficient mice had reduced brain weight and neuronal loss in the CA1 hippocampal region. CNS-TGF-β1-deficient mice showed GluN2B-dependent aberrant synaptic plasticity in the CA1 area of the hippocampus similar to the glutamate transport inhibitor DL-TBOA and these mice were highly sensitive to excitotoxic injury. In addition, hippocampal neurons from TGF-β1-deficient mice had elevated GluN2B-mediated calcium signals in response to extrasynaptic glutamate receptor stimulation, whereas cells treated with TGF-β1 exhibited reduced GluN2B-mediated calcium signals. In summary, our study demonstrates a previously unrecognized function of TGF-β1 in the CNS to control extracellular glutamate homeostasis and GluN2B-mediated calcium responses in the mouse hippocampus.
Keywords: TGF-β1, glutamate uptake, hippocampus, neuronal calcium, extrasynaptic, astrocytes
INTRODUCTION
Transforming growth factor-β (TGF-β) superfamily members exert essential functions during development and in maintaining tissue homeostasis (Abutbul et al. 2012; Massague et al. 2000; Schulz et al. 2009; Stipursky and Gomes 2007). In the central nervous system (CNS), TGF-βs and their receptors are expressed at low levels by both neurons and glial cells (Flanders et al. 1991; Hamby et al. 2010; Unsicker et al. 1991; Unsicker et al. 1996), and their expression is up-regulated in response to a wide range of neuronal insults and during aging (Boche et al. 2003; Henrich-Noack et al. 1996; Klempt et al. 1992; Krieglstein et al. 2002; Wiessner et al. 1993). Numerous studies have demonstrated that TGF-β1 exerts a protective effect against various neuronal insults (Flanders et al. 1998; Prehn et al. 1993a; Prehn et al. 1993b). In accord with those findings, TGF-β1-deficiency causes a widespread increase in degenerating neurons in vivo, suggesting a non-redundant function for TGF-β1 in maintaining neuronal integrity and survival (Brionne et al. 2003). However, mechanistically the effect of TGF-β1 on neuronal survival is not well understood.
Glutamate is considered the predominant excitatory neurotransmitter in the vertebrate nervous system (Dingledine et al. 1999). Glutamate causes calcium influx through N-Methyl-D-Aspartate-type glutamate receptors (NMDARs) to promote neuronal plasticity and survival (Citri and Malenka 2008). However, excessive accumulation of glutamate results in neuronal calcium overload and neuronal death, transforming physiologically regulated excitability into neuronal injury (Waxman and Lynch 2005),(Lau and Tymianski). Clearing of extracellular glutamate is thus essential for limiting neuronal cell death. In the hippocampus, astrocytes are primarily responsible for preventing the accumulation of extracellular glutamate through the high-affinity Na+-dependent GluTs (EAAT2) GLT-1 and GLAST (EAAT1) (Danbolt 2001; Kawahara et al. 2002). GluTs promote synapse independence by limiting glutamate ‘spill-over’ to neighboring synapses (Arnth-Jensen et al. 2002; Takayasu et al. 2006), glutamate ‘spill-out’ to extrasynaptic receptors (Gouix et al. 2009; Huang et al. 2004; Nie and Weng 2010; Sullivan et al. 2004) and they control postsynaptic excitation (Tzingounis and Wadiche 2007). GluTs thereby contribute to synaptic plasticity as well as to learning and memory (Bellesi et al. 2009; Katagiri et al. 2001; Levenson et al. 2002). Under physiological conditions, GLT-1 accounts for the majority of extracellular glutamate removal throughout the brain, except for the cerebellum and the retina where GLAST predominates (Danbolt 2001). In accord, homozygous mice deficient in GLT-1 show spontaneous lethal seizures and increased susceptibility to acute cortical injury (Tanaka et al. 1997).
Here, we investigated the effect of TGF-β1 on extracellular glutamate homoeostasis and synaptic plasticity. We found that deficiency of TGF-β1 in the CNS leads to a loss of the astroglial glutamate transporters GLT-1 and GLAST, which results in reduced glutamate uptake in the hippocampus. Conversely, treatment with TGF-β1 induced the expression of GLT-1 and GLAST in cultured astrocytes and increased glutamate uptake. We show that CNS-TGF-β1-deficient mice display aberrant forms of glutamate-binding NMDA receptor subunit 2B (GluN2B)-dependent synaptic plasticity in the CA1 area of the hippocampus. In addition, we found that TGF-β1 counteracts the consequences of excessive extracellular glutamate accumulation through a distinct mechanism in neurons, as TGF-β1 attenuated GluN2B-mediated calcium signals in hippocampal neurons independent from glutamate uptake. Together, these mechanisms maintain neuronal survival and support synaptic plasticity.
METHODS & MATERIALS
Animals
The Institutional Animal Care and Use Committee (IACUC) at Harvard Medical School approved all experimental procedures involving animals. Adult male and female heterozygous TGF-β1 mice (TGF-β1−/+) were kindly provided by Sharon M. Wahl (National Institutes of Health) and described elsewhere (Kulkarni et al. 1993). Il-2-transgenic mice that express TGF-β1 under the control of an Il-2 promoter were recently generated in our laboratory and described elsewhere (Carrier et al. 2007).
Real-time PCR
Total RNA was extracted from fresh hippocampus tissue or from cultured astrocytes after 10–15 days in vitro using RNAeasy columns (Qiagen). Complementary DNA (cDNA) was prepared as recommended (Applied Biosytems) and used as template for real-time PCR. The expression of TGF-β1, 2 and 3 or otherwise of GLT-1 and GLAST was quantified with specific primers (Applied Biosystems) on the GeneAmp 5500 Sequence Detection System (Applied Biosystems).
ELISA
The concentration of TGF-β1 in the serum was measured on a multiwell plate reader (Tecan) using the Quantikine Mouse/Rat/Porcine/Canine TGFβ1 Elisa (R&D) as recommended.
EAE and FACS
Experimental autoimmune encephalitis (EAE) was induced as described (Bannerman et al. 2007). Brain mononuclear cells were separated through Percoll (GE Healthcare Life Sciences) gradient centrifugation from brain homogenates, incubated with antibodies against surface CD4 and CD8 (1:100, BD Bioscience) and analyzed on a FACSCalibur (BD Biosciences).
Immunohistochemistry (IHC)
8–10 weeks old mice were euthanized by CO2 asphyxation and transcardially perfused with phosphate buffered saline (PBS) and 4% paraformaldehyde (PFA). Free-floating cryosections (40 μm thick) of the hippocampus were blocked with 1% bovine serum albumine (BSA) for 1 hr at room temperature (RT) and incubated with the following primary antibodies (in 1% BSA) overnight: rabbit anti-GLT-1 (Abcam, 1:100), rabbit anti-GLAST (1:100), mouse anti-GFAP (BD, 1:300), rabbit anti-S100B (Abcam, 1:100), mouse anti-NeuN (Millipore, 1:300), rat anti-CD4 (BD, 1:300). Sections were washed 3×5 min in PBS and incubated with Alexa 488- and Alexa 594-conjugated secondary antibodies (Molecular Probes) for 1 hr at RT. The sections were briefly incubated in TO-PRO 3 Iodide (Invitrogen) and mounted on glass slides. Stained sections were evaluated on a Zeiss LSM 510 confocal microscope and images were analyzed in Image J software (NIH).
Evaluation of neuronal apoptosis
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed as recommended (Roche) and tissue sections were subsequently incubated overnight at 4°C with the following primary antibodies diluted in 1% BSA: mouse anti-NeuN (Millipore, 1:300); rabbit anti-cleaved caspase-3 (Cell Signaling, 1:100), rabbit anti-IBA1 (Wako Chemicals, 1:200). Fluorescence-labeled anti-mouse or anti-rabbit secondary antibodies in blocking buffer were used to visualize primary antibodies (Molecular Probes, 1:300) for 60 min at RT. Sections were evaluated on a LSM 510 confocal laser-scanning microscope (Zeiss) using a 63x magnification. Nissl- or Haematoxylin-Eosin (H&E)-stained paraffin-embedded sections were used to evaluate the number of neurons in subregions of the hippocampus. Propidium Iodide (PI) staining was used to quantify cell death 24 hrs after NMDA exposure of cultured hippocampal neurons (DIV10-12) treated with either TGF-β1 or vehicle. Therefore, cells were exposed to PI (Invitrogen, 50 μg ml−1) dissolved in PBS for 10 min, fixed with 4% PFA and cellular outcomes were evaluated under a fluorescent microscope.
Dendritic Spine Imaging
Organotypic hippocampal slices (400 μm thick) were prepared and cultured from postnatal day 6–7 (P6-7) mice as described (Shankar et al. 2007; Tavazoie et al. 2005). Slices were biolistically transfected with the eGFP-N1 construct (Clontech) using a Helios Gene gun (Bio-Rad) after 2 days in vitro (DIV). Bullets were prepared using 12.5 mg of 1.6 μm-gold particles and 60 μg of plasmid DNA. For 2-photon laser microscopy (2PLM), slices were placed in the microscope chamber and perfused with artificial cerebrospinal fluid (ACSF) saturated with 95% O2 and 5% CO2. ACSF consisted of the following (in mM): 127 NaCl, 25 NaHCO3, 1.25 Na2HPO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 glucose. GFP-expressing neurons were imaged using a custom-built two-photon laser-scanning microscope with an excitation wavelength of 910 nm. Images were analyzed using custom software written in Matlab (Mathworks) as described (Shankar et al. 2007).
Tissue Preparation and Western blot
Whole-cell preparations were made from the hippocampus of 8–10 weeks animals or otherwise from cultured astrocytes after 10–15 days in vitro. Mice were euthanized by CO2 asphyxation and their brains quickly removed. The hippocampi were dissected in ice-cold HBSS containing 10 mM HEPES and the tissue was homogenized in ice-cold lysis buffer containing 150 mM NaCl, 25 mM Tris and 1% SDS (pH 7.4). Homogenates were rotated for 30 min at 4°C and centrifuged for 10 min at 12.000xg. The supernatant was removed and stored at −80°C until used and the pellet discarded. Astrocytes were lysed in the same buffer and tissue lysates were stored at −80°C until used. Protein concentration was normalized by BCA protein assay (Pierce). For Western blots, samples containing 25 μg protein were separated on a 4–12% Bis-Tris gel (Invitrogen) and transferred to nitrocellulose membranes (Whatman). Membranes were blocked (60min at RT in 5% dry milk in TBST) and incubated separately overnight with the following primary antibody (diluted in blocking buffer): rabbit anti-GLT-1 (Abcam, 1:1000), rabbit anti-GLAST (Abcam, 1:1000), rabbit anti-EAAC1 (a kind gift from Jeffrey Rothstein, 1:300), rabbit anti-Glutamine Synthase (GS) (Abcam, 1:1000), mouse anti-GFAP (BD, 1:3000), rabbit anti-PSD95 (Cell Signaling, 1:1000), rabbit anti-GluN1, rabbit anti-GluN2A (both Millipore, 1:1000), mouse anti-GluN2B (BD, 1:1000), rabbit anti-Smad2/3 (Cell Signaling, 1:1000), rabbit anti-Phospho-Smad2 (Ser 495/467) and rabbit anti-Phospho-Smad3 (Ser 423/425) (both Cell Signaling, 1:1000). For visualization, HRP-coupled secondary antibodies (Thermo Scientific) (RT, 1h) and enhanced chemiluminescence substrate (Thermo Scientific) was used. Optical density of immunoreactive bands was quantified with Image-J (NIH).
Measurement of Glutamate Uptake
To measure glutamate uptake, hippocampi from 8–10 weeks old mice were dissected in ice-cold ACSF containing the following (in mM): 124 NaCl, 2.8 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 MgSO4, 10 D-glucose. Transverse hippocampal slices (350 μm) were prepared using a tissue slicer (McIlwain) and the slices were recovered in oxygenated ACSF containing 2.5 mM CaCl2 for 1 hr at RT. Slices were incubated for 10 min in ACSF containing nm L-[3H]-glutamate (0.5 μCi) and 30 μM unlabbeled glutamate. Slices were rapidly chilled on ice, throurougly washed and solubilized in buffer containing 25 mM Tris and 1% SDS. When meassuring glutamate uptake in primary astrocytes in vitro, medium was removed and cells were briefly washed with ACSF, incubated for 10 min in ACSF containing nm L-[3H]-glutamate (0.5 μCi) and 30 μM unlabbeled glutamate and solubilized in buffer containing 25 mM Tris and 1% SDS. The radioactivity was meassured in a liquid scintilation counter (Beckman) and normalized to protein concentration. Na+-independent glutamate uptake was meassured by replacing sodium with choline chloride in ACSF.
Astrocyte culture
Primary astrocytes were prepared from 1-day old mice. Briefly, mice were euthanized, decapitated and brains were dissected in in cold HBSS buffer w/o Ca2+ and Mg2+ and the tissue triturated to a single-cell suspension using a papain-based dissociation kit (Miltenyi Biotec). Cells were initially cultured in DMEM + 10% fetal calf serum (FCS) in 75-cm2 tissue culture flasks until confluent after about 10 days in vitro (DIV) and then split and platted on PLL-coated 24 well-plates for further experiments as specified.
Electrophysiology
6–8 weeks old mice were sacrificed and their brain submerged in oxygenated sucrose-replaced artificial cerebrospinal fluid (ACSF) cutting solution containing the following (in mM): 206 sucrose, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 1 CaCl2, 1 MgCl2, 26 NaHCO3, 10 D-glucose, pH = 7.4, 315 mOsm. Transverse slices (350 μm thick) from the middle portion of each hippocampus were cut with a vibroslicer (McIlwain). After dissection, slices were incubated in ACSF that contained the following (in mM): 124 NaCl, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 2.5 CaCl2, 26 NaHCO3, 10 D-glucose, pH = 7.4, 310 mOsm. To record field fEPSPs in the CA1 region of the hippocampus a unipolar stimulating electrode (World Precision Instruments) was placed into the Schaffer collateral pathway of acute hippocampal slices to deliver test and conditioning stimuli. A borosilicate glass recording electrode filled with ACSF was positioned in stratum radiatum of CA1, 200–300 μm distant from the stimulating electrode. fEPSP in the CA1 region of the hippocampus were induced by test stimuli at 0.05 Hz with an intensity that elicited fEPSP amplitudes 50% of maximum. fEPSPs were amplified 100x using Axon 200B amplifier and digitized with Digidata 1322A (Axon Instruments). The data were sampled at 10 kHz and filtered at 2 kHz. Traces were obtained by pClamp9.2 and analyzed using the Clampfit9.2 software. Where specifically stated, treatment was applied 45–60 min prior the conditioning stimulus. Recombinant TGF-β1 (Peprotech) was dissolved PBS. SB-431542 (Sigma), D-AP5, MK-801, NBQX, bicuculline, DL-TBOA, Ro 25-6981, SB-203580 and SIB-1757 (all Tocris) were dissolved in dimethyl sulfoxide (DMSO). None of the used vehicles had a significant effect on fEPSP (ACSF or DMSO) at the concentrations used here.
Kainic-acid induced seizures
Kainic acid (KA) (Sigma) was dissolved in saline and male 6–8 weeks old mice were injected intraperitoneally with 10 mg kg−1 body weight. Mice that did not reach a seizure score > 4 (Ben-Ari and Cossart 2000) were excluded from further analysis.
Primary Hippocampal Neuronal Culture and Calcium Imaging
Primary hippocampal neurons were prepared from embryonic day-18 (E18) mice. Therefore, their hippocampi were dissected in cold HBSS buffer w/o Ca2+ and Mg2+ and the tissue triturated to a single-cell suspension using a papain-based dissociation kit (Miltenyi Biotec). Cells were counted and initially plated (100.000 cells/cm2) in DMEM containing 10% FCS on Poly-L-Lysin-coated (PLL) glass-bottom cell culture dishes (MatTek). One hour after plating, the medium was changed to Neurobasal containing B27 and Glutamax (Invitrogen), and thereafter half of the medium was changed every third day. At DIV 3, cytosin arabinosid (5 μM) was added to the culture medium. Hippocampal cultures at DIV 12–14 were used for all calcium imaging experiments. Cells were loaded with 1 μM Fura-2 (Invitrogen) for 45 min in culture medium. Cells were then washed with ACSF containing (in mM): 145 NaCl, 5 KCl, 2 CaCl, 1 MgCl, 10 Glucose and 10 HEPES and transferred to the perfusion chamber on an EclipseTi microscope (Nikon). A perfusion pencil triggered by the ValveBank8 perfusion system (AutoMate Scientific) was used to apply all reagents. Extrasynaptic NMDARs were pharmacologically isolated as described before (Hardingham 2009). Throughout the treatment, images were acquired every 6 sec by an ExiAqua camera (Photometrics) and analyzed with Elements 3.10 software (Nikon). All neurons that showed an initial response to bicuculline and 4-AP and did not exhibit high calcium levels from the beginning (RatioF340/380 < 0.4; [Kd] = 0.1–1 μM) were used for statistical analysis. As the region of interest (ROI), the visually identified somata of hippocampal neurons were selected. The area under the curve (AUC) was calculated (tsynaptic = 2.0–5.5 min and textrasynaptic = 7.0–8.5 min) using Prism Software (GraphPad)
RESULTS
Generation of adult CNS-TGF-β1 deficient mice
Investigating TGF-β1 signaling in the adult mouse brain is limited since TGF-β1-null mutant mice (TGF-β1−/−) succumb to a lethal autoinflammatory syndrome shortly after birth. After normal growth for the first 2 wks, all animals develop a severe autoinflammatory syndrome and die by 4 wks of age, suggesting a prominent role for TGF-β1 in homeostatic regulation of immune responses (Kulkarni et al. 1993),(Shull et al. 1992). Consistent with this, genetic approaches to inhibit TGF-β1 signaling in T-lymphocytes demonstrated that there is a specific disturbance of T-cell homeostasis in the absence of TGF-β1 and that T-cell-derived TGF-β1 is both necessary and sufficient to prevent autoinflammatory responses in mice (Gorelik et al. 2002; Li and Flavell 2008). Thus, the lethal phenotype of TGF-β1−/− mice is linked to a T-cell specific deficiency of TGF-β1.
We previously generated a TGF-β1-transgenic (Tg) mouse in which TGF-β1 is linked to the IL-2 promoter and T cells transiently overexpress TGF-β1 upon T-cell receptor (TCR) stimulation (Carrier et al. 2007) (Figure S1). These mice were termed TGF-β1-Tg mice. We first crossed TGF-β1-Tg mice with mice heterozygous for the endogenous Tgf-β1 allele (TGF-β1−/+), leading to the generation of [TGF-β1-Tg]/TGF-β1−/+ mice. Intercrossing these mice generated [TGF-β1-Tg]/TGF-β1−/− mice, which bear the transgenic Tgf-β1 gene, but lack both endogenous Tgf-β1 alleles. In [TGF-β1-Tg]/TGF-β1−/− mice, the expression of Tgf-β1 is thus limited to T-lymphocytes. We found that [TGF-β1-Tg]/TGF-β1−/− mice were protected from lethal autoinflammation, (Figure S2 and Table S1) survived into adulthood and were indistinguishable from [TGF-β1-Tg]/TGF-β1+/+ littermates at the age of 4, 8 or 10 wks. We did not find signs of tissue inflammation or gross organ dysfunction in these mice. However, when we investigated the CNS of these mice, we found that these animals did not express TGF-β1 in the brain or spinal cord. We thus termed them CNS-TGFβ1−/− mice (p < 0.001) (Figure 1A and Figure S3A). Furthermore, transgenic T-cells did not secrete TGF-β1 into the serum of CNS-TGFβ1−/− mice since TGF-β1 was absent from the serum of those mice (p < 0.001) (Figure 1B and Figure S3B). Finally, we found no TGF-β1-producing T-cells in the CNS of those animals (Figure 1C and S3C–D). Taken together, those results demonstrate that CNS-TGFβ1−/− mice are deficient in TGF-β1 in the CNS, thus providing a model to study the effect of TGF-β1-deficiency in the adult CNS.
Figure 1. Characterization of CNS-TGF-β1-deficient mice.

(A) TGF-β1 mRNA (expression normalized to GAPDH) is undetectable in the hippocampus of CNS-TGFβ1−/− mice (n=3). (B) TGF-β1 protein is undetectable in the serum of CNS-TGFβ1−/− mice (n=3). (C) CD4+ T-cells are absent in the brains of both CNS-TGFβ1−/− and CNS-TGFβ1+/+ mice. (D) Photomicrographs of Nissl-stained brain sections from CNS-TGFβ1−/− and CNS-TGFβ1+/+ mice. The hippocampus of CNS-TGFβ1−/− animals appears normal in size and shape. Scale bar: 200 μm. (E) The brain of CNS-TGFβ1−/− mice is reduced in weight as compared to CNS-TGFβ1+/+ littermates. (F) The pyramidal-cell layer in TGF-β1-deficient animals appears condensed in distinct regions of the hippocampus. Scale bar: 10 μm. (G) 6–8 day-old CNS-TGFβ1−/− mice display significantly reduced numbers of neurons in the area CA1 and CA3 of the hippocampus. (H) Photomicrographs of dendritic spines on biolistically transfected pyramidal neurons in hippocampal slices from 21-day old CNS-TGFβ1−/− or CNS-TGFβ1+/+ mice. Scale bar: 5 μm. (I–J) Pyramidal cells from CNS-TGFβ1−/− display a reduced density of dendritic spines (8 days in vitro). [*p < 0.05; **p<0.01; ***p<0.001]
CNS-TGF-β1−/− mice have astrogliosis and loss of the astrocyte glutamate transporters GLAST and GLT-1
Previous reports demonstrated a non-redundant function for TGF-β1 in maintaining integrity and survival of CNS neurons (Brionne et al. 2003). We thus investigated the CNS of CNS-TGFβ1−/− mice. We found no gross morphological abnormalities (Figure 1D) and no evidence for ongoing neuronal cell death. CNS-TGFβ1−/− mice animals had equal numbers of TUNEL-/cleaved caspase-3 double-positive neurons as their CNS-TGFβ1+/+ littermates (Figure S4). However, their brains were reduced in weight (CNS-TGFβ1+/+: 455.5±2.19 mg, CNS-TGFβ1−/−: 403.1±10.25 mg, n=5, p<0.01) (Figure 1E). In addition we found a decrease in the number of neurons in the CA1 and CA3 area of the hippocampus (CNS-TGFβ1+/+: CA1: 99.60±5.692, n=10/3 (sections/animals), CA3: 54.40±3.070, n=10/3 (sections/animals), CNS-TGFβ1−/−: CA1: 80.50±5.359, n=10/3 (sections/animals), CA3: 41.10± 3.628, n=10/3 (sections/animals), p<0.05) (Figure 1F–G). Overall hippocampal cytoarchitecture appeared normal, although we found a reduction in the density of dendritic spines on hippocampal CA1 pyramidal cells in CNS-TGFβ1−/− mice (Density: CNS-TGFβ1+/+ 0.74 ± 0.03/μm−1, n=9 (cells) vs. CNS-TGFβ1−/− 0.63±0.03/μm−1, n=9 (cells) analyzed at DIV 8, p < 0.05, Student’s t-test) (Figure 1H–J). Those results are consistent with a non-redundant function for TGF-β1 in neuronal homeostasis.
We next investigated the effect of TGF-β1 deficiency on astrocytes. In contrast to previous reports that investigated 3-wk-old animals (P21) (Brionne et al. 2003), we found that CNS-TGFβ1−/− mice had a marked increase in the number of GFAP- and S100B-positive astrocytes throughout the CNS (Figure 2A–B, Figure S5), suggesting an age-dependent effect of TGF-β1 on astrocyte proliferation and activation (#GFAP+ cells CNS-TGFβ1+/+: 30.33±2.947, n=12/3 (sections/animals), CNS-TGFβ1−/−: 108.0±5.875, n=12/3 (sections/animals), p<0.001). One of the principal functions of astrocytes is to recycle extracellular glutamate (Tzingounis and Wadiche 2007). When we investigated the abundance of the astrocyte glutamate transporter proteins GLT-1 (EAAT2) and GLAST (EAAT1) in the hippocampus of CNS-TGFβ1−/− mice, we found a significant reduction of GLT-1 and GLAST in the hippocampus of CNS-TGFβ1−/− animals compared to CNS-TGFβ1+/+ littermates (Figure 2C–D). At the same time, the amount of the neuronal glutamate transporter EAAC1 (EAAT3) was unchanged. To further investigate this observation, we measured the amount of GLT-1 and GLAST-positive punctae in the stratum radiatum of the hippocampus of CNS-TGFβ1−/− animals using confocal microscopy as described previously (Carmona et al. 2009) (Figure 2E–F). In this region, cellular processes of astrocytes contact the CA3-CA1 synapses of the Schaffer collateral pathway and distinct astrocytic processes bearing a punctuate pattern of GluTs are visible (Chaudhry et al. 1995). Consistent with our observation above by Western blot (Figure 2C–D), we found that in the stratum radiatum of the hippocampus, GLT-1 and GLAST-positive punctae were reduced in CNS-TGFβ1−/− animals compared to their CNS-TGFβ1+/+ littermates (Figure 2G). Astrocytes remove the bulk of glutamate in the forebrain (Danbolt 2001). We thus measured uptake of radioactive glutamate in acute hippocampal slices. We found that 3H-[L]-glutamate uptake was reduced by ~50% in slices from CNS-TGFβ1−/− mice compared to CNS-TGFβ1+/+ littermates (glutamate uptake in fmol/μg/min) CNS-TGβ1+/+: 1325±63.71, n=10/4 (slices/animals), CNS-TGF-β1−/−: 807.8±51.46, n=10/4 (slices/animals), p<0.001) (Figure 2H). Taken together, our results demonstrate that TGF-β1 deficiency in the CNS results in astrogliosis and at the same time it is accompanied by a loss of astrocytic GluTs. This resulted in significant reduction in glutamate uptake in vivo.
Figure 2. CNS-TGF-β1-deficient mice display astrogliosis and loss of astroglial glutamate transporters.


(A) Left panel: Photomicrographs of GFAP showing enhanced GFAP staining in the hippocampus of CNS-TGFβ1−/− mice (10x). White box indicates the area that is shown in high magnification (20x). Scale bar: 100 μm. Right panel: Photomicrographs of GFAP staining in the stratum radiatum (20x). Scale bar: 50 μm. (B) CNS-TGFβ1−/− animals display increased numbers of GFAP+ astrocytes in the hippocampus. (C) Western blot of GLT-1, GLAST, EAAC1, glutamine synthetase (GS) and GFAP in the hippocampus of CNS-TGFβ1+/+ and CNS-TGFβ1−/− (D) The amount of GLT-1 and GLAST protein is reduced in the hippocampus of TGF-β1-deficient mice, whereas the expression of EAAC1 (EAAT3) and Glutamine Syntethase (GS) are unchanged. GFAP is increased in CNS-TGFβ1−/− animals. (E–F) Confocal images stained with GFAP plus either GLT-1 (E) or GLAST (F). Photomicrographs illustrate a loss of GLAST and GLT-1-positive punctae on astrocytes in the hippocampus. Scale bar: 10 μm. (G) Quantification of GLT-1 and GLAST-positive punctae in the stratum radiatum of the hippocampus. (H) Uptake of L-[3H]-glutamate is decreased in hippocampal slices from TGF-β1-deficient animals. Treatment with TBOA (100 μM) or use of sodium-free ACSF (replaced with choline chloride) equalized glutamate uptake. (I) Quantification of GLAST protein in cultured astrocytes from CNS-TGFβ1+/+ and CNS-TGFβ1−/− mice measured by Western blot. Astrocytes from CNS-TGFβ1−/− mice (Lane 2) had lower GLAST protein amounts than astrocytes from CNS-TGFβ1+/+ (Lane 1). Astrocytes from CNS-TGFβ1−/− mice that were treated with TGF-β1 (25 ng ml−1, 72 hrs) had restored amounts of GLAST protein (Lane 3). (J) Quantification of Glt-1 and Glast gene expression. Treatment with TGF-β1 (25 ng ml−1, 24 hrs) induces the expression of Glt-1 and Glast in cultured astrocytes from C57BL/6 wild-type mice. (K) Quantification of GLT-1 and GLAST protein by Western blot. Treatment with TGF-β1 induced the expression of GLT-1 and GLAST in cultured astrocytes from C57BL/6 wild-type mice. Wild-type astrocytes express GLAST (Lane 2) and almost no GLT-1 (Lane 4). Prolonged treatment with TGF-β1 (25 ng ml−1, 72 hrs) induced the expression of GLT-1 (Lane 3) and increased the expression of GLAST (Lane 1). (L) Prolonged treatment with TGF-β1 (25 ng ml−1, 72 hrs) increases glutamate uptake in cultured astrocytes from C57BL/6 wild-type mice. [*p < 0.05; **p<0.01; ***p<0.001]
To further investigate the effect of TGF-β1 on the expression of astrocytic GluTs, we examined cultured astrocytes in vitro. In cultured astrocytes, the expression of GLAST and GLT-1 is differentially regulated. GLAST is consistently expressed in cultured astrocytes in vitro and neuron-conditioned media further increases its expression. In contrast, GLT-1 expression is low and GLT-1 protein is almost absent in astrocytes in culture, but co-culturing astrocytes with neuron-conditioned media induces the expression of GLT-1 in astrocytes (Gegelashvili et al. 1997; Swanson et al. 1997). Those findings demonstrate that the expression of GluTs in cultured astrocytes is regulated by secreted factors.
When we investigated astrocytes (10-14 DIV) from CNS-TGF-β1−/− mice and their CNS-TGF-β1+/+ littermates, we found decreased amounts of GLAST protein in cultured astrocytes from CNS-TGF-β1−/− mice as compared to astrocytes from CNS-TGF-β1+/+ mice (n=6, p<0.001) (Figure 2I). In addition, treatment with TGF-β1 (25 ng ml−1, 72 hrs) restored the expression of GLAST in astrocytes from CNS-TGF-β1−/− mice (Figure 2I). In cultured astrocytes from C57BL/6 wild-type mice, treatment with (25 ng ml−1, 24 hrs) increased gene expression of both, Glt-1 and Glast (n=6, p<0.001) (Figure 2J). Prolonged treatment with TGF-β1 (25 ng ml−1, 72 hrs) also increased the amount of GLT-1 and GLAST protein in cultured astrocytes (n=6, p<0.001) (Figure 2K) as well as glutamate uptake by astrocytes (Figure 2L) (glutamate uptake in pmol/μg/min: vehicle 25.95±0.9398, n=25 vs. TGF-β1 29.47 ± 1.001, n=24, p<0.05). Taken together our results demonstrate that TGF-β1 regulates the expression of astrocytic GluTs and thus suggest a role for TGF-β1 to maintain the concentration of extracellular glutamate in the hippocampus.
CNS TGF-β1-deficient mice have impaired synaptic plasticity in the CA1 area of the hippocampus
GluTs promote synapse independence by limiting glutamate ‘spill-over’ to neighboring synapses (Arnth-Jensen et al. 2002; Takayasu et al. 2006), limiting glutamate ‘spill-out’ to extrasynaptic glutamate receptors (Huang et al. 2004) and by modulating the intensity and duration of postsynaptic excitation (Tzingounis and Wadiche 2007). In addition, GluTs modulate long-term changes in synaptic efficacy and contribute to the expression of normal synaptic plasticity. For example, in GLT-1-deficient mice, long-term potentiation (LTP) in hippocampal Schaffer collateral-CA1 synapses is impaired. Mechanistically, the LTP-impairment in GLT-1-deficient mice depends on NMDA-type glutamate receptors, because application of the NMDAR antagonist D-AP5 restored LTP in those animals to the level of wild-type mice (Katagiri et al. 2001). We thus asked if the loss of GluTs in CNS-TGFβ1-deficient mice translates into similar effects on synaptic plasticity in the hippocampus. To address this question, we recorded field excitatory postsynaptic potentials (fEPSP) in CA1 region of hippocampal slices from 10-wk-old CNS-TGFβ1−/− mice and from their CNS-TGFβ1+/+ littermates. We found that high-frequency stimulus (HFS) induced (2 trains of 1 sec/100 Hz, separated by 20 sec) LTP was significantly inhibited in CNS-TGFβ1−/− mice compared to CNS-TGFβ1+/+ mice (% baseline fEPSP: CNS-TGFβ1−/− 112±6%, n=8 vs. CNS-TGFβ1+/+ 145±4%, n=7, P<0.001) (Figure 3A). Furthermore, a weak low-frequency stimulus (LFS) (300 pulses/1 Hz) induced a strong LTD in CNS-TGFβ1−/− mice (% baseline fEPSP: CNS-TGF-β1−/− 71 ± 4%, n=8 vs. CNS-TGFβ1+/+ 97±7%, n=7, P<0.001) (Figure 3B), further indicating an impairment of synaptic plasticity in CNS-TGFβ1−/− mice. Mechanistically, we found that LTP-inhibition and LTD-facilitation in CNS-TGFβ1−/− mice are unlikely to involve the reduction of presynaptic release and basal synaptic transmission, since TGF-β1-deficiency had no effect on the paired-pulse facilitation (50 ms interstimulus interval) (PPF) or stimulus-response curve under similar conditions (P > 0.05) (Figure 3C–D). In addition, TGF-β1 deficiency did not significantly reduce synaptic responses during HFS or LFS (unpublished observations). Taken together, these results suggest a postsynaptic mechanism for synaptic failure in CNS-TGFβ1−/− mice similar to the GLT-1-deficient mice. This demonstrates a significant role of TGF-β1 in extracellular glutamate homeostasis and synaptic plasticity.
Figure 3. CNS-TGF-β1 deficient mice display altered synaptic plasticity.



(A) In acute hippocampal slices from CNS-TGFβ1−/− mice LTP was reduced compared to slices from CNS-TGFβ1+/+ animals. (B) A weak LFS of 300 pulses at 1 Hz induced a strong LTD in slices from CNS-TGFβ1−/− mice (mean ± s.e.m). (C) TGF-β1-deficient mice display normal field excitatory postsynaptic potentials (fEPSP) to electric stimulation in the hippocampus (mean ± s.e.m). (D) Stimulus-response curves for paired-pulse facilitation (PPF) were not different in hippocampal slices from CNS-TGFβ1−/− or CNS-TGFβ1+/+ mice (mean ± s.e.m). (E) The non-specific NMDAR antagonist D-AP5 (0.5 μM) and the GluN2B-antagonist Ro 25-6981 prevent facilitation of LTD in CNS-TGFβ1−/− mice. (F) The mGluR-specific antagonist SIB1757 (3 μM) had no effect on LTD in hippocampal slices from CNS-TGFβ1−/− mice. (G) Co-perfusion of the GluN2B-specific antagonist Ro 25-6981 (0.5 μM) restored LTP in CNS-TGFβ1−/− mice to the level of CNS-TGFβ1+/+ animals. No effect was observed on LTP in slices from CNS-TGFβ1+/+ animals. (H–I) Repetitive induction of LTD interfered with TBOA-induced LTD in CNS-TGFβ1−/− animals (I) but not in CNS-TGFβ1+/+ mice (H). [*p < 0.05; **p<0.01; ***p<0.001]
In the Schaffer collateral, the induction of LTP typically depends on the activation of ionotropic NMDA-type glutamate receptors (NMDARs) whereas the induction of LTD may require NMDARs or metabotropic glutamate receptors (mGluRs) to be activated, depending on the stimulation protocol and the recording conditions (Citri and Malenka 2008). We thus attempted to further characterize the enhanced LTD in the hippocampus of CNS-TGFβ1−/− mice. We found that the subunit-unspecific NMDAR antagonist D-AP5 rescued LTD in CNS-TGFβ1−/− mice to the level of CNS-TGFβ1+/+ animals (50 μM) (% baseline fEPSP LTD CNS-TGFβ1−/− + D-AP5 89±8%, n=7, P>0.05) (Figure 3E). When we incubated hippocampal slices from CNS-TGFβ1−/− mice and their CNS-TGFβ1+/+ littermates during recording with the GluN2B-specific NMDAR-antagonist Ro 25-6981 (0.5 μM) to block GluN2B-containing NMDARs, we found that Ro 25-6981 restored the facilitated LTD (% baseline fEPSP LTD CNS-TGFβ1+/+ 97±7%, n=6 vs. CNS-TGFβ1−/− + Ro 25-6981 87±3.1%, n=7; P>0.05) in CNS-TGFβ1−/− mice to levels of CNS-TGF-β1+/+ and similar to wild-type animals (Figure 3E). Ro 25-6981 restored LTD in CNS-TGFβ1−/− mice similar to the effect of the subunit-unspecific NMDAR antagonist D-AP5, suggesting that blocking of GluN2B-containing NMDARs is sufficient to prevent LTD in those animals. On the other hand, application of the mGluR5-specific antagonist SIB-1757 (3μM) had no significant effect on LTD in CNS-TGF-β1−/− mice (% baseline fEPSP LTD CNS-TGFβ1−/− + SIB1757 76.6±4.5%, n=7, p > 0.05) (Figure 3F), suggesting that LTD-facilitation in CNS-TGFβ1−/− mice predominantly depends on the activation of GluN2B-containing NMDARs. Interestingly, application of Ro 25-6981 (0.5 μM) equally restored the inhibited LTP in CNS-TGFβ1−/− mice to levels of CNS-TGFβ1+/+ (% baseline fEPSP LTP CNS-TGFβ1+/+ 140±10%, n=6 vs. CNS-TGFβ1−/− + Ro 25-6981: 139±6%, n=7; P>0.05) (Figure 3G). We thus conclude that similar to GLT-1-deficient mice (Katagiri et al. 2001), synaptic plasticity in the hippocampus of CNS-TGFβ1−/− mice is impaired through a mechanism that involves the activation of GluN2B-containing NMDARs. This result is consistent with the concept that restrained glutamate uptake causes ‘spill-out’ onto extrasynaptic GluN2B-containing NMDARs and that activation of GluN2B-containing NMDARs facilitates the induction of LTD and attenuates LTP in the CA1 area of the hippocampus (Brigman et al. 2010; Liu et al. 2004). We found that this effect is not a consequence of increased glutamate receptor expression, since the protein abundance of GluR2, GluN1, GluN2A, GluN2B and PSD95 remained unaffected in CNS-TGF-β1−/− mice (Figure S6).
We have previously shown that the glutamate uptake inhibitor DL-threo-β-Benzyloxyaspartic acid (TBOA) inhibits HFS-induced LTP and facilitates NMDAR-mediated LFS-induced LTD in the CA1 area of the hippocampus (Li et al. 2009) similar to what we found in CNS-TGFβ1−/− mice. We thus sought to investigate more directly, if altered synaptic plasticity in CNS-TGFβ1−/− mice was a consequence of decreased glutamate uptake in these animals. To address this question, we recorded LTD in CNS-TGFβ1−/− mice and their CNS-TGFβ1+/+ littermates in a repetitive 300-pulse LFS paradigm (Figure 3H). This results in saturation of the LTD response in both genotypes after the third LFS. When we then applied TBOA (15 μM), we found that another LFS resulted in a significant LTD in CNS-TGFβ1+/+ animals (CNS-TGFβ1+/+ Vehicle % EPSP: Baseline 100.0±1.54 %; LTD: 91.10±4.36 %, n=5; CNS-TGFβ1+/+ TBOA % EPSP: Baseline 100.23±9.94 %, LTD 62.36±10.96 %, n=6, p<0.001) Application of TBOA failed to induce further LTD in CNS-TGFβ1−/− mice (CNS-TGFβ1−/− Vehicle % EPSP: Baseline 100.0±6.06 %; LTD: 93.80±7.82 %, n=5; CNS-TGFβ1−/− TBOA % EPSP: Baseline 100.6±6.32 %, LTD 91.68±2.83 %, n= 6, p >0.05) (Figure 3I). These results show that LTD in CNS-TGFβ1−/− mice interferes with TBOA-induced LTD, suggesting that LTD in CNS-TGFβ1−/− mice mechanistically resembles TBOA-induced LTD. In summary, these findings further demonstrate that decreased glutamate uptake in CNS-TGFβ1−/− mice is associated with altered synaptic plasticity in those mice.
TGF-β1-deficient hippocampal neurons facilitate GluN2B-mediated calcium signals in response to NMDAR stimulation
Inhibiting astrocyte glutamate uptake results in the accumulation of extracellular glutamate, epileptic seizures and ultimately in neuronal death (Coulter and Eid 2012; Danbolt 2001; Tanaka et al. 1997). Because CNS-TGFβ1-deficient mice have reduced glutamate uptake, we next investigated if those mice are more susceptible to neuronal insults. We found that CNS-TGF-β1-deficient mice displayed enhanced neuronal damage 5 and 30 days after injection with convulsive doses of KA, a well characterized excitotoxic agent (Wang et al. 2005) (Figure 4A–B and Figure S7) (# Nissl+ cells CA1: CNS-TGFβ1+/+: 83.50±3.911, n=10/3 (sections/animals), CNS-TGFβ1−/−: 28.20±5.350, n=10/3 (sections/animals), p<0.001; CA3:CNS-TGFβ1+/+: 30.40±3.772, n=10/3 (sections/animals), CNS-TGFβ1−/−:15.10±2.452, n=10/3 (sections/animals), p<0.01). These results thus demonstrate that similar to homozygous GLT-1−/− mice (Tanaka et al. 1997), TGF-β1-deficient mice display enhanced spontaneous and injury-induced neuronal death in the hippocampus, although the expression of GLT-1 and glutamate uptake is decreased by only ~ 50% in those mice. Because heterozygous GLT-1+/− mice show little spontaneous neuronal degeneration (Kiryk et al. 2008), we examined an additional candidate mechanism besides decreased glutamate uptake to explain neuronal cell death in TGF-β1-deficient mice.
Figure 4. CNS-TGFβ1−/− mice display increased sensitivity to excitotoxic injury.
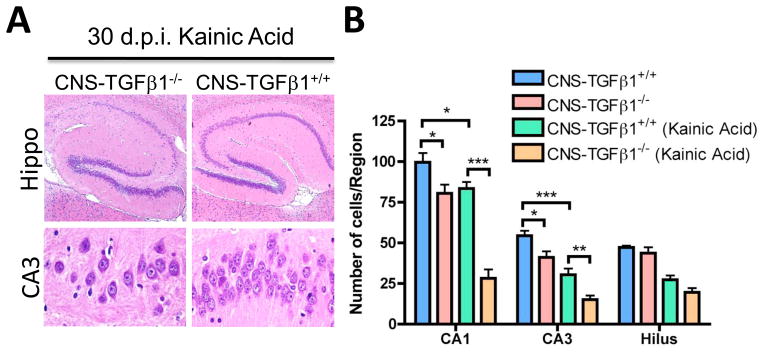
(A) Representative photomicrographs (H&E) from hippocampus tissue sections 30 days after kainic acid (KA) induced seizures. (B) CNS-TGFβ1−/− mice display reduced numbers of neurons in the CA1 and CA3 area of the hippocampus at 30 days following the injection of kainic acid (10 mg kg−1). [*p < 0.05; **p<0.01; ***p<0.001]
Because the cellular downstream effects of high extracellular glutamate levels are ultimately mediated through neuronal calcium overload (Wang et al. 2005; Waxman and Lynch 2005), we investigated glutamate-receptor induced neuronal calcium signals in hippocampal neurons from CNS-TGFβ1−/− mice and their CNS-TGFβ1+/+ littermates in vitro. As decreased glutamate uptake can cause glutamate to ‘spill-over’ to extrasynaptic glutamate receptors (Gouix et al. 2009; Huang et al. 2004; Nie and Weng 2010; Potier et al. 2010), we measured synaptic and extrasynaptic glutamate receptor-mediated calcium signals individually, as described previously (Hardingham 2009). We first tested whether TGF-β1 acutely affects glutamate uptake in neuronal cultures in vitro. We incubated dissociated hippocampal neurons from E18 mice (DIV 10-12) for 2 hrs with TGF-β1 (25 ng ml−1) and measured the uptake of L-[3H]-glutamate. We found that treatment with TGF-β1 had no effect on glutamate uptake in dissociated neuronal cultures (Vehicle 26.37±0.8664, n=16 vs. TGF-β1 24.34±1.503 n=10) (Figure 5A). This result is consistent with our finding that TGF-β1-deficiency has no effect on the expression of the neuronal glutamate transporter EAAC1 (EAAT3) (Figure 2B–C) and therefore does not affect glutamate uptake in clean neuronal cultures without significant astrocytic contamination. We also tested if co-application of the GABA receptor antagonist bicuculine (BIC) and the weak potassium channel blocker 4-aminopyridine (4-AP) affected glutamate uptake in vitro and found that both compounds had no effect on glutamate uptake (unpublished observations). On the other hand, treatment with TBOA decreased glutamate uptake in vitro in a dose-dependent fashion (Figure 5A).
Figure 5. TGF-β1-deficient hippocampal neurons are sensitive to high extracellular glutamate concentrations.

(A) Treatment with TGF-β1 (25 ng ml−1, 2 hrs) does not affect glutamate uptake in dissociated hippocampal neurons. TBOA treatment (2 hrs) reduces glutamate uptake in dissociated neurons. (B) Photomicrographs illustrate calcium-induced fluorescence signals in the somata of dissociated hippocampal neurons (t 1–7.5 min). Heat map indicates fluorescence intensity (Ratio F340/380). (C) Pre-treatment with TGF-β1 (25 ng ml−1, 2 hrs) decreases cytosolic calcium responses in a dose-dependent fashion. (D) Effect of TGF-β1 on BIC/4-AP-induced calcium responses (tAUC = 2.0–5.5 min). (E) Effect of TGF-β1 on NMDA-induced calcium responses (tAUC = 7.0–8.5 min). (F) Effect of TGF-β1 on the ratio of synaptic to extrasynaptic calcium signals. (G) TGF-β1 deficiency does not affect synaptic calcium signals. (H) TGF-β1-deficiency increases extrasynaptic calcium signals. (I) TGF-β1-deficiency decreases the ratio of synaptic to extrasynaptic calcium responses. (J) Treatment with TGF-β1 (25 ng ml−1, 2 hrs) prevents neuronal death induced by stimulation of extrasynaptic NMDARs. (K) Effect of co-perfusion of Ro 25-6189 (2.5 μM) and the subunit-unspecific NMDAR antagonist D-AP5 (100 μM) on extrasynaptic NMDAR-mediated calcium responses in cultured hippocampal neurons. (L) Co-perfusion of low-dose memantine (10 μM) had no effect on synaptic calcium responses blocked NMDA-mediated calcium responses. (M) Memantine blocks extrasynaptic calcium responses. (N) Application of memantine during BIC/4-AP pulses increases the number of BIC-evoked peaks/cell (O) Application of memantine has no effect on the synaptic peak area/#peaks. [*p < 0.05; **p<0.01; ***p<0.001]
Next, we investigated the effect of TGF-β1 on synaptic and extrasynaptic calcium signals in hippocampal neurons. We first applied BIC (10 μM) and 4-AP (2.5 mM) to activate synaptic NMDARs. Once stable calcium responses were achieved, we applied the use-dependent irreversible NMDAR antagonist MK-801 (10 μM), which has been shown to block all active (synaptic) NMDARs (Hardingham et al. 2002). Following washout of MK-801, we applied a brief NMDA pulse (30 μM, 30 sec) to activate the remaining extrasynaptic NMDARs (Figure 5B). Cultured hippocampal neurons from C57BL6 wild-type mice (DIV 12-14) were treated with different concentrations of TGF-β1 (1, 10 or 25 ng ml−1 in ACSF) or vehicle (ACSF) for 2 hrs at 37°C in culture medium before synaptic and extrasynaptic NMDARs were selectively activated (Figure 5C). We found that compared to vehicle (ASCF), pre-treatment with high TGF-β1 doses (25 ng ml−1) reduced the magnitude of both synaptic (AUCt=2–5.5 min Vehicle: 7.277±0.3943, n=123/3 (cells/experiments) vs. TGF-β1 5.461±0.2938, n=144/3 (cells/experiments) and extrasynaptic (AUCt=7–8.5 min Vehicle: 2.619±0.1649, n=123/3 (cells/experiments), TGF-β1: 1.769±0.1198, n=144/3 (cells/experiments) calcium responses, whereas pre-treatment with intermediate TGF-β1 concentrations (10 ng ml−1) had no effect on synaptic calcium signals, but decreased extrasynaptic calcium responses (AUCt=7–8.5 min 1.964±0.1216, n=70, P<0.01). Low doses (1 ng ml−1) had no effect on synaptic or extrasynaptic calcium responses (Figure 5D–E). In addition, high TGF-β1 doses (25 ng ml−1) attenuated synaptic calcium signals less than extrasynaptic calcium signals, and thus continued to increase the ratio of synaptic to extrasynaptic calcium signals (Figure 5F). We then compared cultured neurons from CNS-TGFβ1+/+ and CNS-TGFβ1−/− mice using the same experimental protocol. We found that cells from CNS-TGFβ1−/− mice displayed elevated calcium signals in response to stimulation of extrasynaptic NMDARs compared to neurons from CNS-TGFβ1+/+ littermates (AUCt=7–8.5 min CNS-TGFβ1+/+ 1.421±0.08841, n = 124 vs. CNS-TGFβ1−/− 2.057±0.2102 n=143, P<0.01). At the same time the magnitude of synaptic calcium responses was similar in both (Figure 5G–H). TGF-β1 deficiency enhanced extrasynaptic calcium signals and therefore decreased the ratio of synaptic to extrasynaptic calcium signals (Figure 5I). In summary, these results demonstrate that TGF-β1 distinctly affects the magnitude of synaptic and extrasynaptic neuronal calcium signals and that this effect is independent from the extracellular glutamate concentration. When we isolated extrasynaptic NMDARs as described and exposed cultured cells from wild-type mice to NMDA (30 μM) for 15 min in Mg2+-free ACSF containing glycine (10μM) and counted Propidium Iodide (PI)-positive cells after 24 hrs, we found that the number of PI-positive cells was significantly lower in cultures treated with TGF-β1 (25 ng ml−1) (p<0.01) (Figure 5J). This result is consistent with the concept that neuronal cell death is associated with the activation of extrasynaptic NMDARs, whereas the stimulation of synaptic NMDARs leads to the build-up of a neuroprotective ‘shield’ (Hardingham and Bading 2010).
We then sought to characterize the specific subunit composition of the NMDAR that accounts for those neuronal calcium signals. We found that extrasynaptic NMDAR-mediated calcium responses are predominantly mediated through GluN2B-containing NMDARs, since co-perfusion with Ro 25-6981 (2.5 μM) strongly reduced extrasynaptic cytosolic calcium responses (AUCt=7–8.5 min vehicle: 1.939 ± 0.07102 n = 187/3 (cells/experiments) vs. Ro 25-6189: 0.7134±0.03828, n=175/3 (cells/experiments), P<0.001). This effect is similar to that observed with the subunit-unspecific NMDAR antagonist D-AP5 (100 μM) (AUCt=7–8.5 min 0.4539±0.02841, n=98/3 (cells/experiments)) (Figure 5K). To further investigate the effect of TGF-β1 on extrasynaptic NMDAR-mediated calcium responses, we compared treatment with TGF-β1 to memantine. Memantine is a low-affinity voltage-dependent antagonist at glutamatergic NMDARs (Rogawski and Wenk 2003) that has been shown to preferentially block extrasynaptic NMDARs at low concentrations but preserves physiological synaptic transmission (Lipton 2006; Xia et al. 2010). When we co-incubated cultured hippocampal neurons from wild-type mice with memantine during synaptic and extrasynaptic NMDAR-mediated stimulation, we found that similar to TGF-β1, low concentrations of memantine (10 μM) decreased the magnitude of extrasynaptic calcium responses (AUCt=7–8.5 min AUC vehicle 1.93±0.07, n=187/3 (cells/experiments) vs. memantine 0.97±0.11, n=134/3 (cells/experiments), P<0.0001) (Figure 5L–M). Although treatment with memantine did not change the magnitude of synaptic calcium responses (AUCt=2–5.5 min P>0.05), it significantly affected the kinetics of synaptic calcium responses (Figure 5N–O). In contrast to treatment with vehicle or TGF-β1, memantine increased the number of cytosolic calcium peaks during synaptic NMDAR stimulation (# peaks/cell Memantine 1.35±0.09, n=134/3 (cells/experiments) vs. Vehicle 1.08±0.008, n=123/3 (cells/experiments), P<0.0004) and affected the peak area variability. This result is again consistent with the concept that memantine preferentially blocks extrasynaptic NMDAR and supports the concept that TGF-β1 indeed modulates extrasynaptic GluN2B-mediated calcium responses in cultured hippocampal neurons. Our results thus confirm that TGF-β1 ameliorates neuronal death by limiting calcium signals from extrasynaptic GluN2B-containing NMDARs. Of note, our results demonstrate that this effect is independent from the glutamate concentration in the extracellular space.
DISCUSSION
We describe here a novel mouse model that allows the investigation of TGF-β1-deficiency in the adult mouse brain, something not possible previously since TGF-β1-null mutant mice (TGF-β1−/−) succumb to a lethal autoinflammatory syndrome shortly after birth (Kulkarni et al. 1993),(Shull et al. 1992). Previous reports described genetically modified mouse models, which targeted either TGF-β receptors or distinct components of its downstream signaling pathway, such as the Smad family of transcription factors (Tesseur and Wyss-Coray 2006; Tesseur et al. 2006). Although there is evidence for a role of TGF-β signaling in neuronal maintenance, function, and degeneration, several of these mutant models did not produce overt CNS phenotypes or adult brains were not studied due to embryonic lethality. In addition, because of the complexity and redundancy of TGF-β signaling, previous models addressed different aspects than the present study. This is due to the fact that TGF-β1 is not the sole ligand that signals via TGF-β type I receptors and other ligands, including the TGF-β family members TGF-β2 and 3 and the activins, which act through the same receptor (Schmierer and Hill 2007), are expressed in the brain and have been shown to affect neuronal homeostasis (Ageta et al. 2010). Our work thus demonstrates the importance of examining the role of TGF-β1-deficiency in adult brain, as previous reports that studied young animals did not observe any or different effects of TGF-β1 on astrocytes (Brionne et al. 2003; Reilly et al. 1998).
We report here that TGF-β1-deficiency in the CNS leads to a loss of astroglial GluTs and decreased glutamate uptake in the hippocampus. Treatment with TGF-β1 conversely increased the expression of GluTs and glutamate uptake by astrocytes in vitro, similar to growing astrocytes in the presence of neuron-conditioned media (Gegelashvili et al. 1997; Swanson et al. 1997). Because astroglial GluTs are responsible for clearing most glutamate, restraining rapid removal of glutamate causes accumulation of glutamate in the extracellular space, ‘spill-over’ of glutamate to nearby synapses and ‘spill-out’ to extrasynaptic glutamate receptors. The relative contribution of extrasynaptic NMDARs had been shown to be relevant for neuronal survival: whereas stimulation of synaptic NMDARs leads to the build-up of a neuroprotective ‘shield’, stimulation of extrasynaptic NMDARs promotes cell death (Hardingham and Bading 2010). Consistent with this, TGF-β1-deficient mice display increased sensitivity excitotoxic insults (Brionne et al. 2003). We thus describe a new role for local TGF-β1 to control glutamate homeostasis in the hippocampus by regulating GluT expression. Identifying factors that control the expression of GluTs is of particular relevance to neurodegenerative pathologies where reactive astrogliosis and impaired glutamate uptake contribute to the detrimental effects (reviewed in (Beart and O’Shea 2007; Maragakis and Rothstein 2004).
We demonstrate that TGF-β signaling plays a novel role in the induction and maintenance of synaptic plasticity as TGF-β1 deficiency leads to impaired LTP and facilitated LTD in CA1-CA3 synapses of the hippocampus. Similar to homozygous GLT-1-deficient mice, these impairments depended on the activation of GluN2B-containing NMDARs (Katagiri et al. 2001). With a weak 300-pulse LFS protocol, the glutamate uptake inhibitor TBOA induces similar LTP and LTD impairments in wild-type mice (Li et al. 2009) and interferes with LTD in CNS-TGFβ1−/− mice, further suggesting that the impairments we found in TGF-β1-deficient mice are a consequence of decreased glutamate recycling. Reduced glutamate recycling is associated with glutamate ‘spill-out’ and, as a consequence, the activation of extrasynaptic NMDARs, which may in turn impair synaptic plasticity. Several lines of evidence support this hypothesis. It has been suggested that the subunit composition of NMDARs is crucial to the polarization of synaptic plasticity (LTP vs. LTD) (Hendricson et al. 2002; Hrabetova and Sacktor 1997; Hrabetova et al. 2000; Kutsuwada et al. 1996; Sakimura et al. 1995; Sjostrom et al. 2003; Sprengel et al. 1998; Yoshimura et al. 2003) as studies have found that the induction of LTP was blocked by a GluN2A antagonist but not by GluN2B antagonists (Liu et al. 2004; Massey et al. 2004). Conversely, de novo LTD was unaffected by the GluN2A antagonist but was blocked by GluN2B antagonists. Furthermore, it has been suggested that GluN2A- and GluN2B-containing receptors segregate to synaptic (GluN2A) and extrasynaptic (GluN2B) compartments (Kutsuwada et al. 1996; Liu et al. 2004; Massey et al. 2004; Momiyama 2000; Scimemi et al. 2004; Steigerwald et al. 2000; Stocca and Vicini 1998; Tovar and Westbrook 1999). In addition, GluN2B-containing NMDARs are more sensitive to low concentrations of glutamate and might thus be used to detect extrasynaptic glutamate ‘spill-out’ (Cull-Candy and Leszkiewicz 2004). It is thus possible that in the hippocampus of TGF-β1-deficient mice, the GluN2B-antagonist Ro 25-6189 restores LTP and LTD because it affects extrasynaptic GluN2B-containing NMDARs. On the other hand other reports argue against a simple distinction between synaptic GluN2A and extrasynaptic GluN2B NMDARs and a connection between synaptic LTP and non-synaptic LTD (Morishita et al. 2007). It thus remains unresolved whether the activation of extrasynaptic GluN2B-containing NMDARs is indeed responsible for the impairments we have described. Nonetheless, it seems likely that the effects of TGF-β1 on astrocytes and neurons synergize to prevent the activation of extrasynaptic GluN2B-containing NMDARs.
Like homozygous GLT-1−/− mice (Tanaka et al. 1997) and different from heterozygous GLT-1+/− mice (Kiryk et al. 2008), TGF-β1-deficient mice display enhanced excitotoxic neuronal death in the hippocampus, although the expression of GLT-1 and glutamate uptake is decreased by little more than ~ 50% in those mice. We thus examined additional candidate mechanisms that may synergize with impaired glutamate uptake to explain neuronal degeneration in TGF-β1-deficient mice. Our study provides evidence for a role of endogenous local TGF-β signaling in NMDAR-mediated calcium signaling. TGF-β1 increased the ratio of synaptic to extrasynaptic calcium signals in a dose dependent fashion. We found that in cultured neurons, most extrasynaptic NMDAR-mediated calcium signals depend on GluN2B-containing NMDARs, since Ro 25-6189 almost completely abolished extrasynaptic calcium responses. This result is in accordance with the finding that in hippocampal neurons most extrasynaptic NMDARs contain GluN2B subunits (Tovar and Westbrook 1999). Our results are consistent with previous studies that demonstrated that acute administration of TGF-β1 ameliorates neuronal calcium homeostasis in cultured hippocampal neurons (Prehn et al. 1994). The finding that memantine, which blocks extrasynaptic NMDARs had a similar effect on NMDAR-mediated calcium responses, further supports our finding that TGF-β1 affects extrasynaptic NMDAR-mediated calcium responses, although our results do not resolve, why TGF-β1 has different effects on synaptic and extrasynaptic calcium signals. The fact that treatment with TGF-β1 had no effect on NMDAR-mediated current responses in dissociated neurons (Prehn et al. 1994) raises the question of whether synaptic and extrasynaptic calcium signals can be distinctly modulated downstream from the NMDAR.
We demonstrate here distinct effects of TGF-β1 on astrocytes and neurons, although our results do not clarify the intracellular signal transduction pathway through which TGF-β1 affects astrocytes and neurons, respectively. TGF-β1-mediated downstream cascades are likely to be cell-type specific (e.g. astrocytes vs. neurons). For example, it has been shown that the type I receptor Alk5, which phosphorylates Smad2/3, is expressed in hippocampal neurons and in astrocyes, whereas Alk1, which activates Smad1/5 is predominantly expressed in hippocampal neurons and is up-regulated in response to neuronal injury (Konig et al. 2005). Interestingly, those authors demonstrate that TGF-β1 exerts specific neuroprotective effects through Alk5-mediated signalling by activating Smad1/5 and NF-κB (Konig et al. 2005). Clearly, future experiments in cultured astrocytes and neurons in vitro will be required to dissect the cell-type specific downstream signalling cascades that mediate the effect of TGF-β1 on astrocytic glutamate transport and neuronal calcium homeostasis, respectively. Of note, we found that Smad2/3 protein phosphorylation levels were not affected in the brain of CNS-TGFβ1−/− animals (Figure S3E–F). Several factors could explain these findings. First, it is conceivable that the lacking effect of TGF-β1 on Smad phosphorylation is compensated by other ligands such as TGF-β2 and 3, the activins and nodal, as all of them act on type I receptors to phophorylate Smad2/3 (Schmierer and Hill 2007). Second, as we assessed Smad2/3 phosphorylation levels in whole brain tissue preparations, possible cell-type specific differences might have been masked. Further experiments in cultured astrocytes and neurons in vitro will be required to dissect the cell-type specific impact of TGFβ1 on Smad2/3 phosphorylation in those cells and address the relationship of TGFβ-signalling with other neurotransmitter systems that have been shown to regulate the expression of GluTs, such as metabotropic glutamate receptors (Condorelli et al. 1997; Gegelashvili et al. 2000).
Previous studies indicated that TGF-β1 has neuroprotective effects in a variety of in vivo and in vitro model systems (Boche et al. 2003; Prehn et al. 1993a; Prehn et al. 1993b), although the cellular mechanisms that account for it’s neuroprotective effect remained largely elusive. Prehn and colleagues showed that TGF-β1 prevents neuronal calcium overloading in response to NMDA and leads to a substantial increase in the neuronal expression of the anti-apoptotic Bcl2-protein (Prehn et al. 1994). In line with these results, our results confirm that TGF-β1 reduces neuronal calcium signals that follow the stimulation of NMDA-type glutamate receptors and NMDAR-induced rapidly triggered neuronal cell death. In addition, we demonstrate that this effect is due to reduced extrasynaptic calcium signals mediated by GluN2B-containing NMDARs. Our results thus extend those previous findings and offer further insight into the mechanism by which TGF-β1 exerts neuroprotective effects. In line with those results, we demonstrate that synaptic plasticity in the hippocampus of CNS-TGFβ1−/− mice is impaired through a mechanism that involves the activation of GluN2B-containing NMDARs. In addition, previous research showed that TGF-β1 also enhanced neurotoxicity under certain circumstances (Prehn and Miller 1996). These authors demonstrate that TGF-β1 selectively protects neurons against NMDA-receptor-mediated excitotoxic injury, but significantly potentiates Kainate- and AMPA-receptor-mediated slowly triggered types of necrotic neuronal cell death in a protein synthesis-dependent manner. Those results are different from our own data, showing that CNS-TGFβ1−/− mice display greater number of apoptotic neurons, after being injected with kainic acid. Those discrepancies are likely a consequence of different model systems as Prehn and colleagues obtained their results in vitro using cultured rat hippocampal neurons as a model system, whereas we used kainic acid in mice in vivo. In addition, the contribution of diminished glutamate uptake in CNS-TGFβ1−/− mice following kainic acid induced seizures is unclear.
In general, it has to be taken into account that the preparation of acute hippocampal slices as compared to cultured cells carries the risk of confounding results, since tissue isolation and slicing may induce neuronal death in hippocampal slices cultures. This is a particular problem in long-term cultures (> 1 week in vitro), where tissue preparation may as well induce glial cell activation and proliferation and thus affect GluT expression (Fuller and Dailey 2007). We therefore used only acute hippocampal slices for electrophysiology and glutamate uptake experiments and showed that under those conditions, basal synaptic function remained intact because CNS-TGFβ1−/− mice and CNS-TGFβ1+/+ animals had similar PPF and stimulus-response curve (Figure 3C–D). In addition, we assessed the amount of glutamate transporter protein in acute slices immediately after dissection without any recovery in vitro. We are thus confident that the effects of TGF-β1-deficiency on glutamate uptake and GluN2B-mediated synaptic plasticity are not a result of increased tissue vulnerability in CNS-TGFβ1−/− mice.
In conclusion, our study demonstrates a previously unrecognized function for TGF-β1 in the CNS to control extracellular glutamate homeostasis and extrasynaptic calcium responses in the hippocampus. Both effects may synergize to prevent i) high extracellular glutamate levels and ii) excessive glutamate receptor-induced calcium signals. Because loss of GluTs and the activation of extrasynaptic glutamate receptors play an important role in neurodegenerative diseases (rev. in (Beart and O’Shea 2007; Hu et al. 2009; Maragakis and Rothstein 2004; Okamoto et al. 2009), our findings may provide the basis for new therapeutic strategies to limit neuronal dysfunction and death in neurodegenerative conditions.
Supplementary Material
Footnotes
AUTHOR CONTRIBUTIONS
TK and SL designed and performed the experiments, analyzed the data and wrote the manuscript. CB and JS performed the experiments and analyzed the data. YC generated the CNS-TGFβ1−/− mice. DJS and HLW wrote the paper and provided funding. The authors declare that there is no conflict of interest.
References
- Abutbul S, Shapiro J, Szaingurten-Solodkin I, Levy N, Carmy Y, Baron R, Jung S, Monsonego A. TGF-beta signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia. 2012;60(7):1160–71. doi: 10.1002/glia.22343. [DOI] [PubMed] [Google Scholar]
- Ageta H, Ikegami S, Miura M, Masuda M, Migishima R, Hino T, Takashima N, Murayama A, Sugino H, Setou M, et al. Activin plays a key role in the maintenance of long-term memory and late-LTP. Learn Mem. 2010;17(4):176–85. doi: 10.1101/lm.16659010. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5(4):325–31. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Bannerman P, Hahn A, Soulika A, Gallo V, Pleasure D. Astrogliosis in EAE spinal cord: derivation from radial glia, and relationships to oligodendroglia. Glia. 2007;55(1):57–64. doi: 10.1002/glia.20437. [DOI] [PubMed] [Google Scholar]
- Beart PM, O’Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150(1):5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellesi M, Melone M, Gubbini A, Battistacci S, Conti F. GLT-1 upregulation impairs prepulse inhibition of the startle reflex in adult rats. Glia. 2009;57(7):703–13. doi: 10.1002/glia.20798. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23(11):580–7. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Boche D, Cunningham C, Gauldie J, Perry VH. Transforming growth factor-beta 1-mediated neuroprotection against excitotoxic injury in vivo. J Cereb Blood Flow Metab. 2003;23(10):1174–82. doi: 10.1097/01.WCB.0000090080.64176.44. [DOI] [PubMed] [Google Scholar]
- Brigman JL, Wright T, Talani G, Prasad-Mulcare S, Jinde S, Seabold GK, Mathur P, Davis MI, Bock R, Gustin RM, et al. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci. 2010;30(13):4590–600. doi: 10.1523/JNEUROSCI.0640-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40(6):1133–45. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- Carmona MA, Murai KK, Wang L, Roberts AJ, Pasquale EB. Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc Natl Acad Sci U S A. 2009;106(30):12524–9. doi: 10.1073/pnas.0903328106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier Y, Yuan J, Kuchroo VK, Weiner HL. Th3 cells in peripheral tolerance. I. Induction of Foxp3-positive regulatory T cells by Th3 cells derived from TGF-beta T cell-transgenic mice. J Immunol. 2007;178(1):179–85. doi: 10.4049/jimmunol.178.1.179. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15(3):711–20. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33(1):18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Dell’Albani P, Corsaro M, Giuffrida R, Caruso A, Trovato Salinaro A, Spinella F, Nicoletti F, Albanese V, Giuffrida Stella AM. Metabotropic glutamate receptor expression in cultured rat astrocytes and human gliomas. Neurochem Res. 1997;22(9):1127–33. doi: 10.1023/a:1027317319166. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60(8):1215–26. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004(255):re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51(1):7–61. [PubMed] [Google Scholar]
- Flanders KC, Ludecke G, Engels S, Cissel DS, Roberts AB, Kondaiah P, Lafyatis R, Sporn MB, Unsicker K. Localization and actions of transforming growth factor-beta s in the embryonic nervous system. Development. 1991;113(1):183–91. doi: 10.1242/dev.113.1.183. [DOI] [PubMed] [Google Scholar]
- Flanders KC, Ren RF, Lippa CF. Transforming growth factor-betas in neurodegenerative disease. Prog Neurobiol. 1998;54(1):71–85. doi: 10.1016/s0301-0082(97)00066-x. [DOI] [PubMed] [Google Scholar]
- Fuller L, Dailey ME. Preparation of rodent hippocampal slice cultures. CSH Protoc. 2007 doi: 10.1101/pdb.prot4848. pdb prot4848. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem. 1997;69(6):2612–5. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Dehnes Y, Danbolt NC, Schousboe A. The high-affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochem Int. 2000;37(2–3):163–70. doi: 10.1016/s0197-0186(00)00019-x. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med. 2002;195(11):1499–505. doi: 10.1084/jem.20012076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouix E, Leveille F, Nicole O, Melon C, Had-Aissouni L, Buisson A. Reverse glial glutamate uptake triggers neuronal cell death through extrasynaptic NMDA receptor activation. Mol Cell Neurosci. 2009;40(4):463–73. doi: 10.1016/j.mcn.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Hamby ME, Hewett JA, Hewett SJ. Smad3-dependent signaling underlies the TGF-beta1-mediated enhancement in astrocytic iNOS expression. Glia. 2010;58(11):1282–91. doi: 10.1002/glia.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans. 2009;37(Pt 6):1147–60. doi: 10.1042/BST0371147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11(10):682–96. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5(5):405–14. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hendricson AW, Miao CL, Lippmann MJ, Morrisett RA. Ifenprodil and ethanol enhance NMDA receptor-dependent long-term depression. J Pharmacol Exp Ther. 2002;301(3):938–44. doi: 10.1124/jpet.301.3.938. [DOI] [PubMed] [Google Scholar]
- Henrich-Noack P, Prehn JH, Krieglstein J. TGF-beta 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose-response relationship and potential neuroprotective mechanisms. Stroke. 1996;27(9):1609–14. doi: 10.1161/01.str.27.9.1609. discussion 1615. [DOI] [PubMed] [Google Scholar]
- Hrabetova S, Sacktor TC. Long-term potentiation and long-term depression are induced through pharmacologically distinct NMDA receptors. Neurosci Lett. 1997;226(2):107–10. doi: 10.1016/s0304-3940(97)00252-8. [DOI] [PubMed] [Google Scholar]
- Hrabetova S, Serrano P, Blace N, Tse HW, Skifter DA, Jane DE, Monaghan DT, Sacktor TC. Distinct NMDA receptor subpopulations contribute to long-term potentiation and long-term depression induction. J Neurosci. 2000;20(12):RC81. doi: 10.1523/JNEUROSCI.20-12-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu NW, Klyubin I, Anwy R, Rowan MJ. GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc Natl Acad Sci U S A. 2009;106(48):20504–9. doi: 10.1073/pnas.0908083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Sinha SR, Tanaka K, Rothstein JD, Bergles DE. Astrocyte glutamate transporters regulate metabotropic glutamate receptor-mediated excitation of hippocampal interneurons. J Neurosci. 2004;24(19):4551–9. doi: 10.1523/JNEUROSCI.5217-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri H, Tanaka K, Manabe T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci. 2001;14(3):547–53. doi: 10.1046/j.0953-816x.2001.01664.x. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Hosoya R, Sato H, Tanaka M, Nakajima T, Iwabuchi S. Selective blockade of astrocytic glutamate transporter GLT-1 with dihydrokainate prevents neuronal death during ouabain treatment of astrocyte/neuron cocultures. Glia. 2002;40(3):337–49. doi: 10.1002/glia.10133. [DOI] [PubMed] [Google Scholar]
- Kiryk A, Aida T, Tanaka K, Banerjee P, Wilczynski GM, Meyza K, Knapska E, Filipkowski RK, Kaczmarek L, Danysz W. Behavioral characterization of GLT1 (+/−) mice as a model of mild glutamatergic hyperfunction. Neurotox Res. 2008;13(1):19–30. doi: 10.1007/BF03033364. [DOI] [PubMed] [Google Scholar]
- Klempt ND, Sirimanne E, Gunn AJ, Klempt M, Singh K, Williams C, Gluckman PD. Hypoxia-ischemia induces transforming growth factor beta 1 mRNA in the infant rat brain. Brain Res Mol Brain Res. 1992;13(1–2):93–101. doi: 10.1016/0169-328x(92)90048-g. [DOI] [PubMed] [Google Scholar]
- Konig HG, Kogel D, Rami A, Prehn JH. TGF-{beta}1 activates two distinct type I receptors in neurons: implications for neuronal NF-{kappa}B signaling. J Cell Biol. 2005;168(7):1077–86. doi: 10.1083/jcb.200407027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieglstein K, Strelau J, Schober A, Sullivan A, Unsicker K. TGF-beta and the regulation of neuron survival and death. J Physiol Paris. 2002;96(1–2):25–30. doi: 10.1016/s0928-4257(01)00077-8. [DOI] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90(2):770–4. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, et al. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron. 1996;16(2):333–44. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Levenson J, Weeber E, Selcher JC, Kategaya LS, Sweatt JD, Eskin A. Long-term potentiation and contextual fear conditioning increase neuronal glutamate uptake. Nat Neurosci. 2002;5(2):155–61. doi: 10.1038/nn791. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134(3):392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5(2):160–70. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304(5673):1021–4. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004;15(3):461–73. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103(2):295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24(36):7821–8. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momiyama A. Distinct synaptic and extrasynaptic NMDA receptors identified in dorsal horn neurones of the adult rat spinal cord. J Physiol. 2000;523(Pt 3):621–8. doi: 10.1111/j.1469-7793.2000.t01-1-00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52(1):71–6. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Nie H, Weng HR. Impaired glial glutamate uptake induces extrasynaptic glutamate spillover in the spinal sensory synapses of neuropathic rats. J Neurophysiol. 2010;103(5):2570–80. doi: 10.1152/jn.00013.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15(12):1407–13. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potier B, Billard JM, Riviere S, Sinet PM, Denis I, Champeil-Potokar G, Grintal B, Jouvenceau A, Kollen M, Dutar P. Reduction in glutamate uptake is associated with extrasynaptic NMDA and metabotropic glutamate receptor activation at the hippocampal CA1 synapse of aged rats. Aging Cell. 2010;9(5):722–35. doi: 10.1111/j.1474-9726.2010.00593.x. [DOI] [PubMed] [Google Scholar]
- Prehn JH, Backhauss C, Krieglstein J. Transforming growth factor-beta 1 prevents glutamate neurotoxicity in rat neocortical cultures and protects mouse neocortex from ischemic injury in vivo. J Cereb Blood Flow Metab. 1993a;13(3):521–5. doi: 10.1038/jcbfm.1993.67. [DOI] [PubMed] [Google Scholar]
- Prehn JH, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ. Regulation of neuronal Bcl2 protein expression and calcium homeostasis by transforming growth factor type beta confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci U S A. 1994;91(26):12599–603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehn JH, Miller RJ. Opposite effects of TGF-beta 1 on rapidly- and slowly-triggered excitotoxic injury. Neuropharmacology. 1996;35(3):249–56. doi: 10.1016/0028-3908(96)00001-9. [DOI] [PubMed] [Google Scholar]
- Prehn JH, Peruche B, Unsicker K, Krieglstein J. Isoform-specific effects of transforming growth factors-beta on degeneration of primary neuronal cultures induced by cytotoxic hypoxia or glutamate. J Neurochem. 1993b;60(5):1665–72. doi: 10.1111/j.1471-4159.1993.tb13389.x. [DOI] [PubMed] [Google Scholar]
- Reilly JF, Maher PA, Kumari VG. Regulation of astrocyte GFAP expression by TGF-beta1 and FGF-2. Glia. 1998;22(2):202–10. doi: 10.1002/(sici)1098-1136(199802)22:2<202::aid-glia11>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease. CNS Drug Rev. 2003;9(3):275–308. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373(6510):151–5. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8(12):970–82. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- Schulz R, Vogel T, Mashima T, Tsuruo T, Krieglstein K. Involvement of Fractin in TGF-beta-induced apoptosis in oligodendroglial progenitor cells. Glia. 2009;57(15):1619–29. doi: 10.1002/glia.20875. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Fine A, Kullmann DM, Rusakov DA. NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci. 2004;24(20):4767–77. doi: 10.1523/JNEUROSCI.0364-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27(11):2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359(6397):693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39(4):641–54. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, et al. Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell. 1998;92(2):279–89. doi: 10.1016/s0092-8674(00)80921-6. [DOI] [PubMed] [Google Scholar]
- Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Kohr G. C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci. 2000;20(12):4573–81. doi: 10.1523/JNEUROSCI.20-12-04573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipursky J, Gomes FC. TGF-beta1/SMAD signaling induces astrocyte fate commitment in vitro: implications for radial glia development. Glia. 2007;55(10):1023–33. doi: 10.1002/glia.20522. [DOI] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507 (Pt 1):13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan R, Rauen T, Fischer F, Wiessner M, Grewer C, Bicho A, Pow DV. Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: implications for CNS glutamate homeostasis. Glia. 2004;45(2):155–69. doi: 10.1002/glia.10317. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17(3):932–40. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayasu Y, Iino M, Shimamoto K, Tanaka K, Ozawa S. Glial glutamate transporters maintain one-to-one relationship at the climbing fiber-Purkinje cell synapse by preventing glutamate spillover. J Neurosci. 2006;26(24):6563–72. doi: 10.1523/JNEUROSCI.5342-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276(5319):1699–702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8(12):1727–34. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Wyss-Coray T. A role for TGF-beta signaling in neurodegeneration: evidence from genetically engineered models. Curr Alzheimer Res. 2006;3(5):505–13. doi: 10.2174/156720506779025297. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J Clin Invest. 2006;116(11):3060–9. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19(10):4180–8. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8(12):935–47. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Unsicker K, Flanders KC, Cissel DS, Lafyatis R, Sporn MB. Transforming growth factor beta isoforms in the adult rat central and peripheral nervous system. Neuroscience. 1991;44(3):613–25. doi: 10.1016/0306-4522(91)90082-y. [DOI] [PubMed] [Google Scholar]
- Unsicker K, Meier C, Krieglstein K, Sartor BM, Flanders KC. Expression, localization, and function of transforming growth factor-beta s in embryonic chick spinal cord, hindbrain, and dorsal root ganglia. J Neurobiol. 1996;29(2):262–76. doi: 10.1002/(SICI)1097-4695(199602)29:2<262::AID-NEU10>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol. 2005;31(1–3):3–16. doi: 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11(1):37–49. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- Wiessner C, Gehrmann J, Lindholm D, Topper R, Kreutzberg GW, Hossmann KA. Expression of transforming growth factor-beta 1 and interleukin-1 beta mRNA in rat brain following transient forebrain ischemia. Acta Neuropathol. 1993;86(5):439–46. doi: 10.1007/BF00228578. [DOI] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30(33):11246–50. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Ohmura T, Komatsu Y. Two forms of synaptic plasticity with distinct dependence on age, experience, and NMDA receptor subtype in rat visual cortex. J Neurosci. 2003;23(16):6557–66. doi: 10.1523/JNEUROSCI.23-16-06557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.