

Abstract
Glymes, also known as glycol diethers, are saturated non-cyclic polyethers containing no other functional groups. Most glymes are usually less volatile and less toxic than common laboratory organic solvents; in this context, they are more environmentally benign solvents. However, it is also important to point out that some glymes could cause long-term reproductive and developmental damages despite their low acute toxicities. Glymes have both hydrophilic and hydrophobic characters that common organic solvents are lack of. In addition, they are usually thermally and chemically stable, and can even form complexes with ions. Therefore, glymes are found in a broad range of laboratory applications including organic synthesis, electrochemistry, biocatalysis, materials, and Chemical Vapor Deposition (CVD), etc. In addition, glyme are used in numerous industrial applications, such as cleaning products, inks, adhesives and coatings, batteries and electronics, absorption refrigeration and heat pumps, as well as pharmaceutical formulations, etc. However, there is a lack of comprehensive and critical review on this attractive subject. This review aims to accomplish this task by providing an in-depth understanding of glymes’ physicochemical properties, toxicity and major applications.
Keywords: glyme, ion complexing, synthesis, biocatalysis, electrochemistry, nanomaterial
1. Introduction
Glymes, i.e. glycol diethers, are saturated polyethers containing no other functional groups. When compared with glycols (such as polyethylene glycols, PEGs), glymes do not carry free hydroxyl groups and thus are aprotic polar and chemically inert compounds. There are two major types of glymes: ethylene oxide (EO)-based glymes (also known as PEG-based) and propylene oxide-based glymes (i.e. polypropylene glycol (PPG)-based). The general structure of PEG-based glymes and their common names are illustrated in Scheme 1.
Scheme 1.

Structure of EO-based glymes, and representative common names and symbols.
Most glymes are completely miscible with both water and hydrocarbon solvents, and could solvate alkali cations. In addition, glymes have many other favorable properties including a wide liquid range (typically > 200 °C, except monoglyme), low viscosities, high chemical and thermal stability, relatively low vapor pressure and low toxicity. Due to these excellent solvent properties, glymes have been widely used in many laboratory and commercial applications including reaction media (i.e. organometallic reactions, polymerization, and reactions involving alkali metals, oxidations and reductions), extraction solvents (for metals and organics), gas purification, absorption refrigeration, formulation of adhesives and coatings, textiles, solvents for electronic industry, pharmaceutical formation, batteries, and cleaning solutions, etc.
Despite numerous studies on glymes, there is a lack of a comprehensive and critical review on this subject to meet the continuing interest in glymes; in receny years, there are strong focuses on their applications in electrochemistry, catalysts, Chemical Vapor Deposition (CVD), nanomaterials, and the dissolution of CO2. This review aims to provide a comprehensive set of physicochemical properties of glymes and a systematical overview of their applications, as well as a critical analysis of their structures-properties-applications relationships. To be focused on the subject, glycol monoethers and glycol ether esters are not discussed herein as their physical properties, toxicity and applications have been reviewed elsewhere.1–3
2. Preparations of glymes
Ethylene oxide (EO)-based glymes can be prepared by several common methods at large scales using ethylene epoxide (Scheme 2): Route 1, ethylene oxide reacts with an alcohol to produce glycol monoether, which is further converted to glyme following the Williamson synthesis (i.e. glycol ether reacts with metal sodium to yield sodium alkoxide, which then reacts with alkyl halide to produce glyme); Route 2, methylation of glycol ether with methyl sulfate; Route 3, Lewis acid-catalyzed cleavage of ethylene oxide by ether, typically resulting in glyme mixtures; Route 4, the reaction of ethylene glycol with alcohol catalyzed polyperfluorosulfonic acid resin at high temperature and pressure. Similarly, propylene oxide-based glymes can be prepared by replacing ethylene oxide with propylene oxide in the above methods.
Scheme 2.

Common routes for glyme synthesis.
In addition, Haymore et al.4 described a two-step synthesis of pentaglyme (Scheme 3): the reaction of 2-methoxyethanol with sodium metal to yield an alkoxide followed by a nucleophilic substitution of dichloride. Adcock and Lagow5 prepared perfluoroglyme and perfluorodiglyme as potential high fluids and solvents through a direct fluorination method (fluorine gas).
Scheme 3.

Synthesis of pentaglyme.
3. Toxicity of glymes
In general, glymes exhibit low to moderate acute toxicity (see toxicity data in Tables 1 and 2) when compared with common organic solvents (such as toluene, THF and chloroform). Ethylene glycol dimethyl ether (monoglyme) triggered maternal deaths of pregnant Sprague-Dawley rats at 1000 mg/kg/day and was fetolethal at doses ranging from 120 to 1000 mg/kg/day; a dose of 60 mg/kg/day caused a 7% weight decrease and severe edema in pups surviving to birth.6 When rats were exposed to 200 ppm diglyme vapor for an extended period of time (15 × 6 h), no toxic effect was observed in terms of normal blood and urine tests and normal organs by autopsy; however, at a higher vapor concentration (600 ppm) for the same period of time, irregular weight gain was observed and autopsy suggested atrophied thymus and congested adrenals although the blood and urine tests were normal.7
Table 1.
Estimated toxicity for glymes and common organic solventsa
| Solvent | Oral rat LD50 (mg/kg) |
Bioaccumulation Factor |
Developmental Toxicity |
Mutagenicity |
|---|---|---|---|---|
| Ethanol | (7055b) | 1.28 | (1.00,c toxicant) | −0.10 (+) |
| Toluene | 1083 (636b) | 48.88 | 0.29 (non) | −0.01 (−) |
| Benzene | 2030 (931b) | 30.78 | 0.29 | 0.03 (+) |
| THF | 932 (1652b) | 2.67 | 0.48 | 0.05 (−) |
| Chloroform | 1794 (695b) | 5.07 (6.3019) | 0.67 (toxicant) | 0.45 (−) |
| Monoglyme | 2997 | 1.66 | 0.59 (toxicant17) | 0.59 (1.00d) (+)e |
| Diglyme | 3779 (5404b) | 2.04 | 0.43 (toxicant17) | 0.38 |
| Triglyme | 6496 | 1.73 | 0.36 | 0.18 (1.00d) (+) |
| Tetraglyme | 5146 (5140b) | 6.97 | −0.10 | 0.23 |
| Pentaglyme | 5604 | 2.85 | −0.10 | 0.33 |
| Hexaglyme | 6112 | 6.02 | −0.06 | 0.26 |
| Ethyl glyme | 3949 (3619b) | 4.60 | 0.32 (toxicant17) | 0.20 |
| Butyl glyme | 4427 (3253b) | 24.33 | 0.47 | 0.05 |
| Ethyl diglyme | 6818 (4968b) | 5.82 | 0.26 | 0.16 |
| Butyl diglyme | 3378 (3901b) | 15.57 | 0.05 | 0.23 |
Note:
Toxicity was estimated with the consensus method using Toxicity Estimation Software Tool (TEST) (http://www.epa.gov/nrmrl/std/qsar/qsar.html); disclaimer: these estimated values are for research and evaluation purposes, not for guiding clinical or production applications;
Experimental value from ChemidPlus database (http://chem.sis.nlm.nih.gov/chemidplus/);
Experimental value from the CAESAR database (http://www.caesar-project.eu/index.php?page=results§ion=endpoint&ne=5);
Experimental value from Toxicity Benchmark database (http://doc.ml.tu-berlin.de/toxbenchmark/);
‘+’ means mutagenicity positive, ‘−’ means mutagenicity negative.
Table 2.
Physical and thermodynamic properties of glymes
| Glyme | Freezing point (°C) |
Boiling point (°C) |
Dynamic viscosity (mPa s) at 20 °C |
p (mm Hg) at 20 °Cd |
Density (g/mL) at 25 °Cv |
Flash point (°C)e |
LD50 (mg/Kg)f |
Solubility (20 °C) |
|
|---|---|---|---|---|---|---|---|---|---|
| Monoglyme (G1) | −69.0b,c,h | 85.2,b,h 85,c 84.5mm |
0.4236,o 0.432,q 0.417,bb 0.420,cc 0.455,ll 1.1(20 °C)b,h |
54b,h | 0.86124,j 0.86207,n 0.86132,o 0.8605,q 0.8613,s 0.86114,t,u 0.864,w 0.86260,y 0.86155,z 0.8615,bb 0.8626,cc,dd 0.8612,ii 0.859,ll 0.86370,mm 0.8683 (20 °C),b,h 0.867 (20 °C),c 0.86765 (20 °C),gg |
−6b,c,h | 5370b | miscible with H2O |
|
| Ethyl glyme (G1-Et) | −74.0b,h | 121,b 121.4,h 121.2mm |
0.593,bb 0.7 (20 °C),b 0.65 (20 °C)h |
9,b 9.4h | 0.83607,z 0.8362,aa 0.8360,bb 0.83510,mm 0.8417(20 °C),b,h |
27b | 4400b | 20.4% in H2O, 3.3% H2O inb |
|
| Butyl glyme (G1-Bu) | −69.1h | 203.6,h 206 (dec)mm |
0.09h | 0.83189,mm 0.8374(20 °C)h |
85h (open cup) |
0.2wt% in H2O, 0.6wt% H2O inh |
|||
| Diglyme (G2) | −64.0,a,c −70,g −68h |
162a,c,g | 0.981,m 1.06,x 0.989,bb 0.985,cc 0.991,jj 0.976,nn 2.0 (20 °C),b,h 1.14(20 °C)x |
2,b 3.0 (100 °C)h |
0.93873,j 0.93882,k 0.93924,l 0.9394,m 0.93892,n 0.93871,t 0.93935,u 0.938,w 0.93897,y 0.93875,z 0.9385,bb 0.9389,cc 0.9399,jj 0.9397,kk 0.93961,nn 0.9434 (20 °C),a 0.944 (20 °C),c 0.945 (20 °C),g,h 0.94511 (20 °C)gg |
57,b 51c | 4670b | miscible with H2O, ethanol, diethyl ether |
|
| Ethyl diglyme (G2-Et) | −44.3b | 189b | 1.238,bb 1.241,cc1.4 (20 °C)b |
0.5b | 0.9082(20 °C),b 0.9028,z 0.9021,bb 0.9035cc |
90b | 5000b | miscible with H2O |
|
| Butyl diglyme (G2-Bu) | −60.2,b −60c |
256b,c | 2.122,cc 2.4 (20 °C)b |
<0.01b | 0.8814(20 °C),b 0.884(20 °C),c 0.87830,z 0.8781cc |
118,b 120c |
3900b | 0.3% in H2O, 1.4% H2O inb,c |
|
| Triglyme (G3) | −43.8,a −40,c −45.0h |
218,a 220,c 216h |
1.96,r,nn 2.16,x 1.950,cc 3.8 (20 °C),b,h |
2.41(20 °C)x | 0.02b,h | 0.98001,i 0.98067,j 0.98117,l 0.9795,r 0.98058,t,u 0.981,w 0.98071,y 0.9807,cc 0.98042,ee 0.97981,ff 0.98023,nn 0.986 (20 °C),a,h 0.987 (20 °C),c 0.98569 (20 °C),gg 0.98541 (20 °C)hh |
111,b,h 113c |
5000b | very soluble in H2O, benzene |
| Tetraglyme (G4) | −29.7,b,h −30c |
275b,c,h | 3.295,p 3.67,x 3.40,nn 4.1 (20 °C),b,h 4.18(20 °C)x |
<0.01b,h | 1.00662,i 1.00564,j 1.00627,l 1.0047,p 1.00666,t 1.00668,u 1.00628,y 1.00620,ee 1.00743,nn 1.0132(20 °C),b,h 1.0(20 °C),c 1.0116 (20 °C),gg 1.01115 (20 °C)hh |
141b,c,h | 5100b | miscible with H2O |
|
| Proglyme (P2) | −71b, −80c |
175b,c | 1.1 (20 °C)b | 0.55b | 0.900(20 °C)b,c | 65b,c | — | 35% in H2O, 4.5% H2O inb,c |
|
| PEG-DME 200 | −37c | >300c | 4.3 mm2/s (20 °C)c |
— | 1.01–1.02 (20 °C)c | 154c | — | ||
| PEG-DME 250 | −23c | >300c | 7 mm2/s (20 °C),c 7.215 mm2/s (20 °C),gg |
— | 1.02–1.04(20 °C),c 1.0355 (20 °C)gg |
137c | — | ||
| Polyglyme (MW=236)b | −28b | 275b | 12 (20 °C)b | 0.01b | 1.03(20 °C)b | 135b | — | ||
| Polyglyme (MW=275)b | −23b | 275b | 12 (20 °C)b | <0.01b | 1.04(20 °C)b | >130b | — | miscible with H2O |
|
| Higlyme (MW>400)b | −5–10b | >300b | 34 (20 °C)b | 0.1b | 0.975(20 °C)b | 140b | — | miscible with H2O |
|
| PEG-DME 500 | 13c | >300c | 25 mm2/s (20 °C)c |
— | 1.05(50 °C)c | 220c | — | ||
| PEG-DME 1000 | 36c | >300c | 11 mm2/s (100 °C)c |
— | 1.10(50 °C)c | 260c | — | ||
| PEG-DME 2000 | 50c | >300c | 30 mm2/s (100 °C)c |
— | 1.08(60 °C)c | 254c | — |
Note:
Ref,40
production specifications from Novolyte Technologies (now a part of BASF),
production specifications from Clariant (viscosity is kinematic viscosity),
vapor pressure,
closed cup,
acute toxicity,
Ref41,
Ref42,
Ref,43
Ref,44
Ref45 (this reference also reported other thermodynamic properties of diglyme at 25 °C such as dipole moment μ = 1.87 D, heat capacity Cp = 276.9 J mol−1 K−1, etc.),
Ref,46
Ref,47
Ref,48
Ref49 (densities and viscosities of monoglyme at 308.15 K and 318.15 K were also reported),
Ref,50
Ref,51
Ref52 (this reference also reported dielectric constant of 7.62 as well as refractive index and molar refraction for triglyme),
Ref,53
Ref54,
Ref,55
density data for monoglyme and diglyme were reported at temperatures between 293.15 K and 353.15 K and up to 60 MPa,56
Ref,57
Ref,58
Ref59 (density data at 288.18 and 308.15 K were also reported),
Ref,60
Ref,61
Ref62 (densities and viscosities were reported for several glymes from 288.15 K to 343.15 K),
Ref,63
Ref,64
Ref,65
Ref,66
Ref67 (densities, kinematic viscosities and heat capacities were reported for several glymes including pentaethylene glycol dimethyl ether from 283.15 to 423.15 K),
Ref68 (data on density, isentropic compressibility and isothermal compressibility of triglyme and tetraglyme were reported at 293.15–353.15 K and 0.1–100 MPa),
Ref,69
Ref,70
Ref,71
Ref,72
Ref (densities, refractive indexes, and boiling points of other 1,2-disubstituted ethylene glycol derivatives such as propyl glyme were also reported),73
Ref,74
However, there is a rising concern of glymes that may cause reproductive and developmental harms to exposed workers and consumers using paint, carpet cleaners, inkjet cartridges and other products. McGregor et al.8 studied the exposure of male rats to 250 or 1000 ppm diglyme, and found diglyme was reproductive toxicant causing increased sperm abnormalities. Schuler et al.9 examined fifteen glycol ethers for their adverse reproductive toxic effects using an in vivo mouse screening bioassay; this group found that all mice receiving glycol ethers having terminal methyl groups, i.e., ethylene glycol monomethyl ether, monoglyme, diethylene glycol monomethyl ether, diglyme and triglyme produced few viable litters (0, 0, 16, 0, and 0% respectively); similar results were also observed for ethylene glycol monoether ether and ethyl monoglyme (0 and 11% viable litters respectively). However, other two ethyl ethers (diethylene glycol monoethyl ether and ethyl diglyme), three butyl ethers (ethylene glycol monobutyl ether, diethylene glycol monobutyl ether, butyl diglyme), and three glycol ethers with terminal hydroxyl groups (ethylene glycol, diethylene glycol and triethylene glycol) failed to show this kind of fetotoxicity. They also suggested that: (1) The appending of an alkyl group considerably increased the maternal toxicity of glycols. For example, ethyl glycol monobutyl ether appeared to be more toxic than ethylene glycol monomethyl ether, which was more toxic than ethylene glycol monoethyl ether; but all three showed greater toxicity than ethylene glycol. The diethylene glycol mono-alkyl ethers and (alkyl) diglymes were more toxic than diethylene glycol, and triglyme was more toxic than triethylene glycol. (2) The methyl ethers usually seem more toxic than the ethyl or butyl ethers except ethylene glycol monobutyl ether. Similarly, Johnson et al.10 found that butyl diglyme was more toxic than diethylene glycol, but did not induce significant developmental toxicity to the hydra.
A review11 on the genetic toxicology of glycol ethers suggested that diglyme is lack of genotoxic potential in some mutagenicity tests, but it was reproductive toxicant in mouse sperm test and male rat dominant lethal test. Repeated daily oral doses of diglyme at 684 mg/kg in a subchronic study of Sprague-Dawley rats suggested the onset of testicular pathology, which was similar to the pathology of equal molar doses of 2-methoxyethanol or 2-ethoxyethanol.12 Furthermore, it was confirmed that there were two major metabolites of the testicular toxin (e.g. diglyme): (2-methoxyethoxy)acetic acid (MEAA) (ca. 70%) and methoxyacetic acid (MAA) (ca. 6%), along with several other unidentified metabolites, and two major metabolism pathways; however, only MAA is accounted for the reproductive toxicity of diglyme.13–15 Hardin and Eisenmann16 gave time-mated CD-1 mice oral doses of glycol ethers at a dose of 4 mmol/kg on gestation day 11, and then examined fetuses on gestation day 18 for weight and gross external malformations. All four glycol ethers (ethylene glycol monomethyl ether, monoglyme, diglyme and triglyme) exhibited no treatment-related maternal toxicity and no impact on the intrauterine survival; in addition, no treatment-related gross external malformations other than paw defects. Except triglyme, the other three glycol ethers produced different degrees of paw defects: 87.5% of ethylene glycol monomethyl ether-treated litters (68.5% of fetuses), 86.7% of monoglyme-treated litters (33.8% of fetuses), and 77.8% of diglyme-treated litters (39.7% of fetuses).
In conclusion, although glymes show low to moderate acute toxicity, they have raised concerns regarding chronic exposure and reproductive effects. Therefore, glymes are recommended as benign solvents for industrial applications, not consumer products. The U.S. Environmental Protection Agency (EPA) announced in July 2011 that three glymes pose “high concern to workers, consumers and children” because they may have reproductive or developmental effects: monoglyme and diglyme caused reproductive and developmental damage in rodent studies, and animal studies on ethyl glyme exhibit developmental toxicity and the potential for gene mutation; EPA also plans to restrict new uses of 11 other glymes in the U.S. marketplace.17 Since lower molecular weight glymes have shown reproductive toxicity in rats and mice, only higher molecular weight glymes should be used in pharmaceutical applications. In the European Union, products containing monoglyme or diglyme have been regulated; their labels must indicate “may impair fertility” or “may cause harm to the unborn child.” On the contrary, dipropylene glycol dimethyl ether is known as a very versatile and environmentally friendly solvent. It is not listed as a hazardous air pollutant (HAP), and it has been shown that propylene-based glycol ethers are less toxic than those ethylene-based glycol ethers.18
Although toxicity and biodegradability data are not readily available for many glymes, models based on Quantitative Structure Activity Relationships (QSARs) can be used to predict measures of toxicity from physical characteristics of chemical structures. As shown in Table 1, although there are differences between estimated and experimental values, the estimated toxicity may be used for empirical evaluation of glymes. In general, the estimated toxicity data in Table 1 are consistent with earlier discussion based on experimental data: glymes have low acute toxicity (high oral rat LD50 values when compared with common organic solvents), however, there are concerns of their developmental toxicity and mutagenicity. Except those butyl glymes, methyl- and ethyl glymes seems to have relatively low bioaccumulation factors.
4. Physicochemical and metal complexing properties of glymes
4.1 General properties
Glymes are dipolar aprotic solvents with high chemical stability. They are usually stable under neutral and basic conditions, and only undergo pyrolysis under acidic conditions to form methanol and oxycarbonium ions.20 Some of their common physical and thermodynamic properties are systematically compiled in Table 2. Typically, glymes have high boiling points and a wide range of temperature of being liquids (> 200 °C, or even >300 °C) except monoglyme. In contrast to some volatile organic solvents, most glymes have very low vapor pressures (< 0.5 mmHg at 20 °C) except mono- and diglyme. Many simple glymes have low viscosities in the range of 1–4 mPa s at 20 °C. Most glymes are completely miscible with both water and organic solvents (such as ethanol, acetone, benzene, and octane), and tend to solvate cations like crown ethers. They are also outstanding solvents for many organic materials (including triglycerides21, 22). Different polarity measures of several glymes are listed in Table 3; dipole moments (μ) and dielectric constants increases with the increase in ethylene oxide chain length. The solvatochromic polarity scales in Table 3 also follow the same trend, and confirm that glymes are generally less polar than methanol () and acetone (0.355) but more polar than THF (0.207).23
Table 3.
Polarity and heat capacity of several glymes
| Glyme | Dipole moment (D) at 25 °C | Dielectric Constant at 25 °C |
Solvatochromic polarity23 | Heat capacity at 25 °C (J mol−1 K−1) |
|
|---|---|---|---|---|---|
| ET(30), kcal mol−1 |
|||||
| Monoglyme | 1.62,75 1.61,57 1.59,76 1.71 (in benzene)76 | 7.18,77 7.20,72 7.55,78 | 38.2 | 0.231 | 191.1479 |
| Diglyme | 1.91,75 1.92,57, 76 1.87,45 1.97 (in benzene)76 | 7.480 | 38.6 | 0.244 | 277.76,79 276.9,45 279.0581 |
| Triglyme | 2.16,57, 762.22 (in benzene)76 | 7.6252 | 38.9 | 0.253 | 367.78,79 367.3066 |
| Tetraglyme | 2.44,762.4555, 76 | – | – | – | 457.1079 |
Proton affinity (PA) is a direct measure of gas-phase basicities of organic molecules. Meot-Ner24 calculated the proton affinities (in kcal/mol) from measurements by pulsed high-pressure mass spectrometer as: monoglyme (205), diglyme (219), triglyme (226), 12-crown-4 (220) and 18-crown-6 (221). Sharma et al.25 determined the proton affinities (in kcal/mol) of several glymes and crown ethers by measuring proton-transfer equilibria with a pulsed electron beam high ion source pressure mass spectrometer: Me2O (191), monoglyme (204), diglyme (218), triglyme (224), tetraglyme (227), 12-crown-4 (221), 15-crown-5 (223) and 18-crown-6 (230) [based on PA(NH3) = 204 kcal/mol]. The ab initio calculations of 12-crown-4, 15-crown-5, 18-crown-6, glymes and protonated species suggest that protonated crown ethers share similar moieties with protonated diglyme; the calculated proton affinities (in kcal/mol) are: diglyme (222), 12-crown-4 (221), 15-crown-5 (225), and 18-crown-6 (227).26 Using the B3LYP density functional method, Adötoledo et al.27 found that protonated monoglyme Gl·H+ and protonated monoglyme dimer (Gl)2H+, as well as protonated 12-crown-4-ether (12c4H+), form two internal hydrogen bonds with NH3, CH3OH, CH3NH2, and (CH3)2NH with the exception of (Gl)2H+·NH3 that bear four O⋯H bonds. The insertion energy of NH3, CH3OH and amines into 12c4H+ or (Gl)2H+ increases with increasing proton affinity of the base, while the association energy of CH3OH2+, NH4+, etc., with 12c4 or (Gl)2 decreases with increasing proton affinity (of NH3, CH3OH, etc.). However, in terms of protonation equilibrium constants (Kp), there are considerable differences between macrocyclic polyethers and linear glymes. For example, the Kp constant for the protonation of dicyclohexyl-18-crown-6 (DCC) by HBr at 25 °C is about 106 M−1, while the Kp values for diglyme and triglyme are only 0.17 and 0.20 M−1 respectively.28
Physical properties of aqueous glymes are important to the understanding of solvent properties of glymes. Meot-Ner et al.29 examined the complexing of H+ from the formation of intramolecular or intermolecular hydrogen bonds with glymes (monoglyme, diglyme or triglyme) and 0–3 water molecules by pulsed high-pressure mass spectrometry; they found that the bonding of H3O+ typically involve two or three hydrogen bonds, and complexes of H+ and H3O+ can be stabilized by interactions with bond dipoles of free ether groups of glymes and crown ethers whilst such a stabilization is enhanced by the decreasing constraints on the geometries of polar groups. Anomalous conformational behaviors of short glymes [CH3(OCH2CH2)mOCH3 with m = 2–6] in water were studied by FT-IR30 and Raman spectroscopy;31 both methods indicate that poly(ethylene oxide) chain progressively prefers the gauche (g) conformation for the OCH2–CH2O segment at the first stage, but this direction of the conformational preference is reversed for concentrations lower than a particular solution composition. Such an anomaly can be attributed to specific interactions and/or structures relevant to the glyme-water system. Furthermore, molecular dynamics simulations of monoglyme and diglyme in aqueous solutions32 confirmed that the g population of the O–C—C–O dihedral increases with the increase in water content; a further examination of the composition dependence of diglyme conformational populations implies that the decrease in the O–C—C–O g population in extremely dilute solutions can be attributed to a decrease in the tgt population of the C–O—C—C—O–C conformational triad. An atomistic force field for simulations of monoglyme reveals that the binding of monoglyme to water is comparable to water-water binding in water dimer, suggesting strong hydrogen bonding between monoglyme and water.33 Bedrov and Smith34 found that the properties of monoglyme/water solutions on the particular water model depend on the solution composition; in dilute solutions, structural and conformational properties are almost independent of the water model. However, short glymes [CH3(OCH2CH2)mOCH3 with m = 1–4] in formamide showed a different conformational behavior based on a Raman spectroscopic study:35 for the solutions with solute mole fractions between 0.5 and 0.01, the population of the gauche conformation for the OCH2-CH2O segment increases progressively with increasing solvent fraction. Monte Carlo simulations of monoglyme in water at 298 and 398 K indicate that the anti–anti–anti conformer is the only conformer that increases its probability with temperature; the anti–anti-gauche conformer is the most populated one among all types.36 The Bernal group37, 38 investigated the apparent molar volumes and adiabatic compressibilities of glymes (e.g. triglyme and tetraglyme) and crown ethers (e.g. 18-crown-6, 15-crown-5 and 12-crown-4) in H2O and D2O; their results suggest that the hydration of crown ethers increases with its size and is predominated by the hydrophobic hydration of –CH2– groups; they also indicate that there is a subtle difference between the hydration of the –CH2CH2O– group in crown and straight-chain compounds, and the compressibility data reveal a more negative compressibility due to the addition of a –CH2CH2O– group to a crown ethers than a similar addition to a straight-chain glyme. Douhéret et al.39 measured the densities and ultrasound speeds of aqueous solutions of di-, tri- and tetra-glyme, and determined the excess molar volumes and the excess molar isentropic compressions; the excess molar properties are generally negative values. The standard Gibbs energy changes associated with aggregate formation are negative, implying the aggregation is moderate in aqueous glymes (N = 4–6), although these aggregates are less stable than those formed from self-assembled species containing hydroxyl groups.
Other experimental or theoretic studies of physical properties of glymes or glyme-containing mixtures can be found in literatures: optical anisotropies of monoglyme, diglyme, triglyme and tetraglyme,82 excess heat capacities of liquid mixtures of triglyme and tetraglyme with cyclohexane as well as tetraglyme with n-heptane at 288.15, 298.15 and 308.15 K and at atmospheric pressure,83 weak self-association of glymes based on the evaluation of excess isobaric thermal expansion of glyme and alkane mixtures by an associated mixture model with equation of state contribution,84 excess molar volumes and excess molar isobaric heat capacities of glymes and ethyl acetate,44 excess molar volumes and viscosities of glyme and acetonitrile,85 excess thermodynamic and equilibrium properties of glyme + n-alkane mixtures,45, 65, 66, 71, 81 isobaric vapor-liquid equilibrium for the binary systems of monoglyme + alcohols,53 excess molar volumes of binary mixtures of glymes and 1-propanol,59, 60, 63 calorimetric study of interactions between glyme and alcohol,61 dynamic viscosities of mixtures of refrigerant (HFC-134a) + glyme at different temperatures and pressures,86 excess molar enthalpies of mixtures of methanol or trifluoroethanol + glyme,87, 88 static relative permittivities of the ternary system of 2-methoxyethanol + 1,2-dimethoxyethane + water from −10–80 °C,78 vapor-liquid equilibrium of binary systems consisting of monoglyme with toluene, methylcyclohexane, or (trifluoromethyl)benzene,69 solubility of HFC-134a refrigerant in glymes,89 densities, viscosities, and refractive indices of diglyme + cyclohexane or + 1,2,3,4-tetrahydronaphthalene,70 excess molar heat capacities of mixtures of glymes and various alkanes,79 and excess heat capacities of glyme-dimethylsiloxane systems at 25 °C,90 etc. López et al.91 estimated the densities, isothermal compressibilities, and isobaric thermal expansion coefficients of glymes in the temperature range of 293.15K–353.15K at pressures up to 100 MPa from the P-c-T data (c is the speed of sound in glyme), and found the indirect predictions matched direct experimental values.
4.2 Ion complexing properties
A unique feature about glymes is that they contain multiple ether-type oxygen atoms (similar to crown ethers) and flexible alkoxy chains. Therefore, they often behave like crown ethers in terms of solvating metal ions through oxygen-ion complexing (chelating) property, which leads to many valuable applications. The chain flexibility was demonstrated by the spin-lattice relaxation of methoxy protons in glymes: the experimental T1−l values of glymes are significantly lower than those calculated for a rigid molecule although higher than those for completely free motion of methyl groups.92 This section places a special emphasis on the ion chelating property of glymes while the applications of this property in electrochemistry and chemical reactions will be discussed in later sections.
A vivid illustration of the ion-glyme complexing can be demonstrated by Fig 1, where lithium cation is chelated by a tridentate diglyme molecule. Matsui and Takeyama93 confirmed that Li+ is coordinated with six oxygen atoms in either monoglyme or diglyme; however, monoglyme is a bidentate ligand and diglyme is a tridentate ligand. The trans-cis isomerization of diglyme is necessary for the coordination to a single cation although it typically has a trans-trans conformation of minimum energy.94 Carvajal et al.72 studied the solvation behaviors of [Cs+][BPh4−], [Na+][BPh4−] and [Bu(i-Am)3+][BPh4−] in monoglyme and tetrahydrofuran (THF), and indicated that: (a) free Cs+ ion coordinates with monoglyme but not with THF, (b) in both monoglyme and THF, [Na+][BPh4−] primarily yields solvent-separated pairs which dissociate into solvent-coordinated Na+ ions, (c) however, [Bu(i-Am)3+][BPh4−] forms contact ion pairs in both media and dissociates into free Bu(i-Am)3+ and BPh4− ions not coordinated with the solvent. Using Electron Spin Resonance (ESR) spectroscopy, Hoefelmann et al.95 determined the equilibrium constant of sodium naphthalene and tetraglyme forming loose ion pair as 200–300 M−1 at 27 °C. Collins et al.96 determined the formation constants of tetraglyme separated ion pairs of bolaform electrolytes Na+, −Fl(CH2)nFl−, Na+ (Fl− is a fluorenyl carbanion; n = 2, 3, 4, or 6) in THF and tetrahydropyran (THP) at 25°C. They suggested that it is easier to separate the ion-pair with tetraglyme in THP than in THF. For both monoglyme and diglyme, the lithium-ion solvation-shell coordination number is 4 at room temperature and 5 at a lower temperature based on the matrix-solvation FT-IR data.97 A molecular dynamic simulation study98, 99 on different concentrations of NaI dissolved in dimethyl ether and monoglyme suggests that in diluted solutions, free ions exist as the most common ionic species, followed by ion pairs; with a further increase in salt concentration, ion pairs become the predominate species and many of them are in the form of clusters of 3–6 ions or more ions. Replacing dimethyl ether by monoglyme substantially reduces the ion clustering due to the chelating effect of glyme oxygens with the cation (Na+). At the highest concentration studied in monoglyme (i.e. an oxygen/cation ratio of 16:1), free ions constitute ~50% of the total ion concentration and neutral pairs constitute about 20%. For Na+–glyme systems, the electric current is mainly due to the movement of free ions and the relative movement of ions within loosely bound ion pairs. Based on the ab initio calculations of metal ion–glyme complexes,100, 101 the Lindgren group suggested that the high chain flexibility of glymes enables many stable structures within a narrow energy range with very different geometrical arrangements of the ether oxygens, for example, at least 4 structures of different geometries for M-glyme complexes (M = Li+, Na+, K+, Mg2+ and Ca2+)100 and 11 structures of different geometries for the Li+–tetraglyme systems.101 The coordination numbers of lithium range from 4 to 6 for 1:1 complexes of Li+ with tetra-, penta- and hexaglyme, and the total binding energy increases with the glyme length; the coordination figures mainly consist of the quadratic pyramid, trigonal bipyramid and the trigonal prism type of geometries.101 Johansson et al.102 performed the ab initio calculations of 1:1 (cation : molecule) complexes of lithium ions with linear oligomers, CH3X(CH2CH2X)nCH3, (n=0–5; X=O, NH or S); their results suggest that the total binding energy increases with the increase in glyme chain length and follows this order NH > O > S. Shen et al.103 studied the complexing behavior between alkaline earth cations and crown ethers/triglyme in the gas phase, forming mainly sandwich complexes of doubly charged cations (12-crown-4)2M2+, (12-crown-4)(triglyme)M2+, and (triglyme)2M2+; it was also found that triglyme could be easily replaced by 18-crown-6 than 12-crown-4 possibly due to the rate-limiting step disrupting less cation–ligand interactions for the flexible ligand than for the rigid one. Henderson et al.104 indicated that both [Li2(CF3SO3)2(diglyme)] and [Li3(CF3CO2)3(diglyme)] contain five-coordinate Li+ cations coordinated by a tridentate diglyme molecule and two O atoms (each from separate anions) (see Fig 2). Henderson et al.105 also suggested that the (monoglyme)2:LiClO4 crystals consist of contact ion pairs where the anions have bidentate coordination to the Li+ cations, whilst the (diglyme)2:LiClO4 crystals consist of fully solvated Li+ cations where the cations and anions do not directly interact. Grondin et al.106 further studied the Raman spectra of crystalline complexes of (monoglyme)2:LiClO4, (diglyme)2:LiClO4 and (triglyme)1:LiClO4, and established the vibrational assignment for ClO4− involved in solvent-separated ion pair, contact ion pair and aggregate solvates. Dhuaml and Gejji107 employed the ab initio Hartree Fock and density functional calculations to study the electronic structure, charge distributions and vibrational characteristics of CH3O(CH2CH2O)nCH3 (n = 3–7). They found that the trans- conformation around C–C and C–O bonds of the backbone of tri- to hexaglymes leads the lowest energy conformers; however, for heptaglyme (n = 7), the gauche- conformation around the C–C bonds affords a higher stability to the conformer.
Fig. 1.

Lithium cation chelated by a tridentate diglyme molecule (Molecular geometry obtained via PM3 semi-empirical method in ArgusLab by Richard Terrett at Australian National University).
Fig. 2.

Li+ cation coordination in [Li2(CF3SO3)2(diglyme)].
The Watanabe group108 prepared the equimolar complexes [Li(glyme)1][Tf2N] (Tf2N− or known as TFSA− = bis(trifluoromethane)sulfonimide), which maintain a stable liquid state over a wide temperature range and exhibit a high thermal stability and Li+ ionic conductivity, behaving like room-temperature ionic liquids (ILs). The physicochemical properties (e.g. melting point and viscosity) of the glyme-Li salt complex can be manipulated by the glyme structure. The same group109 further observed a higher oxidative stability of glyme molecules when complexing with Li+ cations. They found that the electrochemical oxidation of [Li(glyme)1][TFSA] occurred at the electrode potential of ~5 V vs Li/Li+, while the oxidation of solutions with excess glyme molecules ([Li(glyme)x][TFSA], x > 1) occurred at a lower potential of ~4 V vs Li/Li+. Further ab initio molecular orbital calculations explained the increased oxidative stability is due to the donation of lone pairs of ether oxygen atoms to the Li+ cation, lowering the highest occupied molecular orbital (HOMO) energy level of glyme molecules. The thermal stability of bis(dipivaloylmethanato)strontium [i.e. Sr(dpm)2] compounds containing glyme adducts [i.e. Sr(dpm)2-triglyme and Sr(dpm)2-tetraglyme] was examined by Cho et al.110 using thermogravimetric analysis, mass spectrometry, and FT-IR spectroscopy. They observed that glyme adducts are decomposed below 200°C, while the Sr-O and the C-C(CH3)3 bonds are dissociated at higher temperatures, and the C-O and the C-C bonds are stable up to 400°C. Glyme molecules in ion complexes weaken the Sr-O bond between the Sr atom and the dpm ligand, leading to the dissociation of the Sr-O bond in Sr(dpm)2-glymes at lower temperatures than the bond in Sr(dpm)2. However, Sr(dpm)2-glymes are less thermally degradable than Sr(dpm)2, resulting in fewer residues at elevated temperatures.
The stability constants (log K, i.e., equilibrium constants for complexing) for 18-crown-6 with Na+, K+, and Cs+ are 3–6 in methanol, and are 1.5–2.2 for pentaglyme.4, 112 The stability constants (log K) of 1:1 complexes of alkali cations M+ (Na+, K+, or Cs+) with glymes CH3O(CH2CH2O)nCH3 in methanol increase with n, which is different from the complexes between M+ and crown-ethers (-CH2-CH2-O-)n: their stability constants reached the maximum at n = 6 (Na+, K+) and 6, 7 (Cs+) (see Table 4).111, 113, 114 The stability constants of glymes are slightly lower than those of corresponding PEGs, but both of them are considerably lower than those of crown ethers.115 The higher stability of the complexes of crown-ethers than those of acyclic analogues (such as glymes) can be attributed to so called “macrocyclic effect” (ME), which is a function of the polyether’s topology and cation’s size.111, 112 The macrocyclic effect was not only observed in solutions, but also in the gas phase formation of alkali metal cation-bound dimers of crown ethers or glymes: the rates for crown ethers were about an order of magnitude higher than those for glymes.116 Varnek et al.111 demonstrated that the Substructural Molecular Fragments method can be used to evaluate the stability constants of these complexes. The Smid group investigated the relative ligand affinities of glymes, crown ethers, polyamines and other cation-binding ligands toward lithium and sodium picrate in toluene117 and dioxane118. The affinity of glymes in binding lithium and sodium picrate dramatically increases with the ether chain length up to tetraglyme, and a further increase in the affinity for longer glymes is primarily due to the increase in the number of binding sites. However, crown ethers generally have much higher binding equilibrium constants toward Li+ and Na+, which could be over 100 times higher than most glymes. Davidson and Kebarle119 indicated that monoglyme forms a stronger complex with K+ than ethylene diamine. Plewa-Marczewska et al.120 determined the formation constants (Ka) of ionic pairs between Li[CF3SO3] (or Li[BF4]) and glymes (monoglyme, diglyme or triglyme) using the anion state sensitive 19F NMR and cation 7Li NMR techniques, and found the Ka values (log Ka = 3–6) are dependent on the salt concentration range used for calculation and are generally a few orders of magnitude lower than those estimated from the conductivity data.
Table 4.
Comparison of stability constants (logK) of glymes [CH3O(CH2CH2O)nCH3] and crown ethers with alkali ions in methanol at 25 °C111
| Glyme, n= |
Na+ | K+ | Cs+ | Crown ether |
Na+c | K+c | Cs+c |
|---|---|---|---|---|---|---|---|
| 3 | 1.18a | 1.38a | 1.17a | 12c4 | 1.41 | 1.58 | 1.6 |
| 4 | 1.28b | 1.72b | 1.45b | 15c5 | 3.30 | 3.35 | 3.58 |
| 5 | 1.47b | 2.20b | 1.85b | 18c6 | 4.36 | 6.07 | 4.79 |
| 6 | 1.60b | 2.55b | 2.17b | 21c7 | 2.54 | 4.41 | 5.01 |
| 7 | 1.67b | 2.87b | 2.41b | 24c8 | 2.35 | 3.53 | 4.15 |
| 8 | 1.83a | 3.28a | 2.77a | 27c9 | 2.14 | 3.47 | 3.95 |
| 9 | 1.96a | 3.66a | 3.09a | 30c10 | 2.14 | 3.98 | 4.15 |
Note:
calculated value,
experimental value,
selected value from the THECOMAC database.
Many studies focus on the ion complexing of glymes in solutions. Chan et al.121–123 studied the coordination of fluorenyllithium, -sodium, and -potassium (carbanion pairs) with various glymes: CH3O(CH2CH2O)nCH3 (1 ≤ n ≤ 6) in dioxane, THF, or tetrahydropyran (THP) using optical and NMR spectroscopy. In the case of fluorenylsodium, 1:1 coordination complexes are formed for glyme-separated ion pairs with glyme-5 (n = 4), glyme-6 (n = 5) and glyme-7 (n = 6), but the separated ion pair contains two glyme molecules with glyme-4 and probably glyme-3. In the case of potassium salt, glyme-separated 1:1 complexes are observed for glyme-6 and glyme-7, but glyme-4 and glyme-5 yield mainly glymated contact ion pairs and a second glyme molecule is required to convert them to separated ion pairs. Therefore, depending on the cation size and glyme chain length, the chelating of glyme with contact ion pairs results in either glymated contact ion pairs or glyme-separated ion pairs or to a mixture of both. A further study on temperature dependence suggests that the reason of glymes being effective complexing agents of alkali ions is mainly due to a small loss in entropy as compared to solvent separated ion-pair formation in THF. Takaki and Smid124 titrated difluorenylbarium with glymes and crown ethers in THF at 25 °C under vacuum to examine the formation of ion pair-glyme and – crown ether complexes, and found that 1:1 complexes are formed for difluorenylbarium with mono- and dibenzo-18-crown-6 as well as with glyme-7 and glyme-9 while a 2:l crown-ion pair complex is formed for monobenzo-15-crown-5. Canters58 analyzed the shift and line widths in 23Na NMR spectra of glyme solutions of NaBPh4 and NaBH4 as a function of temperature; it was found that the transition from solvent separated to contact ion pairs and the type of anions show a considerable impact on the position of alkali NMR signals, while the line width is typically a linear function of the viscosity of pure solvent divided by absolute temperature. Detellier and Laszlo125 observed the tetracoordination of Na+ cation when studying the 23Na NMR chemical shifts for NaClO4 in binary mixtures of glyme and tetrahydrofurfuryl alcohol. Although monoglyme tends to form monocyclic intermediates, diglyme and triglymel form bicyclic intermediates. Gilkerson and Jackson126 noticed that lithium picrate could be dissolved up to 1 mM in the solvent containing 1 equiv. of either triphenylphosphine oxide (TPPO), hexamethylphosphoramide (HMPA), triglyme, or tetraglyme; however, to dissolve 1 mM sodium picrate, at least a tenfold excess of either TPPO, HMPA, or tetraglyme is needed; furthermore, through the conductance and spectrophotometric measurements, they observed a very small extent of dissociation in 1:1 ligand-metal picrate complexes and the formation of 2:1 and 3:1 ligand-cation complexes in the presence of additional ligand. Plewa et al.127 employed 19F NMR spectroscopy and conductivity data to study the ion pair formation of mixtures of salts (LiBF4 and LiCF3SO3) with 1,4-dioxane and glyme (or water). They suggested that mixtures containing monoglyme or diglyme have similar coordinating properties (in terms of donor and acceptor numbers) to liquid PEG dimethyl ether and solid PEG.
The formation of glyme and salt into crystal solid has been used to probe the ion complexing properties of glymes. Smid and Grotens128 reported the crystalline 1:l complexes of NaBPh4 with glyme-5, glyme-6 and glyme-7, as well as a 2:l complex with glyme-4. de Boer et al.129 studied the alkali biphenyl (NaBp, KBp and RbBp) in triglyme or tetraglyme solutions by NMR and the crystal structures of NaBp·(2 triglyme), KBp·(2 tetraglyme) and RbBp·(2 tetraglyme) by X-ray diffraction; these crystals consist of solvent-separated ion pairs in the solid state. Magnetic experiments were performed for single crystals of these three systems, and the susceptibility measurements suggested a ferromagnetic coupling in NaBp·(2 triglyme) and KBp·(2 tetraglyme) while an antiferromagnetic coupling in RbBp·(2 tetraglyme). The Frech group130 analyzed the crystal structure of diglyme:LiCF3SO3 and found the local environment of Li+ ions and the torsional angle sequence of bonds in ethylene oxide units are very similar to those in the high molecular weight poly(ethylene oxide)3:LiCF3SO3. On the other hand, their spectroscopic data of diglyme-LiCF3SO3 solutions suggest that Li+ ion is coordinated by only three oxygen atoms from diglyme and one oxygen atom from a CF3SO3− anion (i.e. five-fold coordinated) while triflate anions are two-fold coordinated. The same group131 further examined the crystal structures of monoglyme:(LiCF3SO3)2 and triglyme:(LiCF3SO3)2, and indicated in the case of monoglyme:(LiCF3SO3)2, triflate anions are three-fold coordinated and lithium ions are four-fold coordinated whilst in the case of triglyme:(LiCF3SO3)2, triflate anions are three-fold coordinated and lithium ions are both four-fold and five-fold coordinated. They also observed the formation of trans-gauche-trans conformations for the bond order -O-C-C-O- in adjacent ethylene oxide sequences interacting with a five-coordinate lithium ion. Furthermore, this group132 characterized the crystalline phases of glyme:NaCF3SO3 using DSC, X-ray diffraction, and vibrational spectroscopy, and suggested Na+ cation coordinates with 5 to 7 ether and triflate oxygens and more ethylene oxide units lead to higher coordination numbers. They also emphasized that these structures are a convoluted effect of ion-ion interactions, ion-chain heteroatom interactions, and packing constraints from the organic chains. Henderson et al.133 characterized the crystalline phases of (triglyme)1:LiX (X = CF3SO3−, BF4−, ClO4− and AsF6−), and found the phases are isostructural and are different from that of (triglyme)1:LiBPh4. The same group134 also studied the crystal structures of lithium salt complexes with tetraglyme including (tetraglyme)1:LiAsF6, (tetraglyme)1/2:LiBF4, and (tetraglyme)2/5:LiCF3CO2; and found a novel form of six-coordinate Li+ cation coordination by glyme oxygen atoms resembling the double-helix dimers. The ionic association strength of LiX salts was investigated in a variety of aprotic solvents including glymes (see a short review in the Supporting Information of Ref135), and is correlated with the crystallization kinetics of glyme-LiX and PEO-LiX mixtures.136, 137 The approximate ionic association strength in aprotic solvents is shown below in an increasing order135, 136:
beti−, Tf2N− < PF6− < ClO4−, I− < SCN− < BF4− < CF3SO3− < Br− < NO3− < CF3COO− < Cl−
This order implies the strength of an anion interacting with solvated cations through ionic attraction.
In addition to alkali metals, the complexing properties of glymes with other metal and organic cations, or even non-ionic molecules have also been studied. Timko et al.138 compared the complexation of t-butylammonium thiocyanate with pentaglyme and 18-crown-6 in chloroform at 24 °C, and found a macrocyclic effect of 18,700. The Bartsch group139 studied the complexing effect of p-tert-butylbenzendiazonium tetrafluoroborate with glymes in 1,2-dichloroethane at 50 °C, and suggested a macrocyclic effect of ~30 when comparing the complexation constants (K) for pentaglyme and 18-crown-6 with aryldiazolium ions. They also observed that the K value is basically constant (~2) for diglyme, triglyme and tetraglyme, followed by a steady increase for pentaglyme (4.78), hexaglyme (7.71), and heptaglyme (11.8) due to an increasing ability of the glyme to assume a pseudo cyclic structure; however, there is a drastic drop in K value for octaglyme (3.81) followed by a gradual increase for nonaglyme (6.32) and decaglyme (13.6). The same group140 further investigated the complexation of PEG and their dimethyl ethers with p-tert-butylbenzendiazonium tetrafluoroborate, and found that complexation constants for PEG 1000 and 1500 and their dimethyl ethers are 12–18% of that for 18-crwon-6. Relying on the 1H NMR spectroscopy, Otera et al.141 suggested the formation of the 1:1 complexes between glymes, CH3O(CH2CH2O)nCH3 (n=2, 3, 4), and dimethyltin dichloride (DMTC) in benzene, as well as both 1:1 and 1:2 glyme/DMTC complexes for glymes (n = 5, 6) in benzene and with all glymes studied in toluene and 1-chloronaphthalene. Hirashima et al.142 suggested the formation of 1:1 complexes between lanthanoid chlorides and triethylene glycol, tetraethylene glycol, pentaethylene glycol, tetraglyme or pentaglyme. The same group143 found that lanthanoid nitrates [i.e. Ln(NO3)3] form solid complexes with polyethylene glycols and glymes at different compositions: 1:1 (Ln:ligand) for triethylene glycol, tetraethylene glycol, pentaethylene glycol and tetraglyme, 1:2 (Ln:ligand) for diethylene glycol, 2:1 for heptaethylene glycol, and 4:3 for pentaglyme and hexaglyme. Inoue and Hakushi144 found the enthalpy-entropy compensation effect for the complexation of cations with glymes/podands, crown ethers, cryptands, and macrocyclic antibiotics in different solvents (such as water and methanol, etc.). Guerra et al.145 observed that bis(trifluorosiyl)cadmium was marginally stable at 20 °C but became very stable in monoglyme by forming alkyl-cadmium-glyme complexes. Markies et al.146 studied the complexation of bis(p-tert-butylphenyl)magnesium with 1,3-xylylene crown ethers and glymes, and indicated that the complex with diglyme magnesium is pentacoordinated whilst in the complex with tetraglyme magnesium is pentacoordinated to three adjacent oxygens of the five available (including the one of methoxy group). In both cases, coordinative saturation has been reached within the limits of steric restraints. Simple ethers are usually associated with a tetrahedrally coordinated magnesium, but polyethers induce higher coordination states, which could result in an improved reactivity of the organomagnesium reagent. Meot-Ner et al.147 determined the binding energies of NH4+ to glymes using pulsed high-pressure mass spectrometry, and suggested that binding energies in these complexes increase with the ligand size and the number of available oxygen groups. The ab initio calculations of complexing with monoglyme result in the order of binding energies of ligands to H3O+ > Na+ > NH4+ ≈ K+. To prepare new inorganic polymers as precursors for thin layer deposition, two different dimensional compounds [Ca(monoglyme)n(H2O)m]I2·(monoglyme)x (1: n=3, m=3, x=1; 2: n=2, m=4, x=0) and [Ca(triglyme)(H2O)4]I2 were formed through metal ion complexation and hydrogen bonding.148 Mishra et al.149 prepared hydroxo-bridged, centrosymmetric dimeric yttrium complexes with glymes: [Y2L2(μ-OH)2(H2O)x(ROH)y]I4 [1: L = triglyme, x = 2, y = 2, R = iPr; 2: L = tetraglyme, x = 2, y = 0; 3·2EtOH: L = diglyme, x = 2, y = 4, R = Et; 4·2EtOH: L = triglyme, x = 4, y = 0; 5: L = tetraglyme, x = 2, y = 2, R = Et]; these ionic derivatives are potential sources of yttrium oxide in high Tc superconductors. Chantooni et al.150 observed two polymorphic forms (i.e. triclinic polymorph I and orthorhombic polymorph II) of crystals of the 1:2:2 pentaglyme : dichloropicric acid : water adduct complex. The IR spectra of the 1:2:2 tri-, tetra-, and pentaglyme : dichloropicric acid : water complexes indicate that the hydrogen-bonding in triglyme complex is as strong as or stronger than that in the pentaglyme complex, but the hydrogen-bonding in tetraglyme complex is weaker than in pentaglyme complex.
The complexing properties of some glyme analogues have been reported in literatures. Jaycox et al.151 examined the insoluble complexes formed in acidic aqueous media upon mixing poly(acrylic acid) (PAA) and poly-(vinylbenzo-18-crown-6) (P18C6) or polyvinylbenzoglymes. The complex formation is a direct result of hydrogen bonding between carboxyl groups and crown ether- or glyme–oxygen atoms and also from hydrophobic interactions; such a precipitation is pH dependent. Saraswathi and Miller152 studied the interaction of proton and alkali metal ions with dinucleotide analogs of acyclic polyethers using the fast-atom bombardment (FAB) mass spectrometry, and suggested the following order of chelation of alkali metal ions, acyclic glymes < dinucleotide analogs (acyclic glymes substituted with nitrogen bases) < crown ethers.
5. Overview of industrial applications
Glymes have a broad range of industrial applications, such as their uses in cleaning products, inks, adhesives and coatings, batteries and electronics, absorption refrigeration and heat pumps, and pharmaceutical formulations, etc. For example, mixtures of methanol or trifluoroethanol + PEG-DME 250 or tetraglyme can be used as working fluids for absorption refrigeration machines;87, 88 triglyme and tetraglyme are lubricants for the automotive air-conditioning (A/C) compressor when mixed with refrigerants (such as HFC-134a).68, 86, 89 Table 5 summarizes some representative commercial applications of common glymes.
Table 5.
Major solvent features and industrial applications of glymes
| Glyme | Solvent Features | Representative industrial applications |
|---|---|---|
| Monoglyme (G1) |
|
Production of active ingredients, metal-organics, electrolyte solvent for sealed lithium batteries, entrainer, chromium electroplating, cyanoacrylate based adhesives solvent, etching of printed circuit boards, treating aluminum surfaces |
| Ethyl glyme (G1-Et) | Solvent in paints, adhesives, coatings, shellacs, resins, detergents, dyes and polycarbonate products | |
| Diglyme (G2) |
|
API production (such as anti-AIDS drug Nevirapine), reaction solvents for organometallic reagents, entrainer for azeotropic distillation, battery electrolyte, conducting thermoplastic paste, solvent for Teflon etchants, a suspension of sodium and naphthalene in G2 used for destruction of polychlorinated biphenyls (PCBs) in transformer oil, solvent in a formulation to improve the bonding of tire cord to rubber, solvent in printing and inkjet inks and inkjet cartridges, brake fluid, paints and other coatings, plastics, adhesives and sealants |
| Butyl diglyme (G2-Bu) |
|
Selective extraction of gold from hydrochloric solutions containing other metals, used in compositions for production of printed circuits and diode fabrication, |
| Triglyme (G3) |
|
Solvent for Teflon etching, high boiling and inert solvent for organic reactions, a solvent in consumer adhesives and paints, a component of consumer brake fluids and paint/graffiti removers |
| Tetraglyme (G4) |
|
Flue gas cleaning systems, solvent for production of binders for paints, coalescing agent in paint formulations, adhesives production, electrodeposition, manufacture of soldering fluxes/solder pastes, adsorption liquid and gas scrubbing, formulations of paint strippers and adhesive removers, extraction of volatile organic compounds from solid wastes, inert additive for the fixation of methylated methylolmelamine resins in durable-press cotton and cellulosic fabrics, an HFC/CFC lubricant |
| Proglyme (P2) |
|
Excellent replacement for NMP (N-methyl pyrrolidone) in many applications, polyurethane dispersion (PUD) formulation and two-pack PU coatings, waterborne coatings and high solid coatings, paint strippers, coatings- and graffiti removers, cleaners for degreasing, electrodeposition coatings, co-solvent in aluminum paste formulations, replacement of xylene for the production of alkyd and polyester resins |
| Polyglymes (MW 236 or 275) |
|
Gas purification by removing CO2, H2S, COS and water from natural gas or ammonia synthesis gas feed stocks, solvent for manufacturing polyester fibers with improved moisture retain and multiporous hydroscopic fibers, formulation in dyes to dye polyester-cotton textiles, consumer paint strippers |
| Higlyme (MW>400) |
|
Formulations of coatings (urethanes and inks) and agro-chemicals |
| PEG-DME 200 |
|
Special solvent, flue gas cleaning systems, delacquing |
| PEG-DME 250 |
|
PTC, special solvent, absorption solvent for H2S, COS, mercaptans and CO2, |
| PEG-DME 500 |
|
PTC, high boiling solvent, electroplating |
| PEG-DME 1000 | High b.p. (> 300 °C) | PTC, high boiling solvent, electrodeposition |
| PEG-DME 2000 |
|
PTC, high boiling solvent, electroplating, depolimerization reactions |
Note: Polyglymes are polyethylene glycol dimethyl ethers with different molecular weights ranging from 200–2000.
In particular, dipropylene glycol dimethyl ether, known as proglyme (P2), is considered as an environmentally friendly solvent and is not listed as a hazardous air pollutant (HAP). Therefore, P2 has versatile applications including the replacement of N-methyl pyrrolidone (NMP). NMP is known to have reproductive toxicity whilst much less toxic P2 has similar physical properties including similar solubility behaviors. Thus, P2 is an excellent solvent for coatings (such as waterborne coatings and high solid coatings) and paints (such as electrodeposition coatings, paint strippers, coatings- and graffiti removers), as well as for industrial cleaners for degreasing (such as formulating hard surface cleaners containing bleach).
6. Electrochemical applications
Due to the unique ion complexing property of glymes (as discussed in 4.2), glymes have been extensively investigated in electrochemistry as electrolyte solvents. Slates and Szwarc153 suggested that when sodium biphenyl coordinates with diglyme or triglyme in a 1:1 stoichiometric ratio, diglyme coordinates with the periphery of the ion pair while two isomeric ion pairs are formed in triglyme (glyme attaching to the periphery of the pair, and glyme separating the ions). Canters et al.154 prepared single crystals of the alkali radical ion pairs of [Li+][biphenyl−] in tetrahydropyran (THP), [Na+][biphenyl−] in triglyme, and [K+][biphenyl−] and [Rb+][biphenyl−] in tetraglyme. They found the presence of solvent molecules in stoichiometric quantities in the crystals, acting as the chelating agents of alkali ions. In particular, Na2[biphenyl]2[tiglyme]5 have shown a strong paramagnetism and electron exchange interaction, and the electron correlation time is in the order of 10−l1 to 10−l2 sec. Smyrl et al.155 suggested that a spontaneous discharge process for doped polyacetylene in a LiI-monoglyme solution could restrict the reversibility of this and other conductive polymers when used as electrode materials. Foos et al.156 examined the conductivity of LiBr dissolved in glymes (mono- and di-) and their mixtures with other ethers, and found the solution conductivity increases with the salt concentration to a maximum. Interestingly, they also discovered that the conductivity of 1.0 M LiBr in 1:1 (v/v) mixture of monoglyme and dioxolane increases with the decrease in temperature; this is particular valuable for battery electrolytes with maximized conductivities at low temperatures.
Sharma and Bhagwat157 measured the conductivities of alkali metal picrates dissolved in tetraglyme, 18-crown-6 and tetraethylene glycol diquinoline ether at 25 °C in methanol/water mixture. The formation constants (KML) of 1:1 complexes of alkali metal ions with these ligands in 70% methanol were found dependent on the anion; the log KML values of alkali metal ions with tetraglyme, 18-crown-6 and quinoline ether are in decreasing orders of (K+ > Na+ > Rb+ ≈ Cs+ > Li+), (K+ >> Na+ > Rb+ > Cs+), and (Na+ > K+ > Rb > CS+ > Li+) respectively. They also observed that the cyclic ligand gives a more stable complex and a higher selectivity than non-cyclic ligands. Pyati and Murray158 found that the microelectrode voltammetry-derived heterogeneous electron transfer kinetic rates kET for the redox couple cobalt tris(bipyridine) [Co(bpy)3]2+/3+ in a series of glyme solvents are inversely proportional to the solvent longitudinal relaxation time and viscosity but directly proportional to the diffusion coefficient of the metal complex. Teeters et al.159 measured the surface tension of tetraglyme-lithium triflate system, and observed a preferential distribution of the triflate ion between the interface and the bulk. They indicated that the surface concentration increases with the concentration of lithium triflate till a high ‘ion aggregation’ concentration. These results further stimulated the infrared study of poly(ethylene oxide)-lithium triflate films, and enabled a more mechanistic understanding of polymer electrolyte materials. To provide an aprotic environment for lithium batteries, Choquette et al.160 studied the phase diagrams, potential windows, conductivities and the lithium interfacial resistances of Li[Tf2N] dissolved in sulfamides and glymes. They suggested that glymes or their mixtures with sulfamides could be useful for batteries whose cathode is not a 2D layer structure. Plewa et al.161 constructed the composite electrolytes comprising polyglyme (Mw = 500), LiX (X = I−, BF4− and CF3SO3−) and calix[6]pyrrole derivative (C6P), and focused on the role of C6P as an anion complexing agent. To probe the potential use of tin as an alternative anode material for rechargeable lithium batteries, Katayama et al.162 examined the charge–discharge property of a tin thin film electrode in an ionic liquid, 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)amide containing 0.1M Li[Tf2N]. To reduce the interfacial resistance in the ionic liquid electrolyte, a small amount (0.2 M) of glymes (mono-, di-, tri- and tetra-) was added to coordinate with Li+ ions.
As discussed in details in section 4.2, glymes dissolve alkali salts and form complexes with them; such unique property has a significant electrochemical implication. Izutsu et al.163 studied the complexing of lithium ion in propylene carbonate (PC) with glymes (mono-, di-, tri-and tetra-) using a univalent cation-sensitive glass electrode. An early study by Aurbach and Granot164 suggested that electrolytes consisting of various lithium salts and glymes (monoglyme, diglyme and ethyl glyme) might not be suitable for rechargeable Li battery systems using Li metal anodes due to a rough morphology of Li upon deposition-dissolution cycling in them causing a low Li cycling efficiency. However, Brouillette et al.165 indicated that glymes (mono-, di-, tri- and tetra-) are electrochemically stable, possess a good redox window, and are analogs of PEOs used in polymer-electrolyte batteries. Therefore, this group measured the conductance and apparent molar volume and heat capacity of Li[Tf2N] at various concentrations in glymes; from these data, they concluded that: (a) at low concentrations, Li[Tf2N] is strongly associated in glymes; (b) at intermediate concentrations, there is a stable solvate of Li[Tf2N] in glymes in the solution state; (c) at high concentrations, the thermodynamic properties of the lithium salt resemble those of molten salts. Hayamizu et al.166 measured the self-diffusion coefficients of Li+, Tf2N−, and the solvent (including glymes) in Li[Tf2N]-solvent systems using the pulse-gradient spin-echo (PGSE) NMR method. They observed that the ionic conductivity and the diffusion coefficients increase with the increase in glyme chain length (monoglyme → diglyme → triglyme), and the degree of dissociation at 30 °C is in the range of 31–38%. To further understand the role of PEO in electrolyte systems, the same group167 employed the pulsed-field gradient spin-echo (PGSE) 1H, 19F, and 7Li NMR to study electrolytes of glymes CH3O(CH2CH2O)nCH3 (n = 3–50) mixing with Li[Tf2N]. They suggested that the segmental motions of the PEO moiety in glymes induce high chain flexibility and enable high solubility and transport of doped Li+ ions; the rate of segmental motions decreases with the increase in PEO chain length. To probe the molecular interactions of lithium salts in glymes used for solid polymer and liquid electrolytes, Henderson et al.168 determined the phase behavior and solvate structures of glyme complexes with Li[Tf2N] and Li[beti] (beti = bis(perfluoroethanesulfonyl)imide) such as (monoglyme)1:Li[beti], (diglyme)2:Li[Tf2N], (diglyme)1/2:Li[Tf2N], and low-temperature (triglyme)1:Li[beti]; most solvates undergo order-disorder solid phase transitions. Kolosnitsyn et al.169, 170 studied the cycling of a sulfur electrode in a mixture of 3-methoxysulfolane and sulfolane with glymes (mono-, di- and tetra-) and lithium triflate (Li[OTf]) as the supporting electrolyte. They found a decrease in the electrode capacity with the increase in PEO units of glymes and in the number of donor centers in sulfone molecules, which is due to the variation of the form taken by lithium polysulfides in solutions and the increase in electrolyte viscosity. Tobishima et al.171 investigated the conductivity, lithium ion solvation state and charge–discharge cycling efficiency of lithium metal anodes in glyme-based electrolytes for rechargeable lithium cells; ethylene carbonate (EC) and methylethylcarbonate (MEC) were added to glymes in order to dissolve 1.0 M LiPF6. They found the conductivity increases with the decrease in PEO chain length and the solution viscosity; they also indicated di- and tri-glyme exhibit a better performance than mono- and tetra-glyme in terms of conductivities at low temperature (below 0 °C) and the charge–discharge cycling at a high current. The same group172 further demonstrated the use of such tertiary electrolyte systems (mixing glymes with 1 M LiPF6-EC/MEC) in rechargeable lithium cells with Si-based anodes: for both Li/Si + C and Li/Si−C + C cells, the discharge capacity appears to be larger than the system without glyme, and the cycling life of the latter cells is also improved. Kaulgud et al.173 carried out ab initio Hartree–Fock calculations to determine the electronic structure and vibrational frequencies of CH3(OCH2CH2)nOCH3–M+–OTf− (n = 2–4, M = Li, Na, and K) complexes. They suggested that the metal ion has various coordinations from 5 to 7 in these complexes, and Li+ ions bind to one of the oxygen atoms of OTf− in tetraglyme–lithium triflate while Na+ or K+ ions show bidentate coordination. In addition, the metal ion tends to bind more strongly to ether oxygens of tetraglyme than its di- or triglyme analogues. The Watanabe group174 determined the physicochemical properties of triglyme and tetraglyme solutions of Li[Tf2N] and observed the formation of complexes [Li(glyme)][Tf2N] in concentrated solutions. The ionic conductivity is concentration-dependent and reaches the maximum at ~1 M. The viscosity increases with the salt concentration while the self-diffusion coefficient of each species in the solutions decreases with the salt concentration. The [Li(glyme)][Tf2N] may be considered as a quasi-ionic liquid in terms of similar ionicity. Orita et al.175 observed that a higher amount of tetraglyme in tetraglyme-Li[Tf2N] complexes decreases the viscosity and increases the ionic conductivity of the mixture; in addition, the mixture has a higher thermal stability than conventional organic electrolytes when the molar ratio of tetraglyme is more than 40 mol%. They further demonstrated the potential of [Li(tetraglyme)][Tf2N] as a replacement of organic electrolytes in lithium ion batteries with appropriate electrode-active materials. Tamura et al.176 prepared glyme–cyclic imide lithium salt (Li[CTFSI]) complexes as thermally stable electrolytes for lithium batteries, i.e. [Li(G3)][CTFSI] (solid) and [Li(G4)][CTFSI] (liquid). The latter complex shows a much higher thermal stability than pure G4, and a high ionic conductivity of 0.8 mS cm−1 at 30 °C despite its high viscosity. They also observed a stable charge–discharge cycling behavior of [LiCoO2|[Li(G4)][CTFSI]|Li metal] cell during 50 cycles, implying the applicability of the [Li(G4)][CTFSI] complex in a 4 V class lithium secondary battery. The Watanabe group177 investigated the physicochemical and electrochemical properties of 1:1 complexing mixture of triglyme and Li[N(SO2F)2] (lithium bis(fluorosulfonyl)amide) as the safe lithium-ion secondary battery electrolyte. This new electrolyte has a relatively high thermal stability, and enabled a stable charge/discharge of Li+ ions with both LiFePO4 positive electrode and graphite negative electrode leading to high coulombic efficiency and long cycles. In addition, they achieved 82% of capacity retention after 100 cycles of charge/discharge operations of [LiFePO4 positive electrode|TG-LiFSI electrolyte|graphite negative electrode] cell. The same group178 further examined the use of 1:1 equimolar complex electrolyte of triglyme:Li[Tf2N] in lithium secondary batteries using lithium metal and two positive electrode materials (LiFePO4 and LiNi1/3Mn1/3Co1/3O2). They observed a relatively favorable surface formed at the electrolyte/lithium metal electrode interface; and they also found an excellent capacity retention with low degradation for both 3V-class [LiFePO4|Li metal] cell and 4V-class [LiNi1/3Mn1/3Co1/3O2|Li metal] cell.
The lithium/sulfur (Li/S) battery has become a promising electrochemical system because of its high theoretical capacity of 1675 mAh g−1.179 However, the present Li/S system has a number of obstacles to overcome such as the poor active material conductivity, active material dissolution, and the highly reactive lithium metal electrode. The Watanabe group180 examined the redox reaction of sulfur supported on inverse opal carbon (IOC) in an [Li(tetraglyme)][Tf2N] molten complex electrolyte. They found that the Li | [Li(tetraglyme)][Tf2N] | sulfur/100 nm IOC cell maintained a large discharge capacity of ca. 800 mA h g−1-sulfur and a high coulombic efficiency of >97% after 50 cycles. They suggested that the [Li(tetraglyme)][Tf2N] molten salt could be a promising electrolyte enabling a high coulombic efficiency of charge–discharge in Li–S batteries. Barchasz et al.181 found that conventional carbonate-based electrolytes cannot be used in Li/S cells and then suggested that Li/S cell electrolytes require solvents with high solvation power such as tetraglyme and ethyl diglyme, but not those solvents (e.g. monoglyme, butyl diglyme and 1,3-dioxolane) with low polysulfide solubility. Polyglyme (average Mn = 250) plays a vital role in Li/S electrolytes, and could prevent fast electrode passivation and extend the length of second discharge plateau, resulting in a discharge capacity about 1100 mAh g−1 for the first discharge and over 550 mAh g−1 remaining after 10 cycles.
In addition to alkaline battery system, other metal battery systems have also been explored using glyme-based electrolytes. Aurbach et al.182 prepared new electrolyte solutions based on glymes (such as mono-, di- and tetra-) and magnesium aluminates, giving electrochemical window of 2.5 V and >99% efficiency of Mg deposition-dissolution cycles. They further developed new rechargeable Mg batteries (1–1.5 V) using these electrolyte solutions and cathodes of the MgxMoSy type (Chevrel phase) for delivering over 1000 charge-discharge cycles. This group183 further constructed the rechargeable Mg battery systems by using Mg(AX4-nRn)2 complexes (A = Al, B, Sb, P, As, Fe, and Ta; X = Cl, Br, and F; and R = butyl, ethyl, phenyl, and benzyl) in several ether solvents and glymes (mono-, di- and tetra-). They found some of these Mg complexes in THF or glymes gave a wide electrochemical window (> 2V) and allowed the reversible deposition of magnesium. This group184 further developed new magnesium batteries comprising Mg metal anodes, an electrolyte with a general structure of Mg(AlX3–nRnR′)2 (R, R′ = alkyl groups, X = halide) in ethereal solutions (such as THF and tetraglyme), and Chevrel phases of MgMo3S4 stoichiometry as highly reversible cathodes. The magnesium battery systems could be cycled thousands of times with little loss in capacity; in addition, they are environmentally benign alternatives to lead-acid and nickel-cadmium batteries and are constructed from abundant, inexpensive, and nonpoisonous materials.
Solid-state electrolytes consisting of poly(ethylene oxide) (PEO) doped with salts have promising applications in batteries, capacitors and fuel cells because these electrolytes exhibit high ionic conductivity. In addition, they are soft solid; and if small glyme molecules are used, they are monodispersed and do not entangle like polymers. Rao and Klemann185 investigated the behavior of Li/TiS2 cells in a low-temperature melt comprising LiI:glyme solvate, and suggested the cells could be discharged at high rates in the low temperature melt and could be stored in solid electrolyte for a long-term. Zhang et al.186 measured the ionic conductivities of several solid electrolytes comprising lithium salts with glymes, and indicated that the (tetraglyme)0.5:LiBF4 electrolyte has the highest conductivity among those small-molecule electrolytes with a lithium transport number of 0.65. The same group187 further observed an increase in conductivity (from around 10−7 to 10−4 S/cm) by substituting PEO molecules in conducting crystalline complexes PEO6:LiXF6 (X = P or As) with glymes (tri- and tetra-). Since the stoichiometric compositions of glyme–salt complexes are lower than 6:1 (4:1 for G3:LiXF6 and 5:1 for G4:LiXF6), there are three phases present, the 6:1 phase, the glyme complex and a liquid containing some salt; and it is believed that the liquid is primarily responsible for the conductivity. This group188 also evaluated two glyme-based solid electrolytes: G3:LiAsP6 and G4:LiAsP6, and reported that they have quite different transport numbers (t+ = 0.8 and 0.1 respectively). This is due to two factors: (a) there are tunnels in the crystal structure of G3 complex for Li+ migration, but not in G4 complex; (b) there is a weaker binding of AsF6− in the structure of G4 than in G3. The Bruce group189 further determined the crystal structures of complexes [CH3O(CH2CH2O)nCH3]:LiAsF6 (n = 8–12), and indicated that Li+ ions are six coordinated if only ether oxygens are involved in coordination and the coordination number becomes five if a fluorine from the AsF6− anion is involved in coordination. They also observed low lithium transport numbers (t+ < 0.3) and lower conductivities in these complexes compared with complexes formed with lower glymes (n = 3, 4). Xia and Smid190 conducted the differential thermal analysis of solid polymer electrolyte complexes of lithium (or sodium) triflate and homopolymers derived from three methacrylate monomers CH2=C(CH3)COO(CH2CH2O)nCH3 (n = 4, 8 or 22 on average), and found the conductivities of these complexes were comparable to those known for alkali complexes of poly(ethylene oxide). Holzer et al.191 described a different type of solid electrolyte containing glyme motif, poly[1,4-(2,5-bis(1,4,7,10-tetraoxaundecyl))phenylene vinylene] (BTEM-PPV, see Scheme 4), for constructing red-orange light-emitting electrochemical cells (LECs). The high electronic conductivity and ionic conductivity of BTEM-PPV could be attributed to its conjugated backbone and glyme-containing side chains. LECs based on this polymer are relatively bright light emitting devices with low response times. Because of the covalent linkage of PEO chains to PPV backbone, an ionochromic effect was also observed in both absorption spectrum and electroluminescence spectrum when BTEM-PPV is complexed with metal ions; this implies its potential application in chemical sensors.
Scheme 4.

Structure of BTEM-PPV.
7. Uses in organic reactions
Glymes play various vital roles in organic synthesis. The most common role is acting as the reaction media (solvent role). Other important roles include reaction additives/metal chelators, catalysts and reagents. The following sections discuss some representative examples in each of these categories.
7.1 Reaction solvents
Glymes have a wide liquid range (Table 2) and are suitable for reactions at low temperatures (low freezing points). For example, monoglyme has a freezing point of ‒69 °C and has been widely used in low-temperature reactions, but it can also be removed easily during the workup (boiling point 85 °C). In addition, glymes have a strong solvating power and can dissolve a variety of compounds, particularly chelating with metal ions. For example, the solvating power of the ether-type solvents increases in the order 2-methyltetrahydrofuran < THF < monoglyme < diglyme < triglyme < tetraglyme; in particular, triglyme and tetraglyme are strong chelating agents for Na+ ions.58 Therefore, glymes (especially monoglyme) have been explored in numerous organic reactions since 1960s, such as reduction, oxidation, substitution, C-C coupling, borane chemistry, and other reactions.
Reduction reactions
When norcamphor and 5-norbornen-2-one were treated with NaH and MeI in monoglyme, reduction products (methyl ethers) were obtained (Scheme 5); it is the carbonyl group that is reduced and not the O-alkylation products, 2-methoxynorbornene and 2-methoxynorbornadiene.192
Scheme 5.

Reduction of norcamphor and 5-norbornen-2-one in monoglyme.
The benzocyclobutadiene radical anion was produced by the addition of trans-1,2-dibromobenzocyclobutene to an excess of solvated electrons in a mixture of monoglyme and diglyme at ‒60 °C (Scheme 6).193 Similarly, treating cyclooctatetraene with potassium mirror in monoglyme at −90 °C (or −80 °C) yielded an anion radical.194, 195 Miller et al.196 treated dibenzonorcaradiene with different electron-transfer reducing agents in anhydrous monoglyme to form dibenzonorcaradiene anion radical, whose cyclopropane isomerizations led to various reduction products; for example, the Na-reduction after 12 h at r.t. produced 9-methylphenanthrene (25.4%), 9-methyl-9,10-dihydrophenanthrene (47.9%), and 6,7-dihydro-5H-dibenzo[a,c]cycloheptene (26.6 %).
Scheme 6.
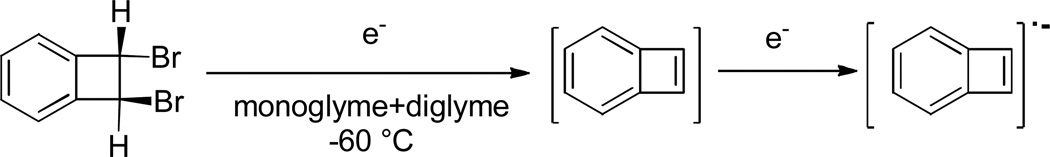
Preparation of benzocyclobutadiene radical anion.
Walborsky et al.197 studied the reductive cleavage of 1,1-biphenylene-2-methylcyclopropane using sodium and lithium in liquid ammonia, sodium in monoglyme, sodium naphthalide in monoglyme, and by controlled potential electrolysis in acetonitrile at a mercury cathode (Scheme 7). The isomer ratio of two products 9-propylfluorene and 9-isopropylfluorene ranged from 96:4 to 81:19. Hwever, the use to monoglyme as the solvent gave a lower product ratio. In the synthesis of asymmetric phosphine macrocycles, Wei et al.198 eliminated the protecting tosyl groups at −78 °C by sodium naphthalenide in monoglyme containing t-butyl alcohol (as a proton source); this method was used by the same group in an earlier detosylation study.199
Scheme 7.

Reductive cleavage of cyclopropane ring.
Partial reduction of a lactam to produce the indole alkaloid roxburghine D was achieved by using di-isobutyl aluminium hydride in monoglyme at −70 °C.200 Dahl et al.201 prepared CH3Ge(PH2)2H and CH3Ge(PH2)3 by the reaction of CH3GeCl3 with LiAl(PH2)4 in triglyme at −23 °C. Saavedra202 carried out the reduction of nitrosoamides to alcohols using NaBH4 in dry monoglyme at room temperature for 2–8 h, achieving 50–82% yields (Scheme 8). Ohsawa et al.203 found that the potassium metal-crown ether-diglyme system could be effective for the reductive removal of sulfonyl group from O-sulfonates or sulfonamides. The Rieke group204, 205 prepared highly reactive metal powders of Fe, Co, Ni, Pd and Pt from the reduction of anhydrous metal halides in monoglyme or THF by lithium in the presence of a small amount of naphthalene. Inaba et al.206 prepared highly reactive metallic nickel by reducing nickel halides with lithium in monoglyme using naphthalene as an electron carrier, and further investigated it as a reductive homocoupling reagent for benzylic mono- and polyhalides (Scheme 9) to produce 1,2-diarylethanes. Specifically, the coupling of benzylic monohalides at room temperature yielded the corresponding ethane derivatives, and the coupling of benzylic di- and trihalides gave mixtures of cis and trans isomers of substituted ethenes. Carbonyl-substituted closo polyhedral boranes with B10 and B12 cages are important derivatives that can be converted to other functional groups such as cyanide, amide, keto, ester, and amine, etc. The same group207 further demonstrated that metallic nickel could be a versatile coupling reagent for ketone preparations by the reaction of benzylic, allylic, vinylic, and pentafluorophenyl halides with acid halides at 85 °C in monoglyme. Reduction of the B12 1,12-dicarbonyl to 1,12-bis(hydroxymethyl)decahydrododecaborate salts was achieved by using LiAlH4 in anhydrous monoglyme at room temperature.208
Scheme 8.
Reduction of nitrosoamides to alcohols.
Scheme 9.

Ni-catalyzed reductive homocoupling of benzylic halides.
Biancon et al.209 described a general method for the reductive coupling of carbonyl ligands in [M(CO)2(dmpe)2Cl] complexes, M = Nb or Ta and dmpe = 1,2-bis(dimethylphosphino)ethane: the reaction of [M(CO)2(dmpe)2Cl] with 40% sodium amalgam in THF or monoglyme, after the filtration, the addition of Me3SiY, Y = Cl or CF3SO3 (triflate), and the recrystallization from pentane results in [M(Me3SiOC≡COSiMe3)(dmpe)2Y] with 40–70% isolated yields. The Walton group210, 211 developed a general method for synthesizing dirhenium octahydrides Re2H8(PR3)4 [PR3 = PMe3, Pet3, P-n-Pr3, PMe2Ph, PEt2Ph, PMePh2, Ph2PCH2PPh2 (dppm), and Ph2PCH2CH2PPh2 (dppe)], via the reduction of triply bonded complexes Re2Cl4(PR3)4 using LiAlH4 in monoglyme and the subsequent hydrolysis step. Taking the advantage of glymes as high-boiling inert solvents, Yang and Pittman212 studied the dechlorination of 4-chlorobiphenyl with NaBH4 in glymes at 120–310 °C (Scheme 10). At comparable reaction conditions, the dechlorination rates decreased in the order of tetraglyme > triglyme > diglyme, and a complete dechlorination was observed in NaBH4/tetraglyme at 310 °C in 1 h. The addition of LiCl to NaBH4 could enhance the dechlorination rate in di-, tri, and tetraglyme respectively at 120–135 °C. On the other hand, the dechlorination of 4-chlorobiphenyl was not successful in the NaBH4/diphenyl ether system. Based on the same methodology, this group213 also accomplished the dechlorinations of pentachlorophenol and 1,2,4-trichlorobenzene using NaBH4 in tetraglyme at 290–315 °C or by NaBH4/LiCl at 125–135 °C in diglyme, triglyme or tetraglyme after premixing at room temperature. Furthermore, this group214 achieved a quantitative dechlorination of a polychlorinated biphenyl (PCB) mixture (Aroclor 1016) using NaBH4 in tetraglyme at 290–310 °C in 2 h in a sealed tube, or using NaBH4/LiCl/glyme solvents (di-, tri-, or tetraglyme) at 125–135 °C.
Scheme 10.
Dechlorination of 4-chlorobiphenyl.
Kanth and Brown215 noted that NaBH4 and NaBF4 have reasonably high solubilities in glymes (particularly triglyme and tetraglyme). As illustrated in Scheme 11, they developed improved procedures for the generation of diborane (B2H6) by the reaction of NaBH4 in triglyme or tetraglyme, followed by the generation of diborane through a reaction of NaBF4 with NaBH4 in triglyme (or tetraglyme) in the presence of Lewis acids such as AlCl3 and BCl3. Triglyme (or tetraglyme) could be easily recovered and recycled.
Scheme 11.

Improved procedures for diborane generation.
Oxidation reactions
Ouellette and Levin216 conducted the oxidation of phenylcyclopropane by Na2PdCl4 in 2:1 (v/v) monoglyme-water, and found the product distribution is dependent on the ratio of phenylcyclopropane to Na2PdCl4 (Scheme 12). At a low ratio of phenylcyclopropane/Pd(II), propiophenone is the major oxidation product; however, at a high ratio, an isomerization to trans-propenylbenzene occurs which is further oxidized to phenylacetone.
Scheme 12.
Oxidation of phenylcyclopropane by Na2PdCl4 in aqueous monoglyme.
Ochiai and Fujita217, 218 synthesized allylic nitrates via the reaction of ent-16-kaurene or ent-15-kaurene with thallium (III) trinitrate in monoglyme. McKillop et al.219 found that thallium(III) nitrate is capable of oxidizing chalcones, deoxybenzoins and benzoins in aqueous glyme-perchloric acid; the same group220 also oxidized diarylacetylenes to benzils in good yields in aqueous acidic glyme or in methanol.
Substitution reactions
Stork and Hudrlik221 synthesized different metal enolates from trialkylsilyl enol ethers using organometallic reagents in monoglyme (Scheme 13). For example, at room temperature, methyllithium completely converts trimethylsilyl enol ether of 2-methylcyclohexanone into lithium enolate in 6 min in monoglyme but in ~1 h in ether. In another study, Stork and Hudrlik222 described the preparation of trialkylsilyl enol ethers from ketone and NaH in monoglyme under the reflux condition followed by the addition of triethylamine and trimethylsilyl chloride and subsequent workups. In the preparation of racemic alkaloid 3-epi-elwesine, piperonyl cyanide was converted to cyclopropane carbonitrile via a two-step substitution reaction using LiNH2 in monoglyme at room temperature with 65–75% yield (Scheme 14),223 although the use of NaNH2/glyme only afforded low yields of the cyclopropane.224
Scheme 13.
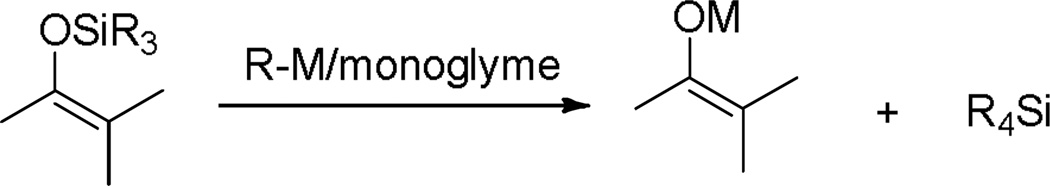
Preparation of metal enolates from trialkylsilyl enol ethers.
Scheme 14.

Conversion of piperonyl cyanide to cyclopropane carbonitrile.
Kice and Kasperek225 examined the hydrolysis of aryl α-disulfones in various Et3N-Et3NH+ buffers in 60% dioxane and 60% monoglyme as solvents; they found that the triethylamine-catalyzed reaction is a general base catalysis (by triethylamine) rather than nucleophilic catalysis (Scheme 15a). The same group226 further investigated the hydrolysis of p-nitrophenyl p-toluenesulfonate in Et3N-Et3NH+ buffers in both 20% acetonitrile and 60% aqueous monoglyme, and found no significant catalysis by triethylamine in this case. However, they observed that N-ethylpyrrolidine (NEP) could catalyze the hydrolysis in 60% monoglyme, which is shown to be nucleophilic catalysis (Scheme 15b).
Scheme 15.

Hydrolysis of (a) aryl α-disulfones and (b) p-nitrophenyl p-toluenesulfonate.
The nucleophilic aromatic substitution of haloaryl sulfones with alkali phenoxides was studied in glymes at 160 °C (Scheme 16).227 The reaction was found to be first order in halo sulfone and of fractional (~0.5) order in phenoxide ion. The longer glyme chain length promoted a faster reaction, and when n ≈ 20 the reaction rate was about 25 times faster than when n = 2 (diglyme). The likely reason for the rate enhancement is that polyglyme is a better cation solvating solvent than diglyme. Pastor and Hessell228 studied the aromatic substitution of hexa-, tetra-, tri-, di-, and monochlorobenzenes with sodium alkanethiolates in several glymes, and found the product yield decreased in the order of tetraglyme > triglyme > diglyme > monoglyme. It was explained that tetraglyme is more effective in solvating the sodium cation, generating a more nucleophilic unsolvated thiolate anion.
Scheme 16.
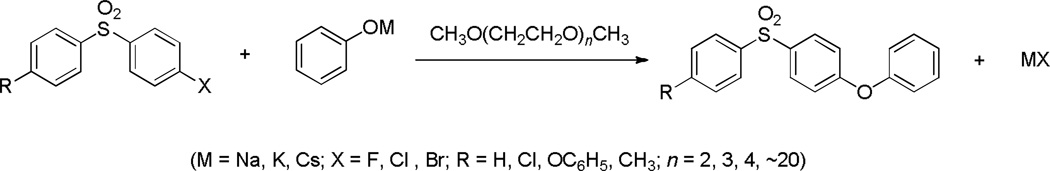
Nucleophilic substitution of haloaryl sulfones with alkali phenoxides.
Bis(h5-cyclopentadienyl)tungsten dichloromethide hydride, Cp2WH(CHCl2), was synthesized as the insertion product from the reaction of bis(h5-cyclopentadieny1)tungsten dihydride and sodium trichloroacetate in monoglyme (Scheme 17); however, in a chlorobenzene-diglyme mixture, the thermal decomposition of sodium chlorodifluoroacetate in the presence of bis(h5-cyclopentadieny1)tungsten dihydride yielded the substitution product, bis(h5-cyclopentadienyl)tungsten bis(chlorodifluoroacetate), Cp2W(O2C2ClF2)2.229
Scheme 17.
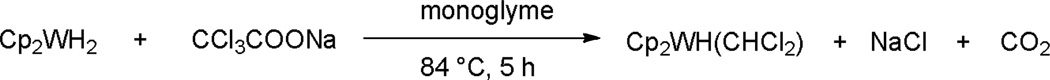
Insertion reaction of bis(h5-cyclopentadieny1)tungsten dihydride.
White and McGillivray230 reported an effective method for azetidine preparation (Scheme 18) via the reductive detosylation using sodium naphthalenide in diglyme, which afforded improved yields and simple procedures (superior to monoglyme and THF). The azetidine-diglyme solution could be further converted to N-aroylazetidines by adding different aroyl chlorides. 6-(Arylalkylamino)uracils and 6-anilinouracils, potent inhibitors of Bacillus subtilis DNA polymerase III, were prepared by the reactions between 6-chlorouracil and appropriate amines in monoglyme instead of aqueous solutions, which considerably reduced the reaction time.231 Izumi and Miller232 examined the nucleophilic attack of carbanions (R3C−) to displace chloride ion from phenylchloroacetylene (Scheme 19); they suggested that reactions in DMSO-KOH are mainly those of ions, however, in aprotic monoglyme, reactions of R3C−Na+ are those of aggregates. The DMSO-KOH medium is only suitable for relatively strong carbon acids, while the Na-glyme condition has broader applications.
Scheme 18.

Preparation of azetidine.
Scheme 19.

Nucleophilic attack of carbanion.
Banitt et al.233 carried out the nucleophilic acyl substitution of 2,2,2-trifluoroethyl 2,5-bis(2,2,2-trifluoroethoxy)benzoate with 2-aminomethylpiperidine in monoglyme (Scheme 20), achieving 76.5% isolated yield. Okamoto et al.234 investigated the reaction of 5-dimethylamino-1-naphthalenesulfonyl chloride with butylamine in chloroform with glymes as oligomer co-solvents, and determined the second-order rate constants by fluorometry. The addition of glymes imposed an acceleration effect especially when their concentrations were lower than 20% (v/v), and such an effect became more pronounced for glymes with longer chain length (tetraglyme > triglyme > diglyme > monoglyme). In addition, the rate acceleration seems correlating with the volume fraction of the co-solvent, which can be attributed to the polymer effects of glymes and is explained by the thermodynamics of polymer solutions.
Scheme 20.

Synthesis of N-(2-piperidylmethyl)-2,5-bis(2,2,2-trifluoroethoxy)benzamide.
Reactions of Li+, Na+, and K+ salts of 2,4,6-trimethyl-s-triazine with 2-halomethyl-4,6-dimethyl-s-triazine (X = Cl, Br) in monoglyme yielded 1,2-bis(4,6-dimethyl-s-triazin-2-yl)ethane (Scheme 21), and other compounds including 1,2-bis(4,6-dimethyl-s-triazin-2-yl)ethene, 1,2,3-tris(4,6-dimethyl-s-triazin-2-yl)cyclopropane, 1,2,3-tris(4,6-dimethyl-s-triazin-2-yl)propane and 1,2,3,4-tetrakis(4,6-dimethyl-s-triazin-2-yl)butane.235 It is suggested that 1,2-bis(4,6-dimethyl-s-triazin-2-yl)ethane is produced through an SN2 mechanism, while other products are formed through carbenoid reactions.
Scheme 21.

Reactions 2,4,6-trimethyl-s-triazine salt with 2-halomethyl-4,6-dimethyl-s-triazine.
Carbon-carbon coupling reactions
Zhu et al.236 employed phenyliodonium zwitterion as an efficient electrophile in the palladium-catalyzed Suzuki-type reaction with aryl boronic acids in monoglyme/water (4/1) (Scheme 22). The mild reaction conditions and commercial accessibility of 4-hydroxycoumarins and boronic acids make this method a versatile tool for the synthesis of 3-aryl-4-hydroxycoumarins.
Scheme 22.

Pd-catalyzed coupling of phenyliodonium zwitterion with aryl boronic acid.
Kim et al.237 indicated that Ni nanoparticles (5.7±3.8 nm) exhibited a moderate catalytic activity in the oxidative addition reaction of benzylchloride and bromoacetonitrile in monoglyme under reflux condition to prepare 3-arylpropanenitrile (Scheme 23), whereas larger Ni particles (3 µm and 100 mesh) showed no activity.
Scheme 23.
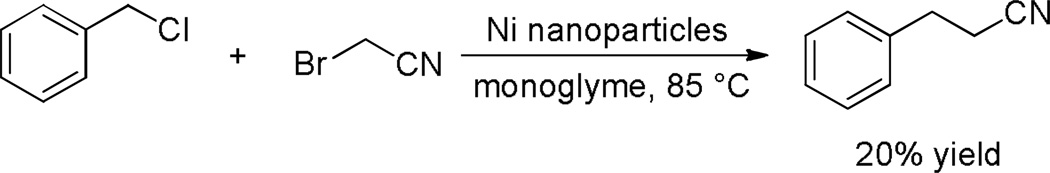
Catalytic activity of Ni nanoparticles in oxidative addition reaction.
Smith and Fu238 developed a stereoconvergent method for the catalytic asymmetric Negishi cross-coupling of various racemic secondary propargylic halides with arylzinc reagents in monoglyme catalyzed by a chiral Ni/pybox complex (Scheme 24). In most cases, they achieved 70–80% yields and ee (enantiomeric excess) near or above 90%. It is important to point out that the catalyst components (NiCl2·glyme and pybox ligand) are commercially available.238, 239
Scheme 24.

Cross-coupling of racemic propargylic halides with arylzinc reagents.
Organometallic compounds are common reagents for C-C coupling reactions. However, Fitt and Gschwend240 pointed out that monoglyme (and perhaps other glymes) should not be used in metalation reactions including those using t-butyllithium (t-BuLi). In the presence of t-BuLi, monoglyme undergoes deprotonation and β-elimination steps even at −70 °C to form lithium methoxide (Scheme 25), which is characteristic of 1,2-diheterosubstituted ethanes. Thus, there is no complex formed between monoglyme and t-BuLi. The reaction rates of different butyllithiums with monoglyme decrease in the order of t-BuLi > sec-BuLi >> n-BuLi.
Scheme 25.
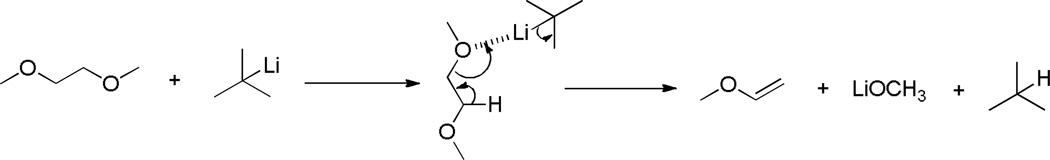
Reactivity of monoglyme with t-butyllithium.
The Delia group241, 242 reported the synthesis of mono-, di- and tri-substituted phenylpyrimidines via Suzuki coupling reactions in various solvents (Scheme 26). They found that the monoglyme/water mixture, t-butanol and polar aprotic solvents (such as acetonitrile, acetone, THF, CHCl3 and CH2Cl2) gave the best results; polar protic solvents (such as methanol and ethanol) induced ether byproducts under the basic conditions while nonpolar solvents (such as hexane) gave more byproducts and 2-substituted isomer.
Scheme 26.

Mono-, di-, and triarylation of the pyrimidine ring.
Borane chemistry
Geanangel and Shore243 firstly prepared NaB5H8 and KB5H8 by reacting B5H9 and metal hydride in monoglyme at room temperature, and then produced B6H10 by the reaction of alkali metal B5H8− salts with diborane (B2H6) at −78 °C in monoglyme; finally, refluxing glyme solutions of B6H10 led to decaborane B10H14 in 20–30% yields. Hosmane et al.244 quantitatively prepared boron hydride species [B11H14]− by reacting K[B9H14] with 0.4 equiv of B5H9 (or directly from B5H9 and metal hydride such as t-BuLi) in monoglyme at 85 °C for 20 h. Lawrence et al.245 synthesized the tetradecahydrononaborate(1−) anion [B9H14]− by the reaction of B5H9 with NaH (or KH) in THF or monoglyme.
Pelter et al.246 prepared a thioester by the reaction of trisethylthioborane and benzoic acid in refluxing monoglyme for 7 h to give 78% isolated yield. Peacock and Geanangel247 conducted the reduction of trimethyl phosphite-borane by sodium naphthalide (which was prepared in monoglyme) to a new type of diphosphine derivative (Scheme 27). Leyden et al.248 carried out the reaction of [(η5-C5H5)2Ni] with nido-(B11H13)2−, (B10H13)−, or (B9H12)− catalyzed by Na/Hg amalgam in monoglyme to produce closo-[(η5-C5H5Ni)B11H11]−, nido-[(η5-C5H5Ni)B10H12]−, as well as isomeric closo-1- and [2-(η5-C5H5Ni)B9H9]− anions, respectively; they also performed the reaction of [(η5-C5H5)2Ni] or [(η5-C5H5NiCO)2] with closo-(B11H11)2−, (B10H10)2−, or (B9H9)2− in monoglyme to yield closo-[(η5-C5H5Ni)B11H11]−, [(η5-C5H5Ni)2B10H10], and isomeric 1- and [2-(η5-C5H5Ni)B9H9]−, respectively. Wermer and Shore249 suggested that pentaborane (B5H9) could be reduced by alkali metal naphthalide in THF or monoglyme to produce the nonahydropentaborate(2−) dianion [B5H9]2−, which could be further protonated to form B5H11 with yields up to 38% (Scheme 28).
Scheme 27.

Reduction of trimethyl phosphite-borane by sodium naphthalide.
Scheme 28.

Preparation of dianion (B5H9)2− and B5H11.
Getman et al.250 synthesized arachno-[B9H13]2− as K+ and Na+ salts via the deprotonation of K[B9H14] by KH in monoglyme and the deprotonation of Na[B9H14] by NaNH2 in liquid ammonia respectively. Kang et al.251 suggested that monoglyme was the best refluxing solvent for converting arachno-S2B7H8− to hypho-S2B7H10−; they further carried out the synthesis of new metalladithiaborane clusters derived from hypho-S2B7H10− in refluxing monoglyme, such as the treatment of hypho-S2B7H10− with Cp(CO)2FeCl to form of C5H5FeS2B7H8, with (CO)5MnBr to form hypho-l-(CO)4Mn-2,5-S2B6H9, and with [Cp*RhCl2]2 to form arachno-7-Cp*Rh-6,8-S2B6H8. Holub et al.252 carried out the reaction between Na2[nido-6,9-C2B8H10] and PCl3 in monoglyme at room temperature for 24 h to prepare phoshadicarbaborane nido-7,8,11-PC2B8H11 (35%), which was converted to [7,8,11-nido-PC2B8H10]− anion by deprotonation; following the same strategy by using PhPCl2 as the phosphorus source, they also synthesized the isomeric compound 7-Ph-7,8,10-nido-PC2B8H10 (64%), and nido-7,8,11-PC2B8H11 (14%) (from an accompanying dephenylation reaction).
Miscellaneous reactions
Rosen and Sworm253 synthesized iodine isocyanate (INCO) in 0.4 to 0.5 N at −30° C in the dark in ether, THF or monoglyme via the reaction of an excess of pure silver cyanate with iodine. The reaction rate was fastest in monoglyme, followed by in THF and then in ether. The decomposition of INCO at −11 °C was also highest in glyme (75% decomposition in 24 h) but much slower in THF and ether (15 to 25% decomposition in 24 h). INCO is a reactive pseudohalogen used in forming C-N bonds from many unsaturated compounds. Gassman et al.254 developed a method for the cleavage of nonenolizable ketones by a mild cleavage reagent: a 10:3 ratio of potassium t-butoxide and water in aprotic solvents including DMSO, monoglyme, hexamethylphosphoramide, hexane, or diethyl ether. A 90% yield was obtained after 4 h at 30 °C in the cleavage of benzophenone to give benzoic acid using potassium t-butoxide-water-monoglyme.
Finucane and Thomson255 oxidized triterpene 12-enes with various substituents in ring A (e.g. taraxeryl acetate and cholesteryl acetate) to their corresponding αβ-unsaturated ketones by treating them with N-bromosuccinimide in moist solvents (dioxane, THF, monoglyme, or diglyme) under the irradiation of visible light. Lateef et al.256 suggested that a series of meta- and para-substituted phenyl carbanilates undergo a rapid reversible dissociation in monoglyme (Scheme 29). They also indicated that the reaction followed the Hammett equation with positive ρ values of 1.49–1.66. As a precursor for preparing 2,5-dihydroxy-3,6-diphenyl-5,6-dihydropyrazine 1,4-dioxide, phenylglyoxal 2-oxime was synthesized by the acid hydrolysis of an α-oximino acetal (Scheme 30) in a mixture of monoglyme and pH 3.5 buffer.257 The oxime substrate is insoluble in the buffer alone or in aqueous methanol, but is soluble in a mixture of glyme and buffer. Pittman et al.258 dissolved polystyrene in a mixture of monoglyme and diglyme (2/1, v/v) for a reaction with Cr(CO)6 to prepare styrenetricarbonylchromiumstyrene copolymers.
Scheme 29.

Thermal dissociation of aryl carbanilates in monoglyme.
Scheme 30.

Hydrolysis of phenylglyoxal dimethyl acetal oxime.
Monoglyme was used as the solvent in the multi-step synthesis of D,L-muscone from cyclododecanone:259 cyclotetradecenone and triethylsilane was refluxed in monoglyme catalyzed by chloroplatinic acid to afford 1-triethylsiloxycyclotetradecene, which was converted to 2-chloro-2-cyclopentadecenone after adding dichlorocarbene in refluxing glyme-tetrachloroethylene (1:4); the final product was obtained by a conjugate addition of dimethylcopper lithium in ether, followed by workup using saturated NH4Cl and then chromium(II) perchlorate reduction of α-chloro ketone in dimethyl formamide. Wilt and Rasmussen260 carried out the Favorskii reaction of bromo ketones in methanol and monoglyme (sodium methoxide as the base) to prepare the epimeric methyl benzonorbornene-2-carboxylate, and found that more polar methanol led to more exo ester (exo:endo 80:20) while the less polar solvent monoglyme increased the endo ester (exo:endo 58.5:41.5). In addition, higher ester yields were reported in monoglyme than in methanol. Stang and Mangum261 prepared alkyl methylenecyclopropenes by the addition of unsaturated carbenes to alkynes in monoglyme with t-BuOK at −55 °C. The same group262 also synthesized 2-indazoles by the reaction of triflate (CH3)2C=CHOTf with azobenzene and tert-butylazobenzene in monoglyme with t-BuOK at −20 °C via unsaturated carbene additions to azo compounds. To encapsulate chalcogen atoms by transition-metal carbonyl clusters, Vidal et al.263 synthesized the [PhCH2N(C2H5)3]3[Rh17(S)2(CO)32] complex through reacting Rh(CO)2acac and alkali carboxylates in tetraglyme, with H2S or SO2 under ~300 atm of CO and H2 at 140–160 °C. Similarly, as an example of the encapsulation of arsenic by transition-metal carbonyl clusters, [PhCH2N(C2H5)3]3[Rh10As(CO)22]·C4H8O was prepared by the reaction of Rh(CO)2acac and alkali carboxylates in tetraglyme with Ph3As under ca 300 atm of CO and H2.264
Collins et al.265 demonstrated that the repeated action of sodium-potassium alloy in glyme-triglyme and the subsequent quenching with CH3I or with water could be an efficient low-temperature method to degrade coal by the cleavage of aliphatic and aromatic-aliphatic carbon-carbon bonds. Ladika and Stang266 carried out the elimination of trifluoromethane-sulfonic aid (CF3SO3H) from RCH=C(OSO2CF3)C≡C–C≡CSiMe3 promoted by different bases in monoglyme to yield unsymmetrical trialkynes R[C≡C]3SiMe3 or R[C≡C]3H (Scheme 31).
Scheme 31.

Preparation of unsymmetrical trialkynes via elimination in monoglyme.
Paxson et al.267 performed cobalt carbonyl-catalyzed reactions of syn gas by using CO-H2 mixture at about 200–250 °C and 200 bar in glymes: in diglyme, the product selectivity for ethanol was 68% while in ethyl diglyme the selectivity for n-propanol increased to 40% (ethanol 28%). It is suggested that the solvent cleavage occurs and the terminal methoxy moieties supplied the methyl group of ethanol. van Tamelen and Seeley268 carried out the catalytic N2-fixation by electrolytic and chemical reduction using monoglyme. Pez et al.269 found that the reaction of titanium metallocene complex, μ-(η1:η5-cyclopentadienyl)-tris(η-cyclopentadienyl)dititanium with N2 (~10 atm) in monoglyme formed a complex giving characteristic ν(N-N) = 1222 cm−1, which was further treated with THF/monoglyme and diglyme to yield a crystalline N2 complex with ν(N-N) = 1282 cm−1. These complex systems could have potential applications in N2-fixation. Budt et al.270 achieved the regioselective NBS-epoxidation of farnesate attached to helical peptides in monoglyme-water (5:1) mixture at 0 °C. Hayward and Shapley271 reacted Re2(CO)10 with sodium dispersion in glymes (diglyme and triglyme) to prepare the rhenium carbonyl clusters Re4(CO)162−, H2Re6C(CO)182−, Re7C(CO)213−, and Re8C(CO)242−, as Et4N+ and (PPh3)2N+ salts. Tee and Enos272 examined the kinetics of hydrolysis of six p-nitrophenyl alkanoates in basic aqueous solutions containing up to 80% (v/v) of the co-solvents: ethylene glycol, 2-methoxyethanol, monoglyme, diglyme, or DMSO; they suggested that the ether-type solvents (2-methoxyethanol, monoglyme and diglyme) are more effective than ethylene glycol or DMSO in reducing and eliminating the hydrophobic aggregation and coiling of longer chain alkanoates. Briggs273 carried out the trimerization of ethylene to hex-1-ene using a homogeneous three-component catalyst of chromium, hydrolyzed alkylaluminium and monoglyme with 74% selectivity; the replacements (such as diglyme, triglyme, THF, and o-dimethoxybenzene) for monoglyme gave less desirable results. A tricyclic ring/cage system of RC6H5·5BNR'2 products were formed by reactions between dehalogenation products of F2BN(i-Pr)2 with monoalkylbenzenes using monoglyme as the co-solvent.274 Hung et al.275 prepared linear and cyclic perfluorinated polyethers through the ionic polymerization of a trifluorovinyl ether alcohol (CF2=CFORfCH2OH, Rf = CF2CF(CF3)OCF2CF2) and the subsequent fluorination of the polyfluorinated polyethers (Scheme 32). They found that without a solvent, linear polymers with Mn values up to 29,000 were produced, while in glyme solution, cyclic oligomers were major products and a cyclic dimer was obtained in yields up to 60%. The reaction of arylcalcium iodides and nitrous oxide (N2O) in monoglyme favored the formation of azobenzenes due to the insertion with N2O into diphenylcalcium.276
Scheme 32.
Preparation of perfluorinated polyethers.
Kirij et al.277 prepared [(CH3)4N][Te(CF3)3] and [(CH3)4N][I(CF3)2] by the reactions of (CH3)3SiCF3/[(CH3)4N]F with Te(CF3)2 and CF3I respectively in THF or monoglyme at −60 °C. Yagupolskii et al.278 carried out an aza Curtius rearrangement by reacting N-(trifluoromethylsulfonyl)carboximidoyl chlorides with sodium azide in monoglyme or acetonitrile at −5 to +10 °C to form carbodiimides RN=C=NSO2CF3. Israelsohn et al.279 reported that the PtCl4-CO catalyst could promote the hydration of internal and terminal alkynes in aqueous monoglyme or diglyme to afford aldehyde-free ketones between 80 and 120 °C. The reaction was found strongly dependent on the electronic and steric nature of the alkynes. Monoglyme or THF was used to dissolve elemental sulfur, Me3SiCF3, and fluoride salts for the preparation of trifluoromethanethiolates, [NMe4]SCF3, CsSCF3 and [(benzo-15-crown-5)2Cs]SCF3 (Scheme 33).280 It is known that SCF3 salts are versatile nucleophilic reagents for synthesizing a variety of organic, organometallic and inorganic molecules. Tyrra et al.281 prepared tetramethylammonium trifluoromethyltellurate(0), [NMe4]TeCF3, with 60% yield from Me3SiCF3, elemental tellurium and [NMe4]F in monoglyme; they further carried out the cation exchange of [NMe4]TeCF3 with [PNP]Br ([PNP] = bis(triphenylphosphoranylidene)ammonium) and [(dibenzo-18-crown-6)K]Br and demonstrated the high nucleophilicity of TeCF3− when coupled with these low coordinating cations.
Scheme 33.
Preparation of trifluoromethanethiolates.
It is also important to know that glymes may be inferior to other organic solvents in some reactions. For example, the alkylation rates of alkali enolates in DMSO were found to be 1,000-fold faster than that in glymes (mono- and di-) although the same reaction was even slower in diethyl ether.282
7.2 Reaction additives/metal chelators
The second important role of glymes in reaction is acting as additives or metal chelators. Shinohara et al.283 observed highly reactive agent-separated ion pairs formed via complexing sodium polystyryl ion pairs with tri- or tetraglyme; by adding glyme to solutions of sodium polystyryl in THF at 25 °C, they found propagation constants of the polymerization of living polymers increasing with the glyme concentration. Trifluorosilyl substituted dialkyl compounds including trans-Pt(SiF3)2(PMe3)2, Pd(SiF3)2(PMe3)2, and Ni(SiF3)2(PMe3)3 were synthesized by reacting an excess amount of Cd(SiF3)2·monoglyme with trimethylphosphine metal dibromides of platinum, palladium, and nickel (Scheme 34).284
Scheme 34.
Synthesis of trifluorosilyl substituted dialkyl compounds.
Liang and Ying285 studied the kinetics and mechanism of anionic equilibrium polymerization of α-methylstyrene in cyclohexane with butyllithium as the initiator and monoglyme or diglyme as the polar additive; kinetic equations of the equilibrium polymerization is dependent on the mole ratio of glyme/butyllithium; the stability of complexes formed by poly(α-methylstyryl)lithium (PnLi) and the ether additive is in a decreasing order of [PnLi (diglyme)] > [PnLi (monoglyme)] > [PnLi (THF)]. Wang et al.286 employed 7Li and/or l3C NMR spectroscopy to investigate the aggregation equilibrium and electronic structure of methyl α-lithioisobutyrate (MIBLi) in THF with the co-existence of various Li+-binding ligands; they found that the addition of ligands to coexisting tetrameric and dimeric MIBLi in THF increases the dimeric population in the order monoglyme < diglyme < 12-crown-4 < hexamethylphosphoric triamide, which is in agreement with the increasing strength of complexation between MIBLi (lithium cation) and the ligands.
Bis(trifluoromethy1)cadmium·glyme [(CF3)2Cd·monoglyme] is a convenient reagent used in the preparation of trifluoromethyl substituted metal compounds.287 For example, (CF3)2Cd·monoglyme exchanges ligands with GeI4, SnI4, or PI3 to form (CF3)4Ge, (CF3)4Sn, or (CF3)3P respectively at room temperature. The reaction of (CF3)2Cd·monoglyme with acyl halides such as CH3C(O)Br formed the acyl fluorides CH3C(O)F in 95% yield at −25 °C. Krause and Morrison288 prepared Lewis base adducts of bis(trifluoromethyl)cadmium (CF3)2Cd by mixing (CF3)2Hg with dimethylcadmium in THF, monoglyme, diglyme, or pyridine; Lewis base exchange is achieved when the glyme adduct is dissolved in a base, for example, (CF3)2Cd·monoglyme in pyridine. They suggested that (CF3)2Cd·base species are more reactive than (CF3)2Hg: (CF3)4Sn (66% yield) and (CF3)4Ge (43% yield) were synthesized by ligand exchanges between SnBr4 and GeI4 with (CF3)2Cd·monoglyme respectively at room temperature; the reaction of acyl halides with (CF3)2Cd·monoglyme at subambient temperature produced the acyl fluoride with ~90% yield, and stereospecific difluorocarbene cis-2-butene at −30 °C. This group289 used the same method to prepare monosubstituted compounds (CF3)BrNi(PEt3)2, (CF3)BrPd(PEt3)2, and (CF3)IPt(PBun3)2 in 60–70% yields by the reaction of (CF3)2Cd·monoglyme with bis(trialkylphosphine) Group 8B dihalides such as Br2Ni(PEt3)2 within 0.5–5 h; however, when an excess amount of (CF3)2Cd·monoglyme was present, disubstituted compounds (CF3)2M(PEt3)2 (M = Ni, Pd, or Pt) were found over extended reaction period. The Morrison group290 also conducted the synthesis of (η5-C5H5)Co(CO)(CF3)2 (63% yield) via the reaction of (CF3)2Cd·monoglyme with (η5-C5H5)Co(CO)I2. This group291 further investigated the ligand exchange reactions of Cd(CF3)2·monoglyme with various aryl-containing Pb, Sn, and Ge halides, acetates, and thioethers in THF or CHCl3; they found that when an excess of trifluoromethylating agent was used, these new compounds PbPh(CF3)3 (51%), PbPh2(CF3)2 (61%), PbPh3CF3 (81%), SnPh2(CF3)2 (55%), SnPh3CF3 (80%), and GePh3CF3 (72%) were obtained, and when the trifluoromethylating agent was the limiting reactant, partially substituted compounds PbPh2(Cl)CF3 (78%), PbPh2(O2CCH3)CF3 (89%), PbPh(O2CCH3)2CF3 (63%), and PbPh(O2CCH3)(CF3)2 (64%) were isolated. Murray et al.292 performed the reaction of [Au(CH2)2PPh2]2Br2 with Cd(CF3)2(monoglyme) in CH2Cl2 to synthesize a dialkyl Au(II) phosphorus ylide dimer [Au(CH2)2PPh2]2(CF3)2, whose X-ray crystal structure was also determined. Nair and Morrison293 prepared TlPh(CF3)2 with 87% yield by reacting Cd(CF3)2·monoglyme with TlPhCl2 after 72 h, and Tl(CF3)2OAc with 46% yield by reacting Cd(CF3)2·monoglyme with Tl(OAc)3 after 45 min. Loizou et al.294 prepared cyclopentadienyldinitrosyl(trifluoromethyl)chromium(0) CpCr(NO)2CF3 (71%), and cyclopentadienyldinitrosyl(trifluoromethyl)molybdenum(0) CpMo(NO)2CF3 (44%) via the reaction of Cd(CF3)2·monoglyme with the corresponding chlorides at 65 °C. Ludovici et al.295 achieved nearly quantitative yields of CnF2n+1NO (n = 1, 2, 3, 6) by the reaction of NOCl with Cd(CnF2n+1)2·monoglyme. Daniele et al.296 synthesized La(OTf)(OC6H3-2,6-Me2)2(glyme) [glyme = triglyme or tetraglyme] derivatives through reactions between lanthanum triflate adducts La(OTf)3(glyme) and 2 equiv. LiOAr (Ar=C6H3-2,6-Me2) in THF. The grafting of La(OTf)(OC6H3-2,6-Me2)2(triglyme) onto silica produced an hybrid material, which was used as a catalyst for the activation of formaldehyde in water for hydroxymethylation of silyl enol ether in mild conditions. The Fu group at MIT has demonstrated that the NiCl2·glyme/pybox ligand is an effective chiral catalyst for the asymmetric Negishi cross-coupling of racemic secondary propargylic halides with alkylzinc239 or arylzinc reagents.238
7.3 Catalysts
Glymes contain multiple ethylene oxide units, which define many glymes as amphiphiles (with both hydrophilic and lipophilic properties). Ideally, these glymes can be used as direct phase-transfer catalysts. The second case for glymes acting as catalysts is that glymes can form complexes with alkali cations like crown ethers. The third category is that glyme oxygens can form hydrogen-bonds with reaction intermediates and thus promote the reaction by stabilizing these intermediates. The fourth category is where glymes are ligands of metal salt catalysts.
Direct phase-transfer catalysts
Gokel et al.115 suggested that shorter-chain PEGs and their ether derivatives are more effective catalysts than longer-chain analogues in phase-transfer reactions such as the nucleophilic substitution of 1-chlorooctane in n-decane with aqueous sodium cyanide to synthesize 1-cyanooctane. Sukata297 synthesized alkyl p-tolyl sulfones (80–95% yields) from sodium p-toluensulfinate monohydrate and different alkyl halides in PEG-400 or PEG-400 diethyl ether, or in methanol containing PEG-1000 or PEG-1000 diethyl ether as a catalyst.
Complexing with alkali cations
Lee and Chang298 found that a mixture of PEG dimethyl ethers could be used as phase-transfer agents to move KMnO4 from aqueous solution into benzene or CH2Cl2; such a system was shown effective for the oxidation of terminal alkenes to corresponding carboxylic acids with one less carbon, and nonterminal alkenes to diones, diols, ketols and carboxylic acids. Paradisi et al.299 carried out the nucleophilic substitution reaction of 1-chloro-4-nitrobenzene with KOH in 2-propanol to synthesize 1-isopropoxy-4-nitrobenzene catalyzed by Bu4NBr or alkali ion complexing agents such as 18-crown-6, Carbowax 20M, MPEG 5000, and Triton X-100; however, tetraglyme was less effective and monoglyme showed no effect. Bergbreiter and Blanton300 reduced alkyl and aryl halides to hydrocarbons by suspensions of NaBH4 in toluene co-catalyzed by tri-n-butyltin chloride and polyether phase-transfer catalysts (PTCs); among three PTCs studied, benzo-15-crown-5, polyethylene-bound benzo-15-crown-5 and poly(ethy1ene glycol) dimethyl ether (MW 1000), the crown ether is generally more active. Sukata and Akagawa301 conducted the phase-transfer reaction of inactivated aryl halides with diphenylamine catalyzed by a series of PEGs and glymes (such as PEG-3000, PEG-400 dimethyl ether and PEG-6000 dimethyl ether) (Scheme 35). Their results suggest that one molecule of high molecular weight PEG or glyme could bind with more than one K+; also, ~9 ethylene oxide units are typically needed to form a crown-shape complex, and PEGs and glymes with 4–9 ethylene oxide units are most effective in complexing with a K+ cation. It was found302, 303 that cyclophosphazenic polypodands are stronger complexing agents of alkali metal salts than glymes, and thus become highly efficient catalysts for solid-liquid and liquid-liquid phase-transfer reactions including nucleophilic substitution, alkylation, reduction and oxidation reactions. The stronger ability of polypodands in binding ion pair aggregates and thus activating the anion by increasing the interionic distance in the single ion pair attributes to the higher catalytic activities of polypodands.304 Lazrek et al.305 achieved the regioselective phase-transfer reaction of N-alkylation with acyclic side chains of pyrimidine and purine heterocycles at N-1 and N-9 respectively catalyzed by 18-crown-6 or tetraglyme in the presence of potassium tert-butoxide at 0°C.
Scheme 35.
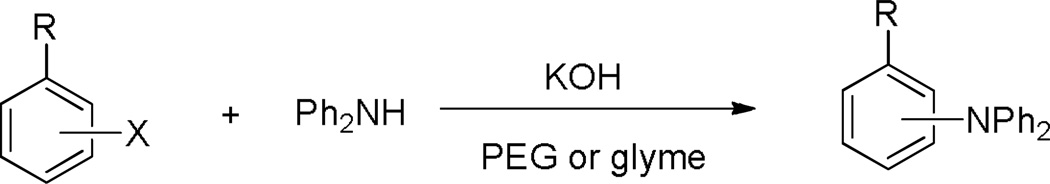
Phase-transfer reaction of aryl halides with diphenylamine.
Kudryavtsev and Zakharov306 investigated the phosphorylation of heptafluorobutanol with phosphorus(V) oxychloride (Scheme 36) catalyzed by complex catalysts based on Li+, Na+, K+, or Cs+ chlorides and polydentate ligands (such as dibenzo-18-crown-6, mono-, di-, and tetraglymes, and PEG-600 and PEG-1000); the addition of polydentate ligands improved the solubility of inorganic salts and increased the reaction rates by a factor of 1.3–2.8. Although the LiCl and PEG systems were shown to be most efficient, the use of glymes with LiCl led to 91–92% yields in 1.9–2.7 h (vs. 94% yield in 3.6 h without the addition of any ligand). Kochergina and Anufriev307 carried out the nucleophilic substitution of 6,7-dichloro-3-ethyl-2-ethoxynaphthazarine to prepare echinochrome trimethyl ether (Scheme 37). The reaction was accelerated by the use of monoglyme and diglyme as both catalysts and solvents; in particular, the reaction in diglyme produced up to 72% yield vs 52% without adding any glyme. It was explained that glymes effectively solvate potassium cation to form ion pairs.
Scheme 36.

Phosphorylation of heptafluorobutanol with phosphorus oxychloride.
Scheme 37.

Conversion of 6,7-dichloro-3-ethyl-2-ethoxynaphthazarine into echinochrome trimethyl ether.
Stabilizing reaction intermediates
Koh et al.308 studied the aminolysis of thiophenyl 4-nitrobenzoate with deuterated 4-chlorobenzylamine in acetonitrile catalyzed by glymes, and found that the kcat/Oxy values (Oxy is the number of oxygens in glymes) increase with the chain length up to triglyme and only up to four oxygens per glyme molecule are used to catalyze the aminolysis (see the interaction between zwitterionic tetrahedral intermediate and triglyme in Scheme 38). They further indicated that this reaction has primary deuterium kinetic isotope effects (PKIEs), kH(cat)/kD(cat) = 1.28–1.62.
Scheme 38.

Binding interaction between glyme and intermediate during aminolysis of thiophenyl 4-nitrobenzoate with 4-chlorobenzylamine.
Hogan and Gandour309 suggested that glymes are more effective catalysts than crown ethers (an inverse macrocyclic effect) in the butylaminolysis of p-nitrophenyl acetate in chlorobenzene; this group310 further observed a break in a plot of the catalytic rate constant vs chain length of catalyst, indicating triglyme gives the optimum catalysis. The same group311 further examined the transition structure complexes for the glyme- and α,ω-dimethoxyalkane-catalyzed butylaminolysis of 4-nitrophenyl acetate in chlorobenzene. They confirmed that the kcat/Oxy values increase with oligomer length up to triglyme and then plateau. The ester aminolysis in aprotic solvents involves a rate-limiting breakdown of a zwitterionic tetrahedral intermediate, which forms a complex with a glyme molecule (Scheme 39). They further concluded that the number of polyether oxygens required for optimum catalysis and the best spacing (in α,ω-dimethoxyalkanes) among these oxygens are strongly dependent on the number of ammonium protons in the transition structure. More recently, Basilio et al.312, 313 studied the butylaminolysis of p-nitrophenyl acetate in chlorobenzene catalyzed by crown ethers or glymes as phase-transfer catalysts, and proposed a reaction pathway to reflect a 1st-order dependence on the catalyst concentration and a 2nd-order dependence on butylamine concentration (Scheme 40). The new pathway (red arrows) involves the complexing of ether-TI (ether-tetrahedral intermediate) with an amine molecule, which is an addition to the mechanisms (green and blue arrows) traditionally accepted for the catalysis by phase-transfer agents of aminolysis reactions in aprotic solvents. The green arrows dictate butylamine attacking the ester to form the tetrahedral intermediate (TI), and the amine complexing with the ether catalyst before binding to TI; the blue arrows indicate butylamine attacking the ester to form the TI and a second butylamine forming hydrogen bonds with the TI followed by glyme binding to both butylamine components. The same group314 further examined a more complex reaction system of butylaminolysis of 4-nitrophenylcaprate in water/AOT/chlorobenzene microemulsions catalyzed by triglyme (Scheme 41), and discussed four simultaneous reaction pathways.
Scheme 39.

Complexing between zwitterionic tetrahedral intermediate and triglyme.
Scheme 40.

Mechanisms of glyme-catalyzed ester aminolysis.
Scheme 41.

Butylaminolysis of 4-nitrophenylcaprate.
Ligands of metal salt catalysts
The Fu group315 developed a nickel (NiCl2·monoglyme)/diamine catalyst (diamine = (S,S)-N,N'-dimethyl-1,2-diamino-1,2-diphenylethane) to carry out the asymmetric Hiyama reactions of α-bromo esters with aryl silanes to yield α-aryl esters in satisfactory ees (Scheme 42).
Scheme 42.
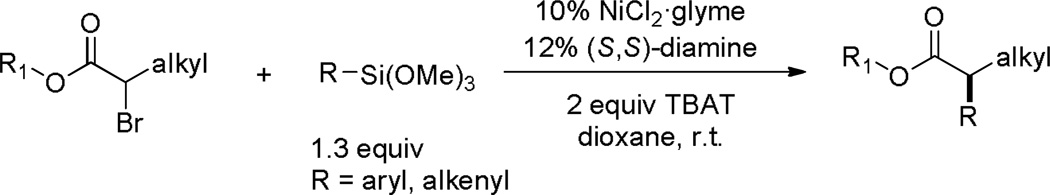
Hiyama reactions of α-bromo esters with aryl silanes.
It is also interesting to notice in some cases, glymes are inactive or less active than PEGs as phase-transfer catalysts, such as reactions of aryldiazonium salts with CBrCl3 or CH3I initiated by potassium acetate,316 dehydrohalogenation of 2-bromooctane with aqueous KOH,317
7.4 Reagents
Typically, glymes are chemically inert; however, they could become reactive under certain conditions. Newman and Liang318 observed that when 3-nitroso-5-methyl-5-tert-butyl-2-oxazolidone was treated with sodium phenoxide, the stereospecific cleavage of monoglyme occurred to form 2-methoxyethyl trans-2,2,3-trimethyl-l-butenyl ether in 46% yield. Ishii et al.319 studied the anodic fluorination of monoglyme and diglyme (Scheme 43) in acetonitrile using a fluoride salt as a supporting electrolyte and a fluoride ion source with an undivided cell; they observed the corresponding monofluoromethyl ethers were obtained as main products in satisfactory yields. However, the anodic fluorination of crown ethers caused the C-C bond cleavage producing selective α,ω-difluoro products with high yields.
Scheme 43.

Anodic fluorination of (a) monoglyme and (b) diglyme.
Grabovskiy et al.320 studied the oxidation of a series of ethers including monoglyme by dimethyldioxirane (DMDO), and suggested a second-order reaction kinetics r = k[DMDO][ether]. They also determined the rate constants between 5 and 50 °C as well as the activation parameters of the reaction. The main oxidation products of monoglyme with DMDO (10:1 molar ratio) in acetone at 25 °C include methanol (32%), 2-methoxyethanal (4%) and methoxyacetic acid (3%). Boron-based compounds including sodium borohydride (NaBH4) and ammonia borane (NH3BH3) have been studied as chemical hydrogen storage materials. Yoon et al.321 developed a safe and efficient method for preparing NH3BH3: iodine oxidation of Bu4N+B3H8− in monoglyme solution yielded (glyme)B3H7, which was converted to NH3BH3 by displacement of the coordinated glyme with anhydrous ammonia.
In addition, there are some reactive glymes (such as polyglycol allyl methyl ethers, polyglycol diallyl ethers and PEG methyl ether methacrylate) commercially available. As a typical application, the allyl-containing glymes can react with siloxanes through a Pt-catalyzed hydrosilylation reaction to produce polyether modified silicones, which have applications as silicone surfactants and polyurethane foam stabilizers.
8. (Co-)Solvents for biocatalysis
Surprisingly, glymes and their aqueous solutions are not commonly used as solvents for enzymatic processes. There have been some conflicting results on the enzyme activity and stability in aqueous solutions of glymes. Some studies suggest the high enzyme activity and/or stability in aqueous glymes. Yoshpe-Besancon et al.322 observed that 55% triglyme solution could shift the equilibrium of aminopeptidase A from amide bond hydrolysis to peptide bond formation, which allowed a selective α-amino protection of derivatives of many amino acids (except Gly and Pro) by the malyl group. Rosell et al.323 found that 50% (v/v) aqueous solutions of diglyme (G2) or tetraglyme (G4) depressed the hydrolytic activity of penicillin acylase by roughly 65%, but boosted its synthetic activity by ~4.8 times. In addition, these two solutions showed negligible impact on the enzyme stability. Berkowitz et al.324 suggested that 10% triglyme in aqueous buffer solution enabled the optimal efficiency for a PPL-catalyzed hydrolysis of a drug intermediate at both lower enzyme loading and higher temperature. Schroën et al.325 studied the enzymatic synthesis of antibiotic cephalexin catalyzed by penicillin G acylase (Scheme 44), and found the enzyme retained 90% activity after incubation in 30–36% (v/v) glymes (mono-, di- and tri-) at 30 °C for 24 h; comparing with the direct synthesis in water, the addition of methanol and triglyme could increase the equilibrium concentration of cephalexin by a factor of 2–3. However, Illanes and Fajardo326 found that in 50/50 (v/v) mixtures of organic solvents and aqueous buffer, Escherichia coli penicillin acylase maintained a high stability in polyols (such as ethylene glycol and glycerol), a low stability in glyme and diglyme, and no activity after 24 h in methanol, DMF and DMSO.
Scheme 44.
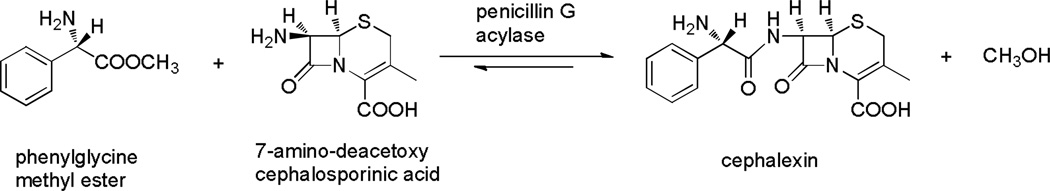
Enzymatic synthesis of antibiotic cephalexin.
On the other hand, there are even limited studies on the enzymatic reactions in neat glymes although PEGs327–329 and PEG-based aqueous biphasic systems (ABS)330 are known media for enzymatic reactions. The Sheldon group331 indicated that cross-linked enzyme aggregates (CLEAs) of penicillin G acylase was not quite active in a mixture of glyme-water (95:5, v/v), resulting in low conversions [10% in monoglyme (log P = ‒0.8) and 5% in triglyme (log P = ‒1.8) after 1 h] for the synthesis of ampicillin. The Zhao group21 found that long-chain glymes are highly compatible with immobilized Candida antarctica lipase B (Novozym 435), resulting in higher enzyme activities and stabilities than t-butanol and some ionic liquids. Furthermore, this group noticed that soybean oil is fully miscible with glymes, which enables a homogeneous reaction mixture for enzymatic preparation of biodiesel; in the presence of glymes, the immobilized lipase showed a very high tolerance to high methanol concentrations (up to 60–70% v/v), and nearly quantitative triglyceride conversions could be obtained under mild reaction conditions.
9. Materials
This section focuses on the applications of glymes as materials or as media to prepare new materials.
9.1 Nanomaterials
A series of metal or nonmetal nanoparticles with functionalized surfaces were synthesized in glyme solvents by the Kauzlarich group. They demonstrated a straightforward and versatile technique to control both crystal size and surface termination of the nanomaterials. Methyl-terminated Ge nanocrystals with an average particle size of around 3.5 nm were produced by the metathesis reaction between the Zintl salt NaGe and GeCl4 in degassed monoglyme or diglyme.332 The Kauzlarich and Taylor group333, 334 further extended this method to prepare alkyl-terminated crystalline Ge nanoparticles by the reactions between GeCl4 and NaGe, KGe, or Mg2Ge followed by surface termination with alkyl Li and Grignard reagents in glymes. It was observed that diglyme and triglyme seemed to support the reaction better than monoglyme and the reactions in triglyme were much faster than in diglyme. Moreover, the largest particles (8–10 nm) of Ge nanoparticles were produced in monoglyme while the smallest particles of 4.5 nm mean size were synthesized in triglyme. Furthermore, alkyl-terminated silicon nanoclusters were also prepared by the reaction of SiCl4 with Mg2Si in monoglyme and surface-terminated with various alkyl groups, R̂-n-Si (R = methyl, ethyl, n-butyl, and n-octyl).335 Based on the same technique, this group336 further synthesized the surface-capped and organic-functionalized tin nanoparticles of Sn/R, Sn/Si-R (R = n-C4H9), and Sn/SiO2 core/shell particles via the reaction of Mg2Sn with SnCl4 or SiCl4 in monoglyme.336
Plasma-deposited PEG-like films have become emerging materials as ‘non-stick’ surfaces toward protein and bacteria. Ratner and co-workers337 designed PEG-like coatings via radio-frequency plasma deposition of short-chain oligoglymes, dioxane, and crown ethers onto glass cover slips; the films were characterized by X-ray photoelectron spectroscopy (XPS), time-of-flight secondary ion mass spectrometry (TOF–SIMS), dynamic contact angle goniometry, and radiolabeled fibrinogen adsorption. The Brétagnol group338, 339 constructed new microstructured surfaces by a spatial arrangement of different functional domains by a combination of plasma polymerization of diglyme (leading to coatings with a high concentration of ethylene oxide groups (>70%)) and photolithography. The high stability of these films in acetone suggests that these coatings could be used in classical lift-off processes involve washing in acetone for designing patterned surfaces.339 The same group340 further developed a straightforward nanoscale writing technique, enabling the fabrication of bio-adhesive patterns directly in a non-bio-adhesive matrix. This method involves two major steps: (a) plasma polymerization of diglyme to form a protein-repelling PEO-like coating, (b) direct electron beam lithography inside the matrix by tuning the ether bond concentration of the coating to produce nanoscale bio-adhesive patterns.
The Stucky group341 developed a time evolution kinetic-dependent crystal growth model to examine the nanocrystal growth of CdS from cadmium acetate and sodium sulfide in different solvents (e.g. ethylene glycol, monoglyme, diglyme, and trioctylphosphine), using trialkylphosphine oxide (alkyl = ethyl or octyl) as a surfactant. They observed that the size of nanoparticles is controllable by the reaction time and temperature; the nanoparticle sizes are suitable for spectroscopic analysis of electron quantum confinement. The same group342 also prepared CdS nanorods through the reaction of cadmium acetate and sodium sulfide at low temperature (25–65 °C) in an aqueous phase using nonionic pluronic amphiphilic triblock copolymers, (EO)x(PO)y(EO)x, as structure-directing agents. However, when the same reaction is refluxed in ethylene glycol and monoglyme without surfactant, new morphology of microrods with flat ends, dumbbell-shaped microrods, and cotton-ball-like microparticles is produced. Chiu and Kauzlarich343 synthesized crystalline germanium nanoparticles by the reduction of GeCl4 with sodium naphthalide in monoglyme within 10 min. Pickering et al.344 prepared octyloxy-capped boron nanoparticles through a reduction of BBr3 with sodium naphthalenide in dry monoglyme followed by the addition of excess octanol at room temperature. The size distribution of the nanoparticles can be controlled by tuning the reaction conditions such as concentration and reducing agent. Similarly, Cho345 performed the reaction of SnCl4 and GeCl4 with sodium naphthalide in monoglyme and RLi (R = butyl, ethyl, methyl) to prepare Sn70Ge30@carbon core–shell nanoparticles. The core sizes and shell thicknesses of these nanoparticles are dependent on the alkyl terminator. Electrochemical studies suggest that nanoparticles synthesized with butyl terminators exhibit the highest capacity retention after 40 cycles (95%) and a first charge capacity of 1040 mAh/g. Shirahata and Sakka346 synthesized highly luminescent Si nanoparticles (NPs) terminated with alkoxy monolayers through the reduction SiCl4 by sodium biphenylide in a toluene + monoglyme mixture using an inverse micelle method; they also observed the size-dependent UV photoluminescence (PL) property for non-oxidized Si NPs at room temperature, as well as a high quantum efficiency of the UV fluorescence. Mishra et al.347 prepared heterometal-organic complexes including NaY(TFA)4(diglyme), [Na(triglyme)2][Y2(TFA)7(THF)2], Na2Y(TFA)5(tetraglyme), NaLn(TFA)4(diglyme) [Ln = Er, Tm, or Yb], and Na2Ln(TFA)5(tetraglyme) [Ln = Er, or Yb] (TFA = trifluoroacetate), and used them as precursors for up-converting NaY(Ln)F4 (Ln = Yb, Er, Tm) nanocrystals and thin films, which have potential applications as lanthanide-doped up-conversion (UC) emission materials.
9.2 Polymeric materials
Shirai et al.348 carried out the radical polymerization to prepare polymers carrying glyme units as alkali cation binding sites and photodimerizable cinnamoyl units. They found that the photodimerization of the cinnamoyl groups with relatively short glyme chains could improve their cation binding ability, and the addition of alkali metal cations as templates enforced the effect of photodimerization on the cation binding properties. Morgado et al.349 found that the orange-emitting PPV-based statistical copolymer with glyme-like side groups (Scheme 45) has a photoluminescence efficiency of ~17%. The presence of glyme units enables the solvation of salts and the ion mobility under the applied electric field, thus this copolymer carries both the ion-coordinating and luminescence moieties.
Scheme 45.

PPV-based statistical copolymer with glyme-like side groups.
Moore et al.350 dissolved poly(enaminonitrile) (PEAN) or miscible blends of PEAN with poly(ethylene oxide) in a number of glymes, and observed the cloud points of these solutions when the temperature increases. The Sneddon and Remsen groups351, 352 found that polyborazylene could be dissolved in monoglyme or THF, and could be further precipitated by the addition of pentane. Monoglyme was used as a solvent for dissolving and mixing two polymeric precursors, namely allylhydridopolycarbosilane (AHPCS) ([Si(CH2CH=CH2)2CH2]0.05[SiH2CH2]0.95) and polyborazylene (PBz) [B3N3H4-x]n, which serve as the sources for the SiC and BN phases respectively.353 Following the removal of solvent, the co-pyrolysis of these two precursors at 1000 °C affords a two-phase SiC-BN ceramic composite whose highly unusual microstructure resembles that of certain polymer blends.
López and Ratner354 produced weakly ionized, radio-frequency and glow-discharge plasmas from the vapor of glyme precursors (mono-, di- and tri-), and then used them to deposit organic thin films on polytetrafluoroethylene (PTFE). Sandner et al.355 performed the free-radical photopolymerization of an oligo(ethylene glycol) dimethacrylate ((EG)23DMA) in two glymes ((EG)3DME and (EG)11DME) as plasticizers in the presence of LiCF3SO3, which was analyzed by differential scanning calorimetry (DSC), FT-Raman spectroscopy and sol-gel analysis. The addition of LiCF3SO3 increased the polymerization rate.
9.3 Inorganic materials
Baker et al.356 prepared homoleptic dicyclohexylphosphide (PCy2) complexes of early transition metals using monoglyme (G1) as the complexing agent for Li+; these new complexes include [Li(G1)][Zr(PCy2)5], [Li(G1)][Hf(PCy2)5], [Li(G1)][Ti(PCy2)4], [Li(G1)][V(PCy2)4], [Li(G1)][Re(PCy2)4], [Li(G1)2][Nb(PCy2)4], Mo(PCy2)4, [Li(G1)3][Cr2(PCy2)5], [Li(G1)3][W2(PCy2)5] and [Li(G1)][Mn2(PCy2)5]. Baxter et al.357 synthesized the adducts of [Gd-(tmhd)3] (tmhd-H = 2,2,6,6-tetramethylheptane-3,5-dione) with a series of glyme ligands from monoglyme to heptaglyme. These complexes were evaluated as precursors for Atmospheric Pressure Chemical Vapor Deposition (APCVD) coatings of Ce0.9Gd0.1O1.95 thick electrolyte films for solid oxide fuel cells (SOFCs). The advantages of using these complexes for metal organic chemical vapor deposition (MOCVD) include: (1) readily available in crystalline solids of fixed stoichiometry; (2) soluble in hydrocarbons (both aliphatic or aromatic) and air stable; and (3) better mass transport properties of more stable complexes can be prepared from longer chain glymes. Drake et al.358 synthesized the monomeric materials [M(β-diketonate)2(L-L)] by reacting oligomeric alkaline earth metal β-diketonate complexes, [M(β-diketonate)2] with a glyme (tri- and tetra-) ligand (L). The same group359 further prepared eight-coordinate triglyme-bridged dimeric complexes, [(Ln(tmhd)3)2L1] (Ln = Eu or Tb, L1 = triglyme, and tmhd = ButCOCHCOBut) through the reaction of hydrated β-diketonate complexes [Ln(tmhd)3(H2O)] with triglyme in hexane. They also prepared the nine-coordinate monomeric compound [La(tmhd)3L2] via the reaction of [La(tmhd)3(H2O)] with tetraglyme (L2) in hexane. These complexes are stable in air with moisture and also have a good volatility and thermal stability. Arunasalam et al.360 used carbonate and hydroxide compounds to prepare a number of Group 2 β-diketonate complexes supported by multidentate glyme ligands (tri-, tetra- or heptaglyme), including a single-crystal structure of a calcium complex of [(Ca(hfpd)2)2(heptaglyme)] where [H-hfpd = 1,1,1,5,5,5-hexafluoropentane-2,4-dione]. These β-diketonate and carboxylate compounds have potential applications as CVD precursors for both MO and MF2 thin films. Arnáiz et al.361 synthesized outer-sphere addition compounds of MoO2Br2(H2O)2 with diethyl ether, dioxane, glyme, diglyme, triglyme and tetraglyme by crystallizing diethyl ether extracts of a solution of sodium molybdate in concentrated hydrobromic acid and the respective ether. They also suggested that the polyether interacts with the MoO2Br2(H2O)2 units through hydrogen bonds based on their X-ray structure analysis for diglyme and tetraglyme adducts. Crochet and Fromm362 prepared and characterized crystalline cobalt, nickel, zinc, and mercury halide adducts with polyethers as ligands including [Co(μ-Cl)2CoCl2(monoglyme)2], cis-[CoI2(H2O)2(monoglyme)2]2+[CoI4]2−, [NiI2(monoglyme)2], [ZnI2(monoglyme)], [HgCl2(monoglyme)], [CoI2(diglyme)], [ZnI2(diglyme)], [HgI2(diglyme)], [CoCl(μ-Cl)(diglyme)]2, [NiI(μ-I)(diglyme)]2, [Co(μ-Cl)(triglyme)]22+[CoCl2(μ-Cl)]22−, and cis-[(NiI2)(triglyme)]n. Some of these adducts exhibit unusual coordination numbers and arrangements.
10. Other applications
10.1 Chemical Vapor Deposition (CVD)
CVD is an important method for producing new materials. When preparing f-element oxides by the CVD procedure, the reproducibility becomes a challenge because regular precursors often form self-association and hydrate and undergo hydrolysis or cleavage of the ligands on storage. As an effort to design new lanthanide complexes with suitable mass transport properties for metal-organic chemical vapor deposition (MOCVD) applications, the Fragalà group363, 364 developed new adducts La(hfac)·monoglyme·H2O, La(hfac)·diglyme and La(hfac)·triglyme (hfac = CF3COCHCOCF3), which possess better volatility and thermal stability than conventional lanthanum CVD precursors. Pollard et al.365 found that monoglyme and diglyme formed neutral complexes of [Y(hfac)3(glyme)], whilst triglyme and tetraglyme produced ionic complexes [Y(hfac)2(glyme)]+ [Y(hfac)4]−; upon CVD at 250–350 °C using oxygen as a carrier gas, these complexes yielded mixed yttrium oxide–fluoride ceramics. The Fragalà group366 prepared and characterized [Y(hfac)3·monoglyme], [Y(hfac)3·diglyme], [Y(hfac)3·(H2O)2·triglyme] and [Y(hfac)2·tetraglyme]+[Y(hfa)4]−; they found these adducts are suitable for MOCVD applications due to their high volatility and high thermal stabilities with a residue lower than 2–4%. In particular, YBaCuO HTc superconductor was produced from the low-pressure MOCVD process of [Y(hfa)3·monoglyme] complex using a multimetal molten single source. Kang et al.367 synthesized thermally stable Ln(hfac)3·monoglyme (Ln=Ho or Y) complexes and indicated their high potential as MOCVD precursors. Pollard et al.368 designed new glyme-adduct precursors of [Ce(hfac)3(glyme)] (glyme = mono-, di-, or tri-) and [(Ce(hfac)3)2(μ-tetraglyme)] and used them in the CVD formation of films of cerium oxides on substrates Si, Pt, and TiN. The Fragalà group369 also prepared Ce(hfac)3·diglyme, Ce(hfac)3·diethyldiglyme and Ce(hfac)3·dibutyldiglyme, and found these complexes are ideal for CeO2 film depositions due to their high volatility and high thermal stability with low residue. Malandrino et al.370 synthesized novel complex precursors Eu(hfac)3·glyme (glyme = mono- or di-) and found that both complexes are thermally stable and could be evaporated with < 4% residue. Another study by this group371 suggested that La(hfac)3·diglyme could be evaporated from the melt up to 130 °C without side decomposition processes. The Malandrino group372 further prepared and characterized Nd(hfac)3·monoglyme·H2O and Nd(hfac)3·diglyme, which exhibit high volatility and good thermal stability with a residue left lower than 3%. In particular, Nd(hfa)3·diglyme was used in a low-pressure MOCVD preparation of NdBa2Cu3O7–δ thin films.
In addition to these lanthanide adducts using glymes, other metal complexes have also been investigated. In an attempt to prepare precursors for Supercritical Fluid Transport (SFT) CVD, Blake et al.373 synthesized new S-donor tetraphenyldithioimidodiphosphinate compounds [Na(Ph2P(S)NP(S)Ph2)(L)] (where L = triglyme or tetraglyme); however, these complexes are not quite soluble in supercritical CO2. The Winter group374 prepared calcium complexes with η2-pyrazolato ligands including those adducts containing glymes [e.g. Ca(tBu2pz)2(triglyme), and Ca(tBu2pz)2(tetraglyme) Ca(Me2pz)2(triglyme), and Ca(Me2pz)2(tetraglyme)], resulting in volatile, thermally stable precursors for CVD applications.
10.2 NMR solvents
Deuterated monoglyme can be used as a co-solvent for low-temperature reactions monitored by a direct NMR analysis to probe the underlying reaction mechanism, as pioneered by the Buncel group. This group375 performed low-temperature (‒40 °C) NMR studies using a new solvent mixture of CD3CN–monoglyme-d10 (1:1, v/v) as the medium for a reaction of 2,4,6-trinitroanisole (TNA) with phenoxide; their results suggest the 1,1 adduct being the only species produced and no 1,3 O-adduct of phenoxide formed either prior to the 1,1 species or later. A further study of this reaction system by this group376 revealed both O- and C-bonded phenoxide σ-complex adducts, implying the formation of the former via kinetic control and of the latter through thermodynamic control. This group377 also used the same solvent system at −40 °C to room temperature to investigate the regioselectivity in Meisenheimer complexation of the reaction of 2,4,6-trimethylphenoxide ion (MesO−) with 2,4,6-trinitroanisole (TNA) via 1H and 13C NMR, and suggest a kinetic preference for C-1 attachment and the σ-adduct from C-3 attack being more thermodynamically stable. The same deuterated solvent system was also employed by this group to examine the reactivity of 4-nitrobenzofuroxan (NBF) with several aryloxide nucleophiles; they observed the formation of a C-7 O-adduct with the ambident (O- and C-) nucleophile phenoxide ion at −40 °C.378
10.3 Chromatography
Schuetz et al.379 synthesized new glyme-substituted polysiloxane (Scheme 46) and 18-crown-6-substituted polysiloxane and applied them as the stationary phase for gas chromatography. The glyme polysiloxane has an operational temperature range of 20–280 °C and a selectivity comparable to that of Carbowax 20M. Schuetz et al.380 found that an eluent of hexane/monoglyme/formic acid (85%) (150:50:3) could be used to purify p-biphenyl-n-hexanoic acid in a silica based column chromatography, achieving 98% purity.
Scheme 46.

Glyme-substituted polysiloxane stationary phase.
10.4 Dissolution of CO2 and other gases
Sciamanna and Lynn381 determined the gas solubilities of H2S, SO2, CO2, propane and n-butane in glymes (di-, tri- and tetra-) and PEG monoethers at the partial pressure of gas solute between 3 and 100 kPa. The temperature dependence of gas solubility was described by Henry’s law coefficients. The presence of a small amount of water (< 6 wt %) decreases the gas solubility in glymes; the hydrogen-bonding property of PEG monoethers also reduces the gas solubility. Henni et al.382 reported the solubilities of CO2 in 14 solvents including glymes, PEG monoethers, selexol® and sulfolane at 25, 40 and 60 °C; they suggested that glymes (particularly diglyme, triglyme and tetraglyme) are outstanding solvents for CO2 removal. Kodama et al.74 measured the solubilities and saturated densities of CO2 in glymes (di-, tri- and tetra-) at 313.15 K as a function of pressure (at high pressures, CO2 mole fraction is 0.857 at 7.126 MPa in diglyme, 0.827 at 7.202 MPa in triglyme and 0.822 at 7.316 MPa in tetraglyme). These data of CO2 solubilities and saturated densities were further correlated with a three parameter pseudo-cubic equation of state.
10.5 Other applications
Monoglyme was used in aqueous solutions to improve the solubility of oxiranes in order to determine their nanomole quantities spectrophotometrically.383 Interestingly, Dasilva-Carbalhal et al.384 studied the effect of glymes on the conductance percolation of AOT/isooctane/water microemulsions. The addition of glymes (mono-, di-, tri-, and tetra-) to the microemulsion led to a decrease in the percolation threshold. This modification promotes the exchange of matter between droplets in the AOT film. More recently, various glymes were used to dissolve triglycerides, and thus were investigated as co-solvents for the CaO-catalyzed transesterification of soybean oil (> 50% v/v loading) into biodiesel; under the optimum conditions, a >98% conversion of triglycerides could be achieved in 4 h using dipropylene glycol dimethyl ether (P2) as the co-solvent.22
11. Perspectives
Recent active research on glymes seems to focus on their applications in electrochemistry, catalysts, CVD, nanomaterials, and the dissolution of CO2. However, there could be renewing interest in exploring their solvent role in organic reactions, biocatalysis and biofuel production. Glymes can also be derived to contain functional groups such as double bonds, and thus become precursors for other applications. Many glymes are less volatile and less toxic than some common laboratory organic solvents; however, there is still lack of a systematic database on their acute and long-term toxicity as well as biodegradability. In addition, despite their versatile roles in industrial and consumer products, glymes are not well studied in terms of their physicochemical properties such as dielectric constant, polarity, hydrophobicity (log P), heat capacity, and phase equilibrium, etc.
In addition to glymes, other solvents carrying polyether or polyol moieties have also been extensively studied; these solvents include glycol monoethers and glycol ether esters,1–3 polyethylene glycol (PEG) and aqueous solutions,330 and glycerol and its derivatives.385–387 New solvents can also be developed to carry the functionality of these moieties, for example, ionic liquids can be functionalized with various glycol groups to become tailored-made solvents.388 As another example, a new family of glycerol derivatives including 1,3-dialkoxy-2-propanols and 1,2,3-trialkoxypropanes were prepared by García et al.;389 these new solvents share some structural features with glymes and have a wide range of polarity properties, implying their high potential for solvent substitution.
Supplementary Material
Acknowledgements
HZ acknowledges the supports by the Henry Dreyfus Teacher-Scholar Award, NIH MBRS-RISE grant (1R25GM096956), NIH NIBIB contract award (HHSN268201200011C), and the National Natural Science Foundation of China (21328601).
References
- 1.Cragg ST. In: Patty’s Toxicology. 6th edn. Bingham E, Cohrssen B, editors. vol. 4. Hoboken, NJ: John Wiley & Sons; 2012. pp. 641–787. [Google Scholar]
- 2.Cragg ST. In: Patty’s Toxicology. 6th edn. Bingham E, Cohrssen B, editors. vol. 4. Hoboken, NJ: John Wiley & Sons; 2012. p.??? [Google Scholar]
- 3.Smith RL. Environ. Health Perspect. 1984;57:1–4. doi: 10.1289/ehp.84571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haymore BL, Lamb JD, Izatt RM, Christensen JJ. Inorg. Chem. 1982;21:1598–1602. [Google Scholar]
- 5.Adcock JL, Lagow RJ. J. Org. Chem. 1973;38:3617–3618. [Google Scholar]
- 6.Leonhardt DE, Coleman LW, Bradshaw WS. Reprod. Toxicol. 1991;5:157–162. doi: 10.1016/0890-6238(91)90044-g. [DOI] [PubMed] [Google Scholar]
- 7.Gage JC. Br. J. Ind. Med. 1970;27:1–18. doi: 10.1136/oem.27.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGregor DB, Willins MJ, McDonald P, Holmström M, McDonald D, Niemeier RW. Toxicol. Appl. Pharmacol. 1983;70:303–316. doi: 10.1016/0041-008x(83)90106-0. [DOI] [PubMed] [Google Scholar]
- 9.Schuler RL, Hardin BD, Niemeier RW, Booth G, Hazelden K, Piccirillo V, Smith K. Environ. Health Perspect. 1984;57:141–146. doi: 10.1289/ehp.8457141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson EM, Gabel BE, Larson J. Environ. Health Perspect. 1984;57:135–139. doi: 10.1289/ehp.8457135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGregor DB. Environ. Health Perspect. 1984;57:97–103. doi: 10.1289/ehp.845797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheever KL, Weigel WW, Richards DE, Lal JB, Plotnick HB. The Toxicologist. 1985;5:140. [Google Scholar]
- 13.Cheever KL, Richards DE, Weigel WW, Lal JB, Dinsmore AM, Daniel FB. The Toxicologist. 1986;6:32. [Google Scholar]
- 14.Cheever KL, Richards DE, Weigel WW, Lal JB, Dinsmore AM, Daniel FB. Toxicol. Appl. Pharmacol. 1988;94:150–159. doi: 10.1016/0041-008x(88)90345-6. [DOI] [PubMed] [Google Scholar]
- 15.Lee KP, Kinney LA, Valentine R. Toxicology. 1989;59:239–258. doi: 10.1016/0300-483x(89)90195-9. [DOI] [PubMed] [Google Scholar]
- 16.Hardin BD, Eisenmann CJ. Teratology. 1987;35:321–328. doi: 10.1002/tera.1420350306. [DOI] [PubMed] [Google Scholar]
- 17. http://www.environmentalhealthnews.org/ehs/news/2011/epa-takes-on-glymes. [Google Scholar]
- 18.Spencer PJ. Toxicol. Lett. 2005;156:181–188. doi: 10.1016/j.toxlet.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 19.Arnot JA, Gobas FAPC. Environmental Reviews. 2006;14:257–297. [Google Scholar]
- 20.Sieck LW, Meot-Ner M. J. Phys. Chem. 1984;88:5324–5327. [Google Scholar]
- 21.Tang S, Jones CL, Zhao H. Bioresour. Technol. 2013;129:667–671. doi: 10.1016/j.biortech.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 22.Tang S, Zhao H, Song Z, Olubajo O. Bioresour. Technol. 2013;139:107–112. doi: 10.1016/j.biortech.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 23.Reichardt C. Chem. Rev. 1994;94:2319–2358. [Google Scholar]
- 24.Meot-Ner M. J. Am. Chem. Soc. 1983;105:4906–4911. [Google Scholar]
- 25.Sharma RB, Blades AT, Kebarle P. J. Am. Chem. Soc. 1984;106:510–516. [Google Scholar]
- 26.Wasada H, Tsutsui Y, Yamabe S. J. Phys. Chem. 1996;100:7367–7371. [Google Scholar]
- 27.Adötoledo D, Aviyente V, Martin JML, Lifshitz C. J. Phys. Chem. A. 1998;102:6357–6365. [Google Scholar]
- 28.Shchori E, Jagur-Grodzinski J. J. Am. Chem. Soc. 1972;94:7957–7962. [Google Scholar]
- 29.Meot-Ner M, Sieck LW, Scheiner S, Duan X. J. Am. Chem. Soc. 1994;116:7848–7856. [Google Scholar]
- 30.Matsuura H, Sagawa T. J. Mol. Liq. 1995;65–66:313–316. [Google Scholar]
- 31.Masatoki S, Takamura M, Matsuura H, Kamogawa K, Kitagawa T. Chem. Lett. 1995;24:991–992. [Google Scholar]
- 32.Bedrov D, Smith GD. J. Chem. Phys. 1998;109:8118. [Google Scholar]
- 33.Bedrov D, Pekny M, Smith GD. J. Phys. Chem. B. 1998;102:996–1001. [Google Scholar]
- 34.Bedrov D, Smith GD. J. Phys. Chem. B. 1999;103:3791–3796. [Google Scholar]
- 35.Begum R, Masatoki S, Matsuura H. J. Mol. Struct. 1996;384:115–120. [Google Scholar]
- 36.Engkvist O, Karlström G. J. Chem. Phys. 1997;106:2411–2417. [Google Scholar]
- 37.Bernal P, Bunn A, Logan J, McCluan J. J. Solution Chem. 2000;29:651–665. [Google Scholar]
- 38.Bernal P, McCluan J. J. Solution Chem. 2001;30:119–131. [Google Scholar]
- 39.Douhéret G, Reis JCR, Davis MI, Fjellanger IJ, Høiland H. Phys. Chem. Chem. Phys. 2004;6:784–792. [Google Scholar]
- 40.Haynes WM, editor. CRC Handbook of Chemistry and Physics. Boca Raton: CRC Press; 2012. [Google Scholar]
- 41.Cheremisinoff NP. Industrial Solvents Handbook. New York: Marcel Dekker, Inc.; 2003. [Google Scholar]
- 42.Flick EW, editor. Industrial solvents handbook. Westwood, NJ: Noyes Data Corporation; 1998. [Google Scholar]
- 43.Treszczanowicz T, Benson GC, Lu BC-Y. Thermochim. Acta. 1990;168:95–102. [Google Scholar]
- 44.Tovar CA, Carballo E, Cerdeiriña CA, Romanı L. J. Chem. Eng. Data. 1997;42:1085–1089. [Google Scholar]
- 45.Nakai H, Soejima H, Tamura K, Ogawa H, Murakami S, Toshiyasu Y. Thermochim. Acta. 1991;183:15–27. [Google Scholar]
- 46.Dethlefsen C, Hvidt A. J. Chem. Thermodyn. 1985;17:193–199. [Google Scholar]
- 47.de Ruiz Holgado ME, de Schaefer CR, Arancibia EL, Katz M. Fluid Phase Equilib. 1994;95:299–312. [Google Scholar]
- 48.Spanedda A, Lepori L, Matteoli E. Fluid Phase Equilib. 1991;69:209–222. [Google Scholar]
- 49.Muhuri PK, Hazra DK. J. Chem. Eng. Data. 1994;39:375–377. [Google Scholar]
- 50.McGee RL, Wallace WJ, Rataiczak RD. J. Chem. Eng. Data. 1983;28:305–307. [Google Scholar]
- 51.Wallace WJ, Mathews AL. J. Chem. Eng. Data. 1963;8:496–498. [Google Scholar]
- 52.Wallace WJ, Shephard CS, Underwood C. J. Chem. Eng. Data. 1968;13:11–13. [Google Scholar]
- 53.Cabezas JL, Beltran S, Coca J. J. Chem. Eng. Data. 1991;36:184–188. [Google Scholar]
- 54.Reis JCR, Iglesias TP. Phys. Chem. Chem. Phys. 2011;13:10670–10680. doi: 10.1039/c1cp20142e. [DOI] [PubMed] [Google Scholar]
- 55.Riadigos CF, Iglesias R, Rivas MA, Iglesias TP. J. Chem. Thermodyn. 2011;43:275–283. [Google Scholar]
- 56.Comuñas MJP, Baylaucq A, Boned C, Fernández J. J. Chem. Eng. Data. 2003;48:1044–1049. [Google Scholar]
- 57.Abboud J-LM, Notari R. Pure Appl. Chem. 1999;71:645–718. [Google Scholar]
- 58.Canters GW. J. Am. Chem. Soc. 1972;94:5230–5235. [Google Scholar]
- 59.Pal A, Kumar A. Int. J. Thermophys. 2003;24:1073–1087. [Google Scholar]
- 60.Serna A, García de la Fuente I, González JA, Cobos JC. Fluid Phase Equilib. 1997;133:187–192. [Google Scholar]
- 61.Villamañan MA, Casanova C, Roux AH, Grolier J-PE. J. Chem. Thermodyn. 1982;14:251–258. [Google Scholar]
- 62.Ku H-C, Tu C-H. J. Chem. Eng. Data. 2000;45:391–394. [Google Scholar]
- 63.Pal A, Sharma S. J. Chem. Eng. Data. 1998;43:532–536. [Google Scholar]
- 64.Pal A, Sharma S. J. Chem. Eng. Data. 1999;44:212–215. [Google Scholar]
- 65.Treszczanowicz T, Cieślak D. J. Chem. Thermodyn. 1993;25:661–665. [Google Scholar]
- 66.Benson GC, Kumaran MK, Treszczanowicz T, D'arcy PJ, Halpin CJ. Thermochim. Acta. 1985;95:59–66. [Google Scholar]
- 67.Conesa A, Shen S, Coronas A. Int. J. Thermophys. 1998;19:1343–1358. [Google Scholar]
- 68.López ER, Daridon JL, Baylaucq A, Fernández J. J. Chem. Eng. Data. 2003;48:1208–1213. [Google Scholar]
- 69.Aizawa K, Kato M. J. Chem. Eng. Data. 1991;36:159–161. [Google Scholar]
- 70.Aminabhavi TM, Gopalakrishna B. J. Chem. Eng. Data. 1995;40:462–467. [Google Scholar]
- 71.Treszczanowicz AJ, Halpin CJ, Benson GC. J. Chem. Eng. Data. 1982;27:321–324. [Google Scholar]
- 72.Carvajal C, Tölle KJ, Smid J, Szwarc M. J. Am. Chem. Soc. 1965;87:5548–5553. [Google Scholar]
- 73.Kusano K. J. Chem. Eng. Data. 1978;23:141–143. [Google Scholar]
- 74.Kodama D, Kanakubo M, Kokubo M, Hashimoto S, Nanjo H, Kato M. Fluid Phase Equilib. 2011;302:103–108. [Google Scholar]
- 75.Lago A, Rivas MA, Legido J, Iglesias TP. J. Chem. Thermodyn. 2009;41:257–264. [Google Scholar]
- 76.Kimura K, Fujishiro R. Bull. Chem. Soc. Jpn. 1966;39:608–610. [Google Scholar]
- 77.Côté J-F, Brouillette D, Desnoyers JE, Rouleau J-F, St-Arnaud J-M, Perron G. J. Solution Chem. 1996;25:1163–1173. [Google Scholar]
- 78.Cocchi M, De Benedetti P, Marchetti A, Menziani MC, Seeber R, Tassi L, Ulrici A. J. Solution Chem. 2001;30:149–169. [Google Scholar]
- 79.Trejo LM, Costas M, Patterson D. J. Chem. Soc., Faraday Trans. 1991;87:3001–3008. [Google Scholar]
- 80.Hefter GT, Salomon M. J. Solution Chem. 1994;23:579–593. [Google Scholar]
- 81.Kimura F, D'arcy PJ, Sugamori ME, Benson GC. Thermochim. Acta. 1983;64:149–154. [Google Scholar]
- 82.Patterson GD, Flory PJ. J. Chem. Soc., Faraday Trans. 2. 1972;68:1111–1116. [Google Scholar]
- 83.Tovar CA, Carballo E, Cerdeiriña CA, Paz Andrade MI, Romaní L. J. Chem. Soc., Faraday Trans. 1997;93:3505–3509. [Google Scholar]
- 84.Treszczanowicz AJ, Treszczanowicz T. Fluid Phase Equilib. 1998;148:209–220. [Google Scholar]
- 85.Pal A, Kumar H. J. Chem. Eng. Data. 1999;44:1330–1334. [Google Scholar]
- 86.Comuñas MJP, Baylaucq A, Boned C, Fernández J. Ind. Eng. Chem. Res. 2004;43:804–814. [Google Scholar]
- 87.López ER, García J, Coronas A, Fernández J. Fluid Phase Equilib. 1997;133:229–238. [Google Scholar]
- 88.López ER, Coxam J-Y, Fernández J, Grolier J-PE. J. Chem. Eng. Data. 1999;44:1409–1413. [Google Scholar]
- 89.Tseregounis SI, Riley MJ. AIChE J. 1994;40:726–737. [Google Scholar]
- 90.Smith J, Andreoli-Ball L, Patterson D. J. Chem. Soc. Faraday Trans. 1992;88:2875–2881. [Google Scholar]
- 91.López ER, Daridon JL, Plantier F, Boned C, Fernández J. Int. J. Thermophys. 2006;27:1354–1372. [Google Scholar]
- 92.Nogrady T, Burgen ASV. J. Am. Chem. Soc. 1969;91:3890–3893. doi: 10.1021/ja01042a035. [DOI] [PubMed] [Google Scholar]
- 93.Matsui T, Takeyama K. Electrochimica Acta. 1998;43:1355–1360. [Google Scholar]
- 94.Andersson M, Karlstrom G. J. Phys. Chem. 1985;89:4957–4962. [Google Scholar]
- 95.Hoefelmann K, Jagur-Grodzinski J, Szwarc M. J. Am. Chem. Soc. 1969;91:4645–4651. [Google Scholar]
- 96.Collins GL, Esch TEH, Smid J. J. Solution Chem. 1978;7:9–18. [Google Scholar]
- 97.Ritzhaupt G, Devlin JP. J. Phys. Chem. 1986;90:1143–1147. [Google Scholar]
- 98.Payne VA, Xu J-H, Forsyth M, Ratner MA, Shriver DF, de Leeuw SW. Electrochimica Acta. 1995;40:2087–2091. [Google Scholar]
- 99.Payne VA, Xu J, Forsyth M, Ratner MA, Shriver DF. J. Chem. Phys. 1995;103:8746–8755. [Google Scholar]
- 100.Johansson P, Gejji SP, Tegenfeldt J, Lindgren J. Solid State Ionics. 1996;86–88:297–302. [Google Scholar]
- 101.Johansson P, Tegenfeldt J, Lindgren J. Polymer. 1999;40:4399–4406. [Google Scholar]
- 102.Johansson P. Polymer. 2001;42:4367–4373. [Google Scholar]
- 103.Shen N, Pope RM, Dearden DV. International Journal of Mass Spectrometry. 2000;195/196:639–652. [Google Scholar]
- 104.Henderson WA, Young VG, Jr, Brooks NR, Smyrl WH. Acta Crystallographica Section C. 2002;C58:m501–m503. doi: 10.1107/s0108270102015135. [DOI] [PubMed] [Google Scholar]
- 105.Henderson WA, Brooks NR, Brennessel WW, Young VG., Jr J. Phys. Chem. A. 2004;108:225–229. [Google Scholar]
- 106.Grondin J, Talaga D, Lassègues J-C, Henderson WA. Phys. Chem. Chem. Phys. 2004;6:938–944. [Google Scholar]
- 107.Dhuaml NR, Gejji SP. Theoretical Chemistry Accounts. 2006;115:308–321. [Google Scholar]
- 108.Tamura T, Yoshida K, Hachida T, Tsuchiya M, Nakamura M, Kazue Y, Tachikawa N, Dokko K, Watanabe M. Chem. Lett. 2010;39:753–755. [Google Scholar]
- 109.Yoshida K, Nakamura M, Kazue Y, Tachikawa N, Tsuzuki S, Seki S, Dokko K, Watanabe M. J. Am. Chem. Soc. 2011;133:13121–13129. doi: 10.1021/ja203983r. [DOI] [PubMed] [Google Scholar]
- 110.Cho YS, Cho S-I, Ryua H-K, Heo JS, Lee DH, Moon SH. J. Electrochem. Soc. 2003;150:F11–F19. [Google Scholar]
- 111.Varnek A, Wipff G, Solov'ev VP, Solotnov AF. J. Chem. Inf. Comput. Sci. 2002;42:812–829. doi: 10.1021/ci010318q. [DOI] [PubMed] [Google Scholar]
- 112.Frensdorff HK. J. Am. Chem. Soc. 1971;93:600–606. [Google Scholar]
- 113.Izatt RM, Bradshaw JS, Nielsen SA, Lamb JD, Christensen JJ, Sen D. Chem. Rev. 1985;85:271–339. [Google Scholar]
- 114.Izatt RM, Pawlak K, Bradshaw JS, Bruening RL. Chem. Rev. 1991;91:1721–2085. [Google Scholar]
- 115.Gokel GW, Goli DM, Schultz RA. J. Org. Chem. 1983;48:2837–2842. [Google Scholar]
- 116.Zhang H, Dearden DV. J. Am. Chem. Soc. 1992;114:2754–2755. [Google Scholar]
- 117.Xu W-Y, Smid J. J. Am. Chem. Soc. 1984;106:3790–3796. [Google Scholar]
- 118.Tsvetanov CB, Petrova EB, Dimov DK, Panayotov IM, Smid J. J. Solution Chem. 1990;19:425–436. [Google Scholar]
- 119.Davidson WR, Kebarle P. Can. J. Chem. 1976;54:2594–2599. [Google Scholar]
- 120.Plewa-Marczewska A, Kalita M, Marczewski M, Siekierski M. Electrochimica Acta. 2010;55:1389–1395. [Google Scholar]
- 121.Chan LL, Smid J. J. Am. Chem. Soc. 1967;89:4547–4549. [Google Scholar]
- 122.Chan LL, Smid J. J. Am. Chem. Soc. 1968;90:4654–4661. [Google Scholar]
- 123.Chan LL, Wong KH, Smid J. J. Am. Chem. Soc. 1970;92:1955–1963. [Google Scholar]
- 124.Takaki U, Smid J. J. Am. Chem. Soc. 1974;96:2588–2593. [Google Scholar]
- 125.Detellier C, Laszlo P. Helv. Chim. Acta. 1976;59:1333–1345. [Google Scholar]
- 126.Gilkerson WR, Jackson MD. J. Am. Chem. Soc. 1982;104:1218–1223. [Google Scholar]
- 127.Plewa A, Kalita M, Siekierski M. Electrochimica Acta. 2007;53:1527–1534. [Google Scholar]
- 128.Smid J, Grotens AM. J. Phys. Chem. 1973;77:2377–2382. [Google Scholar]
- 129.de Boer E, Klaassen AAK, Mooij JJ, Noordik JH. Pure Appl. Chem. 1979;51:73–83. [Google Scholar]
- 130.Rhodes CP, Frech R. Macromolecules. 2001;34:2660–2666. [Google Scholar]
- 131.Frech R, Rhodes CP, Khan M. Macromol. Symp. 2002;186:41–49. [Google Scholar]
- 132.Rhodes CP, Khan M, Frech R. J. Phys. Chem. B. 2002;106:10330–10337. [Google Scholar]
- 133.Henderson WA, Brooks NR, Brennessel WW, Young VGJ. Chem. Mater. 2003;15:4679–4684. [Google Scholar]
- 134.Henderson WA, Brooks NR, Young VGJ. Chem. Mater. 2003;15:4685–4690. [Google Scholar]
- 135.Henderson WA. J. Phys. Chem. B. 2006;110:13177–13183. doi: 10.1021/jp061516t. [DOI] [PubMed] [Google Scholar]
- 136.Henderson WA. Macromolecules. 2007;40:4963–4971. [Google Scholar]
- 137.Henderson WA, Brooks NR. Inorg. Chem. 2003;42:4522–4524. doi: 10.1021/ic034518q. [DOI] [PubMed] [Google Scholar]
- 138.Timko JM, Helgeson RC, Newcomb M, Gokel GW, Cram DJ. J. Am. Chem. Soc. 1974;96:7097–7099. doi: 10.1021/ja00830a042. [DOI] [PubMed] [Google Scholar]
- 139.Bartsch RA, Juri PN. Tetrahedron Lett. 1979;20:407–410. [Google Scholar]
- 140.Bartsch RA, Juri PN, Mills MA. Tetrahedron Lett. 1979;20:2499–2502. [Google Scholar]
- 141.Otera J, Shiomi T, Murakami K, Kawasaki Y. Bull. Chem. Soc. Jpn. 1981;54:2964–2967. [Google Scholar]
- 142.Hirashima Y, Ito K, Shiokawa J. Chem. Lett. 1983;12:9–10. [Google Scholar]
- 143.Hirashima Y, Kanetsuki K, Yonezu I, Kamakura K, Shiokawa J. Bull. Chem. Soc. Jpn. 1983;56:738–743. [Google Scholar]
- 144.Inoue Y, Hakushi T. J. Chem. Soc., Perkin Trans. 2. 1985:935–946. [Google Scholar]
- 145.Guerra MA, Bierschenk TR, Lagow RJ. J. Am. Chem. Soc. 1986;108:4103–4105. [Google Scholar]
- 146.Markies PR, Akkerman OS, Bickelhaupt F, Smeets WJJ, Spek AL. Organometallics. 1994;13:2616–2627. [Google Scholar]
- 147.Meot-Ner M, Sieck LW, Liebman JF, Scheiner S. J. Phys. Chem. 1996;100:6445–6450. [Google Scholar]
- 148.Fromm KM, Goesmann H, Bernardinelli G. Polyhedron. 2000;19:1783–1789. [Google Scholar]
- 149.Mishra S, Daniele S, Hubert-Pfalzgraf LG, Jeanneau E. Eur. J. Inorg. Chem. 2007;2007:2208–2215. [Google Scholar]
- 150.Chantooni MKJ, Britton D, Kolthoff IM. Journal of Crystallographic and Spectroscopic Research. 1993;23:497–503. [Google Scholar]
- 151.Jaycox GD, Sinta R, Smid J. J. Polym. Sci. Polym. Chem. Ed. 1982;20:1629–1638. [Google Scholar]
- 152.Saraswathi M, Miller JM. J. Am. Soc. Mass Spectrom. 1996;7:42–49. doi: 10.1016/1044-0305(96)84506-5. [DOI] [PubMed] [Google Scholar]
- 153.Slates RV, Szwarc M. J. Am. Chem. Soc. 1967;89:6043–6050. [Google Scholar]
- 154.Canters GW, Klassen AAK, de Boer E. J. Phys. Chem. 1970;74:3299–3302. [Google Scholar]
- 155.Smyrl WH, Kurtz SR, Zeigler JM, Ginley DS. J. Chem. Soc., Chem. Commun. 1983:1155–1156. [Google Scholar]
- 156.Foos JS, Stolki TS, Beebe X. J. Electrochem. Soc. 1989;136:2748–2749. [Google Scholar]
- 157.Sharma U, Bhagwat VK. Asian J. Chem. 1992;4:758–763. [Google Scholar]
- 158.Pyati R, Murray RW. J. Am. Chem. Soc. 1996;118:1743–1749. [Google Scholar]
- 159.Teeters D, Neuman RG, Tate BD. Solid State Ionics. 1996;85:239–245. [Google Scholar]
- 160.Choquette Y, Brisard G, Parent M, Brouillette D, Perron G, Desnoyers JE, Armand M, Gravel D, Slougui N. J. Electrochem. Soc. 1998;145:3500–3507. [Google Scholar]
- 161.Plewa A, Chyliński F, Kalita M, Bukat M, Parzuchowski P, Borkowska R, Siekierski M, Żukowska GZ, Wieczorek W. J. Power Sources. 2006;159:431–437. [Google Scholar]
- 162.Katayama Y, Miyashita S, Miura T. J. Power Sources. 2010;195:6162–6166. [Google Scholar]
- 163.Izutsu K, Nakamura T, Miyoshi K, Kurita K. Electrochimica Acta. 1996;41:2523–2527. [Google Scholar]
- 164.Aurbach D, Granot E. Electrochimica Acta. 1997;42:697–718. [Google Scholar]
- 165.Brouillette D, Perron G, Desnoyers JE. J. Solution Chem. 1998;27:151–182. [Google Scholar]
- 166.Hayamizu K, Aihara Y, Arai S, Martinez CG. J. Phys. Chem. B. 1999;103:519–524. doi: 10.1021/jp9825664. [DOI] [PubMed] [Google Scholar]
- 167.Hayamizu K, Akiba E, Bando T, Aihara Y. J. Chem. Phys. 2002;117:5929–5939. [Google Scholar]
- 168.Henderson WA, McKenna F, Khan MA, Brooks NR, Young VGJ, Frech R. Chem. Mater. 2005;17:2284–2289. [Google Scholar]
- 169.Kolosnitsyn VS, Karaseva EV, Seung DY, Cho MD. Russ. J. Electrochem. 2003;39:1089–1093. [Google Scholar]
- 170.Kolosnitsyn VS, Karaseva EV, Shakirova NV, Seung DY, Cho MD. Russ. J. Electrochem. 2002;38:1360–1363. [Google Scholar]
- 171.Tobishima S, Morimoto H, Aoki M, Saito Y, Inose T, Fukumoto T, Kuryu T. Electrochimica Acta. 2004;49:979–987. [Google Scholar]
- 172.Inose T, Watanabe D, Morimoto H, Tobishima S. J. Power Sources. 2006;162:1297–1303. [Google Scholar]
- 173.Kaulgud TV, Dhumal NR, Gejji SP. J. Phys. Chem. A. 2006;110:9231–9239. doi: 10.1021/jp062431v. [DOI] [PubMed] [Google Scholar]
- 174.Yoshida K, Tsuchiya M, Tachikawa N, Dokko K, Watanabe M. J. Phys. Chem. C. 2011;115:18384–18394. [Google Scholar]
- 175.Orita A, Kamijima K, Yoshida M, Dokko K, Watanabe M. J. Power Sources. 2011;196:3874–3880. [Google Scholar]
- 176.Tamura T, Hachida T, Yoshida K, Tachikawa N, Dokko K, Watanabe M. J. Power Sources. 2010;195:6095–6100. [Google Scholar]
- 177.Seki S, Takei K, Miyashiro H, Watanabe M. J. Electrochem. Soc. 2011;158:A769–A774. [Google Scholar]
- 178.Seki S, Serizawa N, Takei K, Dokko K, Watanabe M. J. Power Sources. 2013;243:323–327. [Google Scholar]
- 179.Ellis BL, Lee KT, Nazar LF. Chem. Mater. 2010;22:691–714. [Google Scholar]
- 180.Tachikawa N, Yamauchi K, Takashima E, Park J-W, Dokko K, Watanabe M. Chem. Commun. 2011;47:8157–8159. doi: 10.1039/c1cc12415c. [DOI] [PubMed] [Google Scholar]
- 181.Barchasz C, Leprêtre J-C, Patoux S, Alloin F. Electrochimica Acta. 2013;89:737–743. [Google Scholar]
- 182.Aurbach D, Gofer Y, Lu Z, Schechter A, Chusid O, Gizbar H, Cohen Y, Ashkenazi V, Moshkovich M, Turgeman R, Levi E. J. Power Sources. 2001;97–98:28–32. [Google Scholar]
- 183.Aurbach D, Gizbar H, Schechter A, Chusid O, Gottlieb HE, Gofer Y, Goldberg I. J. Electrochem. Soc. 2002;149:A115–A121. [Google Scholar]
- 184.Aurbach D, Weissman I, Gofer Y, Levi E. The Chemical Record. 2003;3:61–73. doi: 10.1002/tcr.10051. [DOI] [PubMed] [Google Scholar]
- 185.Rao BML, Klemann LP. J. Electrochem. Soc. 1980;127:761–762. [Google Scholar]
- 186.Zhang C, Andreev YG, Bruce PG. Angewandte Chemie International Edition. 2007;46:2848–2850. doi: 10.1002/anie.200604934. [DOI] [PubMed] [Google Scholar]
- 187.Zhang C, Staunton E, Andreev YG, Bruce PG. J. Mater. Chem. 2007;17:3222–3228. [Google Scholar]
- 188.Zhang C, Ainsworth D, Andreev YG, Bruce PG. J. Am. Chem. Soc. 2007;129:8700–8701. doi: 10.1021/ja073145f. [DOI] [PubMed] [Google Scholar]
- 189.Zhang C, Lilley SJ, Ainsworth D, Staunton E, Andreev YG, Slawin AMZ, Bruce PG. Chem. Mater. 2008;20:4039–4044. [Google Scholar]
- 190.Xia DW, Smid J. Journal of Polymer Science: Polymer Letters Edition. 1984;22:617–621. [Google Scholar]
- 191.Holzer L, Wenzl FP, Tasch S, Leising G, Winkler B, Dai L, Mau AWH. Appl. Phys. Lett. 1999;75:2014–2016. [Google Scholar]
- 192.McConaghy JSJ, Bloomfield JJ. J. Org. Chem. 1968;33:3425–3428. [Google Scholar]
- 193.Rieke RD, Hudnall PM. J. Am. Chem. Soc. 1969;91:3678–3679. [Google Scholar]
- 194.Moshuk G, Petrowski G, Winstein S. J. Am. Chem. Soc. 1968;90:2179–2181. [Google Scholar]
- 195.Rieke R, Ogliaruso M, McClung R, Winstein S. J. Am. Chem. Soc. 1966;88:4729–4730. [Google Scholar]
- 196.Miller LL, Jacoby LJ. J. Am. Chem. Soc. 1969;91:1130–1134. [Google Scholar]
- 197.Walborsky HM, Aronoff MS, Schulman MF. J. Org. Chem. 1971;36:1036–1040. [Google Scholar]
- 198.Wei L, Bell A, Ahn KH, Holl MM, Warner S, Williams ID, Lippard SJ. Inorg. Chem. 1990;29:825–837. [Google Scholar]
- 199.Wei L, Bell A, Warner S, Williams ID, Lippard SJ. J. Am. Chem. Soc. 1986;108:8302–8303. [Google Scholar]
- 200.Riesner H, Winterfeldt E. J. Chem. Soc., Chem. Commun. 1972:786–787. [Google Scholar]
- 201.Dahl AR, Heil CA, Norman AD. Inorg. Chem. 1975;14:1095–1098. [Google Scholar]
- 202.Saavedra JE. J. Org. Chem. 1979;44:860–861. [Google Scholar]
- 203.Ohsawa T, Takagaki T, Ikehara F, Takahashi Y, Oishi T. Chem. Pharm. Bull. 1982;30:3178–3186. [Google Scholar]
- 204.Kavaliunas AV, Taylor A, Rieke RD. Organometallics. 1983;2:377–383. [Google Scholar]
- 205.Rochfort GL, Rieke RD. Inorg. Chem. 1986;25:348–355. [Google Scholar]
- 206.Inaba S, Matsumoto H, Rieke RD. J. Org. Chem. 1984;49:2093–2098. [Google Scholar]
- 207.Inaba S, Rieke RD. J. Org. Chem. 1985;50:1373–1381. [Google Scholar]
- 208.King GT, Miller NE. Inorg. Chem. 1986;25:4309–4311. [Google Scholar]
- 209.Bianconi PA, Vrtis RN, Rao CP, Williams ID, Engeler MP, Lippard SJ. Organometallics. 1987;6:1968–1977. [Google Scholar]
- 210.Fanwick PE, Root DR, Walton RA. Inorg. Chem. 1989;28:395–397. [Google Scholar]
- 211.Fanwick PE, Root DR, Walton RA. Inorg. Chem. 1989;28:3203–3209. [Google Scholar]
- 212.Yang C, Pittman CUJ. Tetrahedron Lett. 1997;38:6561–6564. [Google Scholar]
- 213.Yang C, Pittman CUJ. Synthetic Communications. 1998;28:517–525. [Google Scholar]
- 214.Pittman CUJ, Yang C. Journal of Hazardous Materials. 2001;82:299–311. doi: 10.1016/s0304-3894(01)00175-3. [DOI] [PubMed] [Google Scholar]
- 215.Kanth JVB, Brown HC. Inorg. Chem. 2000;39:1795–1802. doi: 10.1021/ic0000911. [DOI] [PubMed] [Google Scholar]
- 216.Ouellette RJ, Levin C. J. Am. Chem. Soc. 1971;93:471–476. [Google Scholar]
- 217.Ochiai M, Fujita E. J. Chem. Soc., Chem. Commun. 1975:967–968. [Google Scholar]
- 218.Fujita E, Ochiai M. J. Chem. Soc., Perkin Trans. 1. 1977:1948–1953. [Google Scholar]
- 219.McKillop A, Swann BP, Ford ME, Taylor EC. J. Am. Chem. Soc. 1973;95:3641–3645. [Google Scholar]
- 220.McKillop A, Oldenziel OH, Swann BP, Taylor EC, Robey RL. J. Am. Chem. Soc. 1973;95:1296–1301. [Google Scholar]
- 221.Stork G, Hudrlik PF. J. Am. Chem. Soc. 1968;90:4464–4465. [Google Scholar]
- 222.Stork G, Hudrlik PF. J. Am. Chem. Soc. 1968;90:4462–4464. [Google Scholar]
- 223.Stevens RV, DuPree LE. J. Chem. Soc. D: Chem. Commun. 1970:1585–1586. [Google Scholar]
- 224.Stevens RV, Wentland MP. J. Am. Chem. Soc. 1968;90:5580–5583. doi: 10.1021/ja01022a048. [DOI] [PubMed] [Google Scholar]
- 225.Kice JL, Kasperek GJ. J. Am. Chem. Soc. 1970;92:3393–3397. [Google Scholar]
- 226.Kice JL, Walters CA, Burton SB. J. Org. Chem. 1974;39:346–351. [Google Scholar]
- 227.Reichle WT. J. Org. Chem. 1972;37:4254–4257. [Google Scholar]
- 228.Pastor SD, Hessell ET. J. Org. Chem. 1985;50:4812–4815. [Google Scholar]
- 229.Chen KS, Kleinberg J, Landgrebe JA. Inorg. Chem. 1973;12:2826–2828. [Google Scholar]
- 230.White J, McGillivray G. J. Org. Chem. 1974;39:1973–1974. [Google Scholar]
- 231.Brown NC, Gambino J, Wright GE. J. Med. Chem. 1977;20:1186–1189. doi: 10.1021/jm00219a015. [DOI] [PubMed] [Google Scholar]
- 232.Izumi T, Miller SI. J. Org. Chem. 1978;43:871–875. [Google Scholar]
- 233.Banitt EH, Conard GJ. Journal of Labelled Compounds and Radiopharmaceuticals. 1981;18:713–720. [Google Scholar]
- 234.Okamoto A, Hayashi T, Mita I. Polymer Journal. 1983;Vol.15(No.6 (1983)):423–427. [Google Scholar]
- 235.Stephens CL, Nyquist HL, Hardcastle KI. J. Org. Chem. 2002;67:3051–3056. doi: 10.1021/jo0111412. [DOI] [PubMed] [Google Scholar]
- 236.Zhu Q, Wu J, Fathi R, Yang Z. Org. Lett. 2002;4:3333–3336. doi: 10.1021/ol020159b. [DOI] [PubMed] [Google Scholar]
- 237.Kim S, Yoo BK, Chun K, Kang W, Choo J, Gong M-S, Joo S-W. J. Mol. Catal. A: Chem. 2005;226:231–234. [Google Scholar]
- 238.Smith SW, Fu GC. J. Am. Chem. Soc. 2008;130:12645–12647. doi: 10.1021/ja805165y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Smith SW, Fu GC. Angewandte Chemie International Edition. 2008;47:9334–9336. doi: 10.1002/anie.200802784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Fitt JJ, Gschwend HW. J. Org. Chem. 1984;49:209–210. doi: 10.1021/jo00435a027. [DOI] [PubMed] [Google Scholar]
- 241.Schomaker JM, Delia TJ. J. Org. Chem. 2001;66:7125–7128. doi: 10.1021/jo010573+. [DOI] [PubMed] [Google Scholar]
- 242.Delia TJ, Schomaker JM, Kalinda AS. Journal of Heterocyclic Chemistry. 2006;43:127–131. [Google Scholar]
- 243.Geanangel RA, Shore SG. J. Am. Chem. Soc. 1967;89:6771–6772. [Google Scholar]
- 244.Hosmane NS, Wermer JR, Hong Z, Getman TD, Shore SG. Inorg. Chem. 1987;26:3638–3639. [Google Scholar]
- 245.Lawrence SH, Wermer JR, Boocock SK, Banks MA, Keller PC, Shore SG. Inorg. Chem. 1986;25:367–372. [Google Scholar]
- 246.Pelter A, Levitt T, Smith K. J. Chem. Soc. D: Chem. Commun. 1969:435–436. [Google Scholar]
- 247.Peacock LA, Geanangel RA. Inorg. Chem. 1976;15:244–246. [Google Scholar]
- 248.Leyden RN, Sullivan BP, Baker RT, Hawthorne MF. J. Am. Chem. Soc. 1978;100:3758–3765. [Google Scholar]
- 249.Wermer JR, Shore SG. Inorg. Chem. 1987;26:1644–1645. [Google Scholar]
- 250.Getman TD, Krause JA, Niedenzu PM, Shore SG. Inorg. Chem. 1989;28:1507–1510. [Google Scholar]
- 251.Kang C-H, Kim S-J, Ko J-J, Lee K-B, Kang SO. Bull. Korean Chem. Soc. 1993;14:537–539. [Google Scholar]
- 252.Holub J, Ormsby DL, Kennedy JD, Greatrex R, Štíbr B. Inorganic Chemistry Communications. 2000;3:178–181. [Google Scholar]
- 253.Rosen S, Sworm D. Anal. Chem. 1966;38:1392–1397. [Google Scholar]
- 254.Gassman PG, Lumb JT, Zalar FV. J. Am. Chem. Soc. 1967;89:946–952. [Google Scholar]
- 255.Finucane BW, Thomson JB. J. Chem. Soc. D: Chem. Commun. 1969:1220–1220. [Google Scholar]
- 256.Lateef AB, Reeder JA, Rand L. J. Org. Chem. 1971;36:2295–2298. [Google Scholar]
- 257.Karpetsky TP, White EH. J. Org. Chem. 1972;37:339–341. [Google Scholar]
- 258.Pittman CUJ, Grube PL, Ayers OE, McManus SP, Rausch MD, Moser GA. Journal of Polymer Science Part A-1: Polymer Chemistry. 1972;10:379–386. [Google Scholar]
- 259.Stork G, MacDonald TL. J. Am. Chem. Soc. 1975;97:1264–1265. [Google Scholar]
- 260.Wilt JW, Rasmussen RR. J. Org. Chem. 1975;40:1031–1036. [Google Scholar]
- 261.Stang PJ, Mangum MG. J. Am. Chem. Soc. 1975;97:3854–3856. [Google Scholar]
- 262.Stang PJ, Mangum MG. J. Am. Chem. Soc. 1977;99:2597–2601. [Google Scholar]
- 263.Vidal JL, Fiato RA, Cosby LA, Pruett RL. Inorg. Chem. 1978;17:2574–2582. [Google Scholar]
- 264.Vidal JL. Inorg. Chem. 1981;20:243–249. [Google Scholar]
- 265.Collins CJ, Hombach HP, Maxwell B, Woody MC, Benjamin BM. J. Am. Chem. Soc. 1980;102:851–853. [Google Scholar]
- 266.Ladika M, Stang PJ. J. Chem. Soc., Chem. Commun. 1981:459–460. [Google Scholar]
- 267.Paxson TE, Reilly CA, Holecek DR. J. Chem. Soc., Chem. Commun. 1981:618–619. [Google Scholar]
- 268.van Tamelen EE, Seeley DA. J. Am. Chem. Soc. 1969;91:5194–5194. [Google Scholar]
- 269.Pez GP, Apgar P, Crissey RK. J. Am. Chem. Soc. 1982;104:482–490. [Google Scholar]
- 270.Budt K-H, Vatele J-M, Kishi Y. J. Am. Chem. Soc. 1986;108:6080–6082. doi: 10.1021/ja00279a089. [DOI] [PubMed] [Google Scholar]
- 271.Hayward C-MT, Shapley JR. Organometallics. 1988;7:448–452. [Google Scholar]
- 272.Tee OS, Enos JA. Can. J. Chem. 1988;66:3027–3030. [Google Scholar]
- 273.Briggs JR. J. Chem. Soc., Chem. Commun. 1989:674–675. [Google Scholar]
- 274.Maringgele W, Seebold U, Heine A, Stalke D, Noltemeyer M, Sheldrick GM, Meller A. Organometallics. 1991;10:2097–2098. [Google Scholar]
- 275.Hung M-H, Farnham WB, Feiring AE, Rozen S. J. Am. Chem. Soc. 1993;115:8954–8959. [Google Scholar]
- 276.Hays ML, Hanusa TP. Tetrahedron Lett. 1995;36:2435–2436. [Google Scholar]
- 277.Kirij NV, Yagupolskii YL, Maggiarosa N, Tyrra W, Naumann D. Journal of Fluorine Chemistry. 2001;112:213–218. [Google Scholar]
- 278.Yagupolskii LM, Shelyazhenko SV, Maletina II, Petrik VN, Rusanov EB, Chernega AN. Eur. J. Org. Chem. 2001;2001:1225–1233. [Google Scholar]
- 279.Israelsohn O, Vollhardt KPC, Blum J. J. Mol. Catal. A: Chem. 2002;184:1–10. [Google Scholar]
- 280.Tyrra W, Naumann D, Hoge B, Yagupolskii YL. Journal of Fluorine Chemistry. 2003;119:101–107. [Google Scholar]
- 281.Tyrra W, Kirij NV, Naumann D, Yagupolskii YL. Journal of Fluorine Chemistry. 2004;125:1437–1440. [Google Scholar]
- 282.Zook H, Miller J. J. Org. Chem. 1971;36:1112–1116. [Google Scholar]
- 283.Shinohara M, Smid J, Szwarc M. J. Am. Chem. Soc. 1968;90:2175–2177. [Google Scholar]
- 284.Guerra MA, Lagow RJ. J. Chem. Soc., Chem. Commun. 1990:65–66. [Google Scholar]
- 285.Liang L, Ying S. Makromol. Chem. 1993;194:581–600. [Google Scholar]
- 286.Wang JS, Jerome R, Warin R, Zhang H, Teyssie P. Macromolecules. 1994;27:3376–3382. [Google Scholar]
- 287.Krause LJ, Morrison JA. J. Chem. Soc., Chem. Commun. 1980:671–672. [Google Scholar]
- 288.Krause LJ, Morrison JA. J. Am. Chem. Soc. 1981;103:2995–3001. [Google Scholar]
- 289.Krause LJ, Morrison JA. J. Chem. Soc., Chem. Commun. 1981:1282–1283. [Google Scholar]
- 290.Ontiveros CD, Morrison JA. Organometallics. 1986;5:1446–1448. [Google Scholar]
- 291.Galiotos JK, Morrison JA. Organometallics. 2000;19:2603–2607. [Google Scholar]
- 292.Murray HH, Fackler JPJ, Porter LC, Briggs DA, Guerra MA, Lagow RJ. Inorg. Chem. 1987;26:357–363. [Google Scholar]
- 293.Nair HK, Morrison JA. Inorg. Chem. 1989;28:2816–2820. [Google Scholar]
- 294.Loizou DC, Castillo J, Oki AR, Hosmane NS, Morrison JA. Organometallics. 1992;11:4189–4193. [Google Scholar]
- 295.Ludovici K, Naumann D, Siegemund G, Tyrra W, Varbelow H-G, Wrubel H. Journal of Fluorine Chemistry. 1995;73:273–274. [Google Scholar]
- 296.Daniele S, Hubert-Pfalzgraf LG, Vaissermann J. Polyhedron. 2003;22:127–132. [Google Scholar]
- 297.Sukata K. Bull. Chem. Soc. Jpn. 1984;57:613–614. [Google Scholar]
- 298.Lee DG, Chang VS. J. Org. Chem. 1978;43:1532–1536. [Google Scholar]
- 299.Paradisi C, Quintily U, Scorrano G. J. Org. Chem. 1983;48:3022–3026. [Google Scholar]
- 300.Bergbreiter DE, Blanton JR. J. Org. Chem. 1987;52:472–473. [Google Scholar]
- 301.Sukata K, Akagawa T. J. Org. Chem. 1989;54:1476–1479. [Google Scholar]
- 302.Landini D, Maia A, Corda L, Maccioni A, Podda G. Tetrahedron Lett. 1989;30:5781–5784. [Google Scholar]
- 303.Landini D, Maia A, Corda L, Maccioni A, Podda G. Tetrahedron Lett. 1991;47:7477–7488. [Google Scholar]
- 304.Varnek AA, Maia A, Landini D, Gamba A, Morosi G, Podda G. J. Phys. Org. Chem. 1993;6:113–121. [Google Scholar]
- 305.Lazrek HB, Taourirte M, Barascut J-L, Imbach J-L. Nucleosides and Nucleotides. 1991;10:1285–1293. doi: 10.1080/15257779908041535. [DOI] [PubMed] [Google Scholar]
- 306.Kudryavtsev IY, Zakharov LS. Russian Chemical Bulletin. 2001;50:1457–1460. [Google Scholar]
- 307.Kochergina TY, Anufriev VF. Russian Journal of Organic Chemistry. 2002;38:534–537. [Google Scholar]
- 308.Koh HJ, Han KL, Lee I. J. Org. Chem. 1999;64:4783–4789. doi: 10.1021/jo990115p. [DOI] [PubMed] [Google Scholar]
- 309.Hogan JC, Gandour RD. J. Am. Chem. Soc. 1980;102:2865–2866. [Google Scholar]
- 310.Hogan JC, Gandour RD. J. Org. Chem. 1991;56:2821–2826. [Google Scholar]
- 311.Hogan JC, Gandour RD. J. Org. Chem. 1992;57:55–61. [Google Scholar]
- 312.Basilio N, García-Río L, Leis JR, Mejuto JC, Pérez-Lorenzo M. Chem. Commun. 2005:3817–3819. doi: 10.1039/b504629g. [DOI] [PubMed] [Google Scholar]
- 313.Basilio N, García-Río L, Mejuto JC, Pérez-Lorenzo M. J. Org. Chem. 2006;71:4280–4285. doi: 10.1021/jo060389u. [DOI] [PubMed] [Google Scholar]
- 314.García-Río L, Mejuto JC, Pérez-Lorenzo M. J. Phys. Chem. B. 2007;111:11149–11156. doi: 10.1021/jp0743323. [DOI] [PubMed] [Google Scholar]
- 315.Dai X, Strotman NA, Fu GC. J. Am. Chem. Soc. 2008;130:3302–3303. doi: 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]
- 316.Bartsch RA, Yang IW. Tetrahedron Lett. 1979;20:2503–2504. [Google Scholar]
- 317.Kimura Y, Regen SL. J. Org. Chem. 1982;47:2493–2494. [Google Scholar]
- 318.Newman MS, Liang WC. J. Org. Chem. 1973;38:2438–2441. [Google Scholar]
- 319.Ishii H, Hou Y, Fuchigami T. Tetrahedron. 2000;56:8877–8881. [Google Scholar]
- 320.Grabovskiy SA, Timerghazin QK, Kabal’nova NN. Russian Chemical Bulletin. 2005;54:2384–2393. [Google Scholar]
- 321.Yoon CW, Carroll PJ, Sneddon LG. J. Am. Chem. Soc. 2009;131:855–864. doi: 10.1021/ja808045p. [DOI] [PubMed] [Google Scholar]
- 322.Yoshpe-Besancon I, Auriol D, Paul F, Monsan P, Gripon JC, Ribadeau-Dumas B. Biotechnol. Appl. Biochem. 1993;18:93–102. [PubMed] [Google Scholar]
- 323.Rosell CM, Terreni M, Fernandez-Lafuente R, Guisan JM. Enzyme Microb. Technol. 1998;23:64–69. [Google Scholar]
- 324.Berkowitz DB, Hartung RE, Choi S. Tetrahedron: Asymmetry. 1999;10:4513–4520. [Google Scholar]
- 325.Schroën CGPH, Nierstrasz VA, Kroon PJ, Bosma R, Janssen AEM, Beeftink HH, Tramper J. Enzyme Microb. Technol. 1999;24:498–506. [Google Scholar]
- 326.Illanes A, Fajardo A. J. Mol. Catal. B: Enzym. 2001;11:587–595. [Google Scholar]
- 327.Ohno H, Yamaguchi N. Bioconjugate Chem. 1994;5:379–381. doi: 10.1021/bc00029a001. [DOI] [PubMed] [Google Scholar]
- 328.Kawahara NY, Ohno H. Bioconjugate Chem. 1997;8:643–648. doi: 10.1021/bc9701196. [DOI] [PubMed] [Google Scholar]
- 329.Kurusu F, Ohno H. Electrochimica Acta. 2000;45:2911–2915. [Google Scholar]
- 330.Chen J, Spear SK, Huddleston JG, Rogers RD. Green Chem. 2005;7:64–82. [Google Scholar]
- 331.Cao L, van Rantwijk F, Sheldon RA. Organic Letters. 2000;2:1361–1364. doi: 10.1021/ol005593x. [DOI] [PubMed] [Google Scholar]
- 332.Taylor BR, Kauzlarich SM. Chem. Mater. 1998;10:22–24. [Google Scholar]
- 333.Taylor BR, Kauzlarich SM. Chem. Mater. 1999;11:2493–2500. [Google Scholar]
- 334.Taylor BR, Fox GA, Hope-Weeks LJ, Maxwell RS, Kauzlarich SM, Lee HWH. Materials Science and Engineering: B. 2002;96:90–93. [Google Scholar]
- 335.Yang C-S, Bley RA, Kauzlarich SM, Lee HWH, Delgado GR. J. Am. Chem. Soc. 1999;121:5191–5195. [Google Scholar]
- 336.Yang C-S, Liu Q, Kauzlarich SM. Chem. Mater. 2000;12:983–988. [Google Scholar]
- 337.Johnston EE, Bryers JD, Ratner BD. Langmuir. 2005;21:870–881. doi: 10.1021/la036274s. [DOI] [PubMed] [Google Scholar]
- 338.Brétagnol F, Ceriotti L, Lejeune M, Papadopoulou-Bouraoui A, Hasiwa M, Gilliland D, Ceccone G, Colpo P, Rossi F. Plasma Processes and Polymers. 2006;3:30–38. [Google Scholar]
- 339.Brétagnol F, Lejeune M, Papadopoulou-Bouraoui A, Hasiwa M, Rauscher H, Ceccone G, Colpo P, Rossi F. Acta Biomaterialia. 2006;2:165–172. doi: 10.1016/j.actbio.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 340.Brétagnol F, Sirghi L, Mornet S, Sasaki T, Gilliland D, Colpo P, Rossi F. Nanotechnology. 2008;19:125306. doi: 10.1088/0957-4484/19/12/125306. [DOI] [PubMed] [Google Scholar]
- 341.Yang C-S, Awschalom DD, Stucky GD. Chem. Mater. 2001;13:594–598. [Google Scholar]
- 342.Yang C-S, Awschalom DD, Stucky GD. Chem. Mater. 2002;14:1277–1284. [Google Scholar]
- 343.Chiu HW, Kauzlarich SM. Chem. Mater. 2006;18:1023–1028. [Google Scholar]
- 344.Pickering AL, Mitterbauer C, Browning ND, Kauzlarich SM, Power PP. Chem. Commun. 2007:580–582. doi: 10.1039/b614363f. [DOI] [PubMed] [Google Scholar]
- 345.Cho J. Electrochimica Acta. 2008;54:461–466. [Google Scholar]
- 346.Shirahata N, Sakka Y. J. Ceram. Soc. Jpn. 2010;118:932–939. [Google Scholar]
- 347.Mishra S, Ledoux G, Jeanneau E, Daniele S, Joubert M-F. Dalton Trans. 2012;41:1490–1502. doi: 10.1039/c1dt11070e. [DOI] [PubMed] [Google Scholar]
- 348.Shirai M, Ishida H, Tanaka M. Journal of Polymer Science: Part B: Polymer Physics. 1988;26:2075–2091. [Google Scholar]
- 349.Morgado J, Cacialli F, Friend RH, Chuah BS, Rost H, Moratti SC, Holmes AB. Synthetic Metals. 2001;119:595–596. [Google Scholar]
- 350.Moore JA, Kim J-H, Seidel PR. Chem. Mater. 1991;3:742–745. [Google Scholar]
- 351.Fazen PJ, Remsen EE, Beck JS, Carroll PJ, McGhie AR, Sneddon LG. Chem. Mater. 1995;7:1942–1956. [Google Scholar]
- 352.Fazen PJ, Beck JS, Lynch AT, Remsen EE, Sneddon LG. Chem. Mater. 1990;2:96–97. [Google Scholar]
- 353.Moraes K, Vosburg J, Wark D, Interrante LV. Chem. Mater. 2004;16:125–132. [Google Scholar]
- 354.López GP, Ratner BD. Plasmas and Polymers. 1996;1:127–151. [Google Scholar]
- 355.Sandner B, Kotzian N, Tubke J, Wartewig S, Lange O. Macromol. Chem. Phys. 1997;198:2715–2727. [Google Scholar]
- 356.Baker RT, Krusic PJ, Tulip TH, Calabrese JC, Wreford SS. J. Am. Chem. Soc. 1983;105:6763–6765. [Google Scholar]
- 357.Baxter I, Drake SR, Hursthouse MB, Malik KMA, McAleese J, Otway DJ, Plakatouras JC. Inorg. Chem. 1995;34:1384–1394. [Google Scholar]
- 358.Drake SR, Miller SAS, Williams DJ. Inorg. Chem. 1993;32:3227–3235. [Google Scholar]
- 359.Drake SR, Lyons A, Otway DJ, Slawin AMZ, Williams DJ. J. Chem. Soc., Dalton Trans. 1993:2379–2386. [Google Scholar]
- 360.Arunasalam V-C, Baxter I, Drake SR, Hursthouse MB, Malik KMA, Miller SAS, Mingos DMP, Otway DJ. J. Chem. Soc., Dalton Trans. 1997:1331–1336. [Google Scholar]
- 361.Arnáiz FJ, Aguado R, Pedrosa MR, Mahía J, Maestro MA. Polyhedron. 2001;20:2781–2785. [Google Scholar]
- 362.Crochet A, Fromm KM. Zeitschrift für anorganische und allgemeine Chemie. 2010;636:1484–1496. [Google Scholar]
- 363.Malandrino G, Licata R, Castelli F, Fragalà IL, Benelli C. Inorg. Chem. 1995;34:6233–6234. [Google Scholar]
- 364.Malandrino G, Benelli C, Castelli F, Fragalà IL. Chem. Mater. 1998;10:3434–3444. [Google Scholar]
- 365.Pollard KD, Vittal JJ, Yap GPA, Puddephatt RJ. J. Chem. Soc., Dalton Trans. 1998:1265–1268. [Google Scholar]
- 366.Malandrino G, Lo Nigro R, Fragalà IL, Benelli C. Eur. J. Inorg. Chem. 2004;2004:500–509. [Google Scholar]
- 367.Kang S-J, Jung YS, Suh I-H. Bull. Korean Chem. Soc. 1999;20:95–98. [Google Scholar]
- 368.Pollard KD, Jenkins HA, Puddephatt RJ. Chem. Mater. 2000;12:701–710. [Google Scholar]
- 369.Malandrino G, Lo Nigro R, Benelli C, Castelli F, Fragalà IL. Chem. Vap. Deposition. 2000;6:233–238. [Google Scholar]
- 370.Malandrino G, Bettinelli M, Speghini A, Fragalà IL. Eur. J. Inorg. Chem. 2001;2001:1039–1044. [Google Scholar]
- 371.Condorelli GG, Gennaro S, Fragalà IL. Chem. Vap. Deposition. 2000;6:185–192. [Google Scholar]
- 372.Lo Nigro R, Toro RG, Fragalà ME, Rossi P, Dapporto P, Malandrino G. Inorganica Chimica Acta. 2009;362:4623–4629. [Google Scholar]
- 373.Blake AJ, Darr JA, Howdle SM, Poliakoff M, Li W-S, Webb PB. Journal of Chemical Crystallography. 1999;29:547–554. [Google Scholar]
- 374.Pfeiffer D, Heeg MJ, Winter CH. Inorg. Chem. 2000;39:2377–2384. doi: 10.1021/ic991049c. [DOI] [PubMed] [Google Scholar]
- 375.Buncel E, Dust JM, Jonczyk A, Manderville RA, Onyido I. J. Am. Chem. Soc. 1992;114:5610–5619. [Google Scholar]
- 376.Buncel E, Manderville RA. J. Phys. Org. Chem. 1993;6:71–82. [Google Scholar]
- 377.Manderville RA, Buncel E. J. Am. Chem. Soc. 1993;115:8985–8989. [Google Scholar]
- 378.Manderville RA, Buncel E. J. Chem. Soc., Perkin Trans. 2. 1993:1887–1894. [Google Scholar]
- 379.Rouse CA, Finlinson AC, Tarbet BJ, Pixton JC, Djordjevic NM, Markides KE, Bradshaw JS, Lee ML. Anal. Chem. 1988;60:901–905. [Google Scholar]
- 380.Schuetz AJ, Weller MG, Niessner R. Fresenius J. Anal. Chem. 1999;363:777–782. [Google Scholar]
- 381.Sciamanna SF, Lynn S. Ind. Eng. Chem. Res. 1988;27:492–499. [Google Scholar]
- 382.Henni A, Tontiwachwuthikul P, Chakma A. The Canadian Journal of Chemical Engineering. 2005;83:358–361. [Google Scholar]
- 383.Mishmash HE, Meloan CE. Anal. Chem. 1972;44:835–836. doi: 10.1021/ac60312a049. [DOI] [PubMed] [Google Scholar]
- 384.Dasilva-Carbalhal J, García-Río L, Gómez-Díaz D, Mejuto JC, Pérez-Lorenzo M. J. Colloid Interface Sci. 2005;292:591–594. doi: 10.1016/j.jcis.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 385.Gu Y, Jérôme F. Green Chem. 2010;12:1127–1138. [Google Scholar]
- 386.Wolfson A, Dlugy C, Shotland Y. Environ. Chem. Lett. 2007;5:67–71. [Google Scholar]
- 387.Behr A, Eilting J, Irawadi K, Leschinski J, Lindner F. Green Chem. 2008;10:13–30. [Google Scholar]
- 388.Tang S, Baker GA, Zhao H. Chem. Soc. Rev. 2012;41:4030–4066. doi: 10.1039/c2cs15362a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.García JI, García-Marín H, Mayoral JA, Pérez P. Green Chem. 2010;12:426–434. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.