

Abstract
We report a case of congenital oligomeganephronia unexpectedly imaged with computed tomography (CT). Oligomeganephronia is a form of renal hypoplasia that leads to renal failure in childhood or adolescence. If encountered, its CT features should suggest the diagnosis and prompt renal biopsy.
Key words: oligomeganephronia, renal hypoplasia, renal failure, pediatrics, computed tomography
Introduction
Congenital oligomeganephronia is a renal hypoplasia in which nephrons are congenitally deficient in number but enlarged and overworked.1 Nephron number may be reduced by up to 80% while the remaining nephrons undergo hypertrophy, with glomeruli reaching 12-15 times normal volume and tubules reaching up to 17 times normal volume.1,2 Hyperfiltration and excessive stress on the glomerular capillary wall cause progressive glomerular injury and eventual glomerulosclerosis, with renal failure typically occurring during childhood or adolescence.1-4 We provide the first description of contrast-enhanced computed tomography (CT) findings in this disease, discovered unexpectedly in a 16-year old boy with late onset of renal insufficiency.
Case Report
A 16-year old male presented with non-bilious emesis. His emesis began 5 days earlier, upon discharge from the hospital after posterior spinal fusion for scoliosis. Born in Korea, he was adopted and brought to the United States as a baby. His birth history and biological family medical history are unknown. He underwent left congenital diaphragmatic hernia repair soon after birth, orchiopexy for undescended testis at age 5 years, Ladd procedure for small bowel malrotation and obstruction at age 6 years, and additional laparotomy for small bowel volvulus with lysis of peritoneal adhesions and appendectomy at age 14 years. His medical history also included left lung hypoplasia, ostium secundum atrial septal defect (ASD), left pulmonary vein stenosis, and chronic asthma, for which he took fluticasone proprionate/salmeterol and trimethoprim/sulfamethoxazole. He had no known history of renal disease, vesicoureteral reflux, urinary tract infection (UTI), or embolic disease. He was a junior in high school, taking advanced placement courses.
The patient’s height and weight were 170 cm (between 25th and 50th percentiles for age) and 35.9 kg (below 5th percentile for age).5 He had an oral temperature of 36.1°C, blood pressure of 127/76 mmHg, and heart rate of 81 beats per minute. His precordium was hyperdynamic, and a II/VI systolic murmur was heard. He had a midline abdominal wall scar, normal bowel sounds and no rebound tenderness or guarding. Scoliosis and a healing incision from recent spine fusion were apparent. Micrognathia was also present, but no other orofacial, ocular, aural, or limb deformities were noted. Laboratory results obtained in evaluation of emesis included normal serum electrolytes, amylase and lipase.
Chest and abdomen radiographs showed mixed bubbly lucencies and curvilinear opacities in the left lower hemithorax, and the possibility of a recurrent diaphragmatic hernia with bowel migration into the chest was raised. Due to complexity of the patient’s anatomy and his past surgical history, abdominopelvic CT with oral and intravenous contrast was pursued in lieu of ultrasound for further evaluation of the diaphragm and bowel. CT revealed evidence of prior left diaphragmatic hernia repair and Ladd procedure with bowel malrotation. No recurrent diaphragmatic hernia was seen; pleuroparenchymal scarring and atelectasis at the left lung base, alternating with areas of relatively well-aerated lung, explained the plain radiographic abnormalities. There were no CT findings of bowel obstruction, mass, or inflammation. However, renal craniocaudal dimensions of 7.5 cm on the right and 8.7 cm on the left were below the 5th percentile for age.6 In addition, both kidneys were abnormal in appearance (Figure 1), providing impetus for further investigation into the patient’s renal status.
Figure 1.
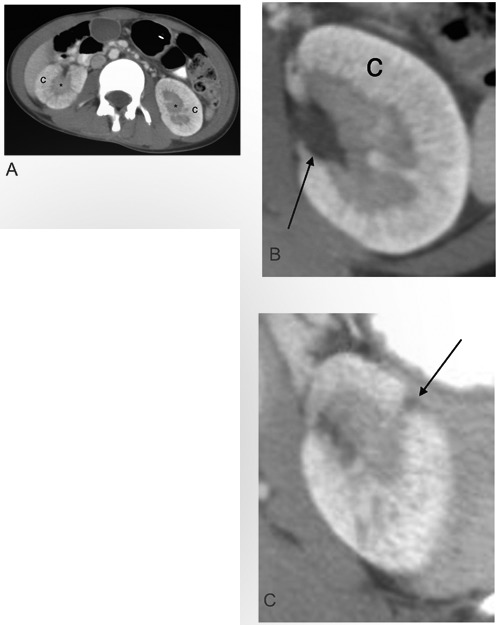
Computed tomography (CT) appearance of oligomeganephronia. A) Axial contrast-enhanced CT (CECT) of the abdomen shows thickening of bilateral renal cortex (c) and medulla (*), reflecting nephron hypertrophy. Normal conical shape of the medullary pyramids (*) is absent, and the pyramids appear confluent. B) Axial CECT image of the left kidney at the level of the renal pelvis (arrow) shows finely striated enhancement of thickened renal cortex (c), perhaps due to urine stasis and contrast hyperconcentration in hypertrophied tubules. There was no pelvocaliectasis or renal malformation. Renal veins (not shown) enhanced normally. C) One of two small cortical scars In the left kidney upper pole is shown (arrow), with volume loss and capsular retraction. Macroscopic scarring was confined to the left kidney upper pole and was not extensive enough to account for bilateral small renal size, marked nephron hypertrophy, and progressive renal failure. No underlying calyceal blunting was seen.
Serum creatinine was 1.2 mg/dL (reference range: 0.6-1.1 mg/dL), and blood urea nitrogen was 13 mg/dL (reference range: 6-20 mg/dL). Estimated creatinine clearance was 99.2 mL/min/1.73 m2 [calculated by the original Schwartz equation, before isotope dilution mass spectrometry (IDMS)-traceable standardization of creatinine calibration].7 Hemoglobin (12.7 g/dL) and hematocrit (36%) were normal. Although urine dip testing showed no protein seven days prior to the CT scan, protein was present in urine (30 mg/dL) on repeat dip testing done the day after CT scanning.
Open surgical biopsy of the right kidney was performed. The surgeon did not report any visible scarring on the exposed surface of the kidney. Histopathological examination of the biopsy specimen revealed findings consistent with oligomeganephronia, namely normal parenchymal organization but hypertrophy of glomeruli and tubules (Figure 2). There were very few globally sclerotic glomeruli, but many tufts contained segmental zones of perihilar sclerosis. These changes were accompanied by streaks of fibrosis, tubular atrophy, and chronic inflammation. There was no evidence of renal dysplasia, medullary nephronophthisismedullary cystic disease complex or polycystic kidney disease. Electron microscopy demonstrated minimal foot process effacement and was negative for glomerular basement membrane changes of hereditary nephritis. Liver biopsy, performed in conjunction with renal biopsy, was normal.
Figure 2.
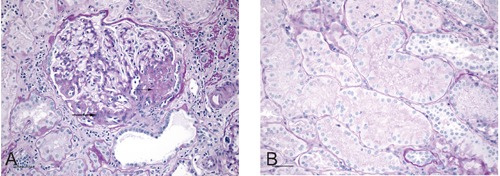
Histopathology of oligomeganephronia. A) Virtually all glomeruli in the sample were at least twice normal size. This glomerulus contains segmental zones of perihilar glomerulosclerosis (arrows) and a voluminous juxtaglomerular apparatus. Fibrosis, tubular atrophy and chronic inflammation are evident in parts of the adjacent parenchyma. The scale bar represents 50 µm [Periodic Acid Schiff (PAS), x200]. B) As shown here, all intact tubules in the sample were hypertrophic. Diameters of the tubules were two to three times normal. The scale bar represents 50 µm (PAS, x200).
The patient’s emesis was attributed to postoperative ileus and narcotic effect. Symptoms resolved as narcotics were weaned. With improvement in nutrition, along with sodium, potassium, and phosphorus restriction, the patient gained ten pounds in five months. Lowdose lisinopril was initiated for mild hypertension and proteinuria. In addition, vitamin D3 therapy was begun. With time, serum creatinine increased (Figure 3), and the patient became progressively fatigued. Prior to renal transplantation for renal failure at age 20 years, serum creatinine was 5.4 mg/dL and creatinine clearance was 13.6 mL/min/1.73 m2 (calculated for an adult using the IDMS-traceable modification of diet in renal disease study equation).8 Urine protein:creatinine ratio peaked at 1.9 (reference range: <0.2).
Figure 3.
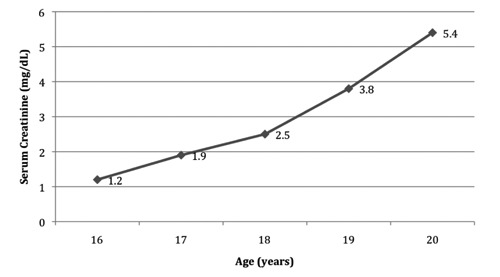
Serum creatinine. Progressive increase in serum creatinine occurred between presentation of disease at age 16 years and pre-transplantation evaluation at age 20 years.
Ultrasound of the native kidneys (Figure 4) was first performed after renal transplantation and showed small, echogenic kidneys with poor corticomedullary differentiation, in keeping with presence of chronic renal parenchymal disease. The sonographic appearance had no specific features to indicate etiology of the renal disease. No renal cysts or calcifications were seen.
Figure 4.
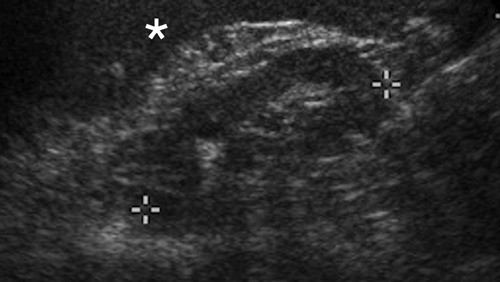
Sonographic appearance of oligomeganephronia. Left kidney (between cursors) is small (6.3 cm craniocaudal length) and isoechoic to spleen (asterisk), with poor corticomedullary differentiation, an appearance that is shared with many chronic renal diseases. Normal reniform shape is maintained. The right kidney (not shown) measured 6.1 cm in length and had a similar, non-specific sonographic appearance.
Discussion
Congenital oligomeganephronia was first described in 1962 by Royer et al.9 The hallmark of the disease is small kidneys with nephrons that are reduced in number but increased in size.1 Renal lobes and calyces may be reduced in number as well.4 Pathogenesis of oligomeganephronia is not fully understood but is believed to reflect premature interruption of nephrogenesis.2,10 Most cases are sporadic, but familial occurrence has been described.2,4 Although oligomeganephronia may be an isolated anomaly, it also occurs in genetic syndromes such as renal-coloboma syndrome (renal hypoplasia and optic nerve coloboma associated with PAX2 gene mutations on chromosome 10) and branchio-oto-renal syndrome [branchial cysts or fistulae, hearing impairment, and ear and renal anomalies associated with EYA1 (chromosome 8) and other gene mutations].4,10,11 Oligomeganephronia has also been associated with orofacial anomalies, developmental delay, seizures, and limb deformities.1,2,10,11 Concurrent congenital heart defects, such as the ASD seen in our patient, have been reported.10,11 Associated urinary tract malformations are uncommon.2,10 Interestingly, oligomegane phronia, micrognathia, congenital diaphragmatic hernia, congenital heart defects, cryptorchidism, and scoliosis are all known to occur in Wolf-Hirschhorn syndrome (4pmonosomy). 11 Although our patient had each of these abnormalities, he lacked other dysmorphic craniofacial features of Wolf-Hirschhorn syndrome (microcephaly, prominent glabella with broad nasal bridge, hypertelorism, down-slanting palpebral fissures, short philtrum, down-turned mouth, cleft lip and/or palate, and low-set dysplastic ears).11 Genetics evaluation did not yield a specific diagnosis and testing revealed normal chromosomes.
In the majority of patients, clinical manifestations develop in the first two years of life and may include anorexia, vomiting, polyuria, polydipsia, and fever.1,4 Azotemia, proteinuria, metabolic acidosis, and growth retardation are usually present.1 Anemia, renal sodium wasting and defective renal concentrating ability are also common.1 Late development of renal insufficiency in adolescence, as in our patient, is unusual.1 The natural history of oligomeganephronia is progression to renal failure.1,3 Management is supportive, with maintenance of fluid and electrolyte balance and promotion of growth.4 Treatment with angiotensin-converting-enzyme inhibitors has been shown to slow nephron damage.4 However, survival into adulthood without renal transplantation is unusual.4
If the entire oligomeganephronic kidney is available for tissue examination, an extreme paucity of nephrons may be inferred. However, in a biopsy specimen, diagnosis is made by correlating histologic findings with imaging evidence of small renal size and grossly normal renal morphology.4 Nephrons are dramatically hypertrophic, with enlargement of glomeruli, tubules, and juxtaglomerular apparatuses.2 Interstitial fibrosis generally begins at a very young age, with eventual development of focal segmental glomerulosclerosis, correlating with increasing proteinuria.2,3 In our patient, development of proteinuria within 24 h of the CT scan may indicate an element of acute contrast-induced renal injury, but gradual increase in proteinuria over subsequent years is consistent with the expected evolution of oligomeganephronia.
There have been few prior reports of imaging findings in oligomeganephronia. As in our patient, ultrasound shows symmetrically small and echogenic kidneys, with maintenance of a normal reniform shape.2,12 This appearance is shared by other chronic renal diseases.12,13 No pathognomonic sonographic features have been reported. Serial sonograms in children show growth of the kidneys over time, but the growth rate is slower than normal.2 Intravenous urography (IVU) is similarly lacking in specificity, showing symmetric, small, reniform kidneys with faint nephrograms and delayed contrast excretion.1,2,4,13 On radionuclide renal scans with iodine-131 hippuran or technetium-99m diethylenetriaminepentaacetic acid, the kidneys are small and poorly functioning, another appearance that is held in common with other chronic renal diseases.1,14 To our knowledge, the CT appearance of oligomeganephronia has not been described before. CT findings in our patient with oligomeganephronia are summarized in Table 11,12,13,15-18 along with features that differentiate oligomeganephronia from other conditions with small renal size and renal insufficiency, as well as from other causes of cortical striation on CT. Thickening of cortical and medullary layers in our patient was likely due to hypertrophy of glomeruli and tubules and may have been responsible for crowding and apparent confluence of the medullary pyramids. Because of this latter finding, the number of lobes in each kidney could not be reliably estimated. Finely striated appearance of the renal cortex may have been due to urine stasis and contrast hyperconcentration in enlarged tubules.15 CT manifestations of scarring were isolated to two small foci in the left kidney upper pole. Scarring, though common in reflux nephropathy, chronic pyelonephritis, and old renal infarction,16 has not been reported as a typical imaging finding in congenital oligomeganephronia. Rather, previous reports emphasize a smooth and non-scarred appearance of the kidneys on ultrasound, IVU, and renal scintigraphy.1-3 Etiology of renal scarring in our patient and its relationship to his oligomeganephronia are unknown. He had no known history of reflux, UTI, or embolic disorder. The interstitial fibrosis that was present on renal biopsy was diffuse, not scar-like, and was probably insufficient to cause the focal cortical volume loss and capsular retraction seen on CT. Likewise, the small amount of unilateral macroscopic scarring shown on CT in our patient was not extensive enough to explain bilateral small renal size, marked nephron hypertrophy, and progressive renal failure.
Table 1.
Computed tomography (CT) features of oligomeganephronia and differential diagnosis: i) CT findings of oligomeganephronia in our patient are summarized, along with features that may differentiate oligomeganephronia from ii) other causes of small kidneys with renal insufficiency and iii) other conditions with fine cortical striation on CT.
| i) Contrast-enhanced CT findings in patient with oligomeganephronia | |
|---|---|
| Bilaterally small renal size | |
| Cortical and medullary thickening | |
| Confluent-appearing renal pyramids with loss of normal conical shape | |
| Finely striated renal cortex | |
| Scant cortical scarring | |
| Differential diagnosis | |
| ii) Other causes of small renal size and renal insufficiency | Differentiating features |
| Diffuse chronic renal parenchymal disease1,13 | Diffuse parenchymal atrophy on pathology and imaging History and histologic features of underlying chronic disease |
| Medullary nephronophthisis-medullary cystic disease complex1,17 | Parenchymal atrophy on pathology and imaging Progressive chronic tubulointerstitial nephritis on histology ±Cysts, predominantly medullary Positive genetic testing |
| Old renal infarction or chronic ischemia1,12,16,18 | Diffuse or segmental parenchymal atrophy on pathology and imaging Interstitial fibrosis on histology ±Clinical history of embolic disease, vasculitis, or renal artery stenosis |
| Reflux nephropathy or chronic pyelonephritis1,12,15,18 | Irregular, asymmetric parenchymal atrophy with blunting of papillae and dilatation of calyces on pathology and imaging Fibrosis and chronic inflammation on histology ±Clinical history of vesicoureteral reflux or urinary tract infection |
| Renal dysplasia1,12 | Frequent urinary tract malformations Frequent renal cysts Dysplastic histologic features |
| iii) Other causes of fine cortical striations on CT16 | Differentiating features |
| Acute pyelonephritis | Large kidney(s) with areas of hypoenhancement and perinephric stranding Fever and flank pain |
| Acute ureteral obstruction | Large kidney(s) with delayed nephrogram and hydroureteronephrosis Acute flank pain and hematuria |
| Autosomal recessive polycystic kidney disease | Large kidneys Cystic dilatation of collecting tubules |
| Renal vein thrombosis | Large kidney(s) with delayed nephrogram Filling defect in enlarged renal vein |
CT, computed tomography.
Conclusions
Given that renal function is poor in oligomeganephronia, most patients with the disease will never undergo contrast-enhanced CT for evaluation. However, when disease presentation is indolent or mild, findings on CT done for other reasons may provide the first indication of renal pathology, since renal function is not universally tested in children and adolescents with no known history of renal disease. In such cases, the unusual CT appearance of oligomeganephronia should lead to renal biopsy for diagnosis. Suggestive CT findings include bilaterally small kidneys that retain their reniform shape but have thickened cortex and medulla and striated nephrograms in the absence of pelvocaliectasis and renal vein thrombosis. Because deterioration of renal function is predictable, correct diagnosis is important, as it allows appropriate monitoring for renal failure and early planning for renal transplantation.
References
- 1.Adelman RD, Shapiro S. Bilateral renal hypoplasia with oligomeganephronia. Urology 1977;9:571-5 [DOI] [PubMed] [Google Scholar]
- 2.Broyer M, Soto B, Gagnadoux MF, et al. Oligomeganephronic renal hypoplasia. Adv Nephrol 1997;26:47-63 [PubMed] [Google Scholar]
- 3.Rennke HG, Klein PS. Pathogenesis and significance of nonprimary focal and segmental glomerulosclerosis. Am J Kidney Dis 1989;13:443-56 [DOI] [PubMed] [Google Scholar]
- 4.Niaudet P. Renal hypoplasia. In: Mattoo TK, Kim MS. UpToDate®. Philadelphia, PA: Wolters Kluwer Health; 2011. Available from: http://www.uptodate.comAccessed: 13 Sept 2012 [Google Scholar]
- 5.CDC growth charts: United States: staturefor-age percentiles: boys, 2 to 20 years. Atlanta, GA: Centers for Disease Control; 2000. Available from: http://www.cdc.gov/growthchartsAccessed: 29 Aug 2012 [Google Scholar]
- 6.Han BK, Babcock DS. Sonographic measurements and appearance of normal kidneys in children. AJR 1985;145:611-6 [DOI] [PubMed] [Google Scholar]
- 7.Schwarz GJ, Brion LP, Spitzer A. The use of plasma creatinine concentration for estimating glomerular filtration rate in infants, children, and adolescents. Pediatr Clin North Am 1987;34:571-90 [DOI] [PubMed] [Google Scholar]
- 8.Levey AS, Coresh J, Greene T, et al. Chronic kidney disease epidemiology collaboration. Using standardized serum creatinine values in the Modification of Diet in Renal Disease study equation for estimating glomerular filtration rate. Ann Intern Med 2006;145:247-54 [DOI] [PubMed] [Google Scholar]
- 9.Royer P, Habib R, Mathieu H, et al. L’hypoplasie rénale bilatérale congénitale avec reduction du nombre et hypertrophie des nephrons chez l’enfant. Ann Pediatr (Paris) 1962;9:133-46 [PubMed] [Google Scholar]
- 10.Kusayama Y, Tsukino R, Oomori H, et al. Familial occurrence of oligomeganephronia. Acta Pathol Jpn 1985;35:449-57 [DOI] [PubMed] [Google Scholar]
- 11.Park SH, Chi JG. Oligomeganephronia associated with 4p deletion type chromosomal anomaly. Pediatr Pathol 1993;13:731-40 [DOI] [PubMed] [Google Scholar]
- 12.Risdon RA, Young LW, Chrispin AR. Renal hypoplasia and dysplasia: a radiological and pathological correlation. Pediatr Radiol 1975;3:213-25 [DOI] [PubMed] [Google Scholar]
- 13.Huntington DK, Hill SC, Hill MC. Sonographic manifestations of medical renal disease. Semin Ultrasound CT MR 1991;12:290-307 [PubMed] [Google Scholar]
- 14.Keeton GR, Pillay GP. Intravenous urography in acute and chronic renal failure. Urol Radiol 1986;8:72-6 [DOI] [PubMed] [Google Scholar]
- 15.Sherman RA, Byun KJ. Nuclear medicine in acute and chronic renal failure. Semin Nucl Med 1982;12:265-79 [DOI] [PubMed] [Google Scholar]
- 16.Saunders HS, Dyer RB, Shifrin RY, et al. The CT nephrogram: implications for evaluation of urinary tract disease. Radiographics 1995;15:1069-85 [DOI] [PubMed] [Google Scholar]
- 17.Niaudet P. Clinical manifestation, diagnosis, and treatment of nephronophthisis. In: Mattoo TK, Perrone RD, Kim MS.UpToDate®. Philadelphia, PA: Wolters Kluwer Health; 2013. Available from: http://www.uptodate.comAccessed: 1 Sept 2013 [Google Scholar]
- 18.Moreau J-F, Grenier P, Grünfeld J-P, Brabant J. Renal clubbing and scarring in adults: a retrospective study of 110 cases. Urol Radiol 1980;1:129-35 [DOI] [PubMed] [Google Scholar]