

Abstract
Rhabdomyosarcomas are among the most common soft-tissue tumors in children. These tumors are derived from mesenchymal tissue with a tendency toward myogenic differentiation that probably originates from immature and highly invasive satellite cells associated with the embryogenesis of skeletal muscle. Some of these tumors are associated with high rates of recurrence and metastasis. The diagnosis is made by microscopic analysis and auxiliary techniques such as immunohistochemistry, electron microscopy, cytogenetic analysis, and molecular biology. We report a case of 28-year-old man who presented with a painless progressive swelling of gingiva since 3 months, which was gradually increasing in size without any systemic symptoms or signs of any metastatic spread. An incisional biopsy was done and histopathology reported an alveolar variant of rhabdomyosarcoma. Immunohistochemistry with panel of markers was done which showed positivity for CD99, vimentin and negative for desmin and myogenin. So the characteristic immunohistological expression was negative in present case. Hence we conclude that haematoxylin and eosin morphology and ultrastructure are needed to classify rhabdomyosarcoma and immunohistochemistry act only as an auxiliary.
Key words: rhabdomyosarcoma, soft-tissue tumors, children, skeletal muscle neoplasm, head and neck tumors, exophytic growth.
Introduction
Rhabdomyosarcomas (RMS) are the malignant tumor of the striated skeletal muscles consisting of cells derived from primitive mesenchyme that exhibit a profound tendency to myogenesis. It is the most common malignant soft-tissue tumor in children representing approximately 4–8% of all cases of malignant disease in those younger than 15 years.1 About 35% of rhabdomyosarcomas arise in head and neck. Other sites include genitourinary tract, retro-peritoneum and to a lesser extent extremities. In head and neck, the most frequently affected sites are orbit, paranasal sinuses, soft tissues of cheek and neck. Oral rhabdomyosarcoma is rare, and when occurring, it is more frequent in the soft palate.2 According to their anatomical location and propensity for invasion of the central nervous system, the rhabdomyosarcomas are divided into orbital, parameningeal and non orbital non parameningeal forms. Parameningeal tumors carry worst prognosis.3 Rhabdomyosarcomas are classified histologically into Embryonal, Botryoid, Alveolar and Pleomorphic varieties.4
Case Report
A 28-year-old male patient was reported to the department of oral medicine and radiology, with a chief complaint of swelling in the extra-oral region of lower jaw since 3 months. On examination there was an exophytic growth on gingiva, which was diffuse and irregular in shape extending from 41 to 47, reddish in color, soft in consistency, tender and not fixed to underlying bone (Figure 1A and 1B). There was no significant cervical lymphadenopathy. Orthopantomograph showed no major pathognomonic findings (Figure 1C). Computed tomography showed 3×3 cm sized, expansile, osteolytic lesion involving the alveolus and body of right side of the mandible (Figure 1D and 1E). On the basis of these findings a provisional diagnosis of giant cell granuloma and lymphoma was given. Incisonal biopsy was done and tissue was prepared for microscopic examination. Hematoxylin and Eosin stained sections showed small round to oval cells arranged in an alveolar pattern on a mucinous background with a sparse inflammatory infiltrate. The tumor cells were predominantly single in arrangement with some showing attachment to fibrous septae and several exhibiting nucleoli. All these findings were suggestive of alveolar rhabdomyosarcoma. To confirm diagnosis we decided to en route with panel of antibodies for immunological expression. Immunohisto chemical staining was carried out and tumor cells showed immunopositivity for vimentin, pancytokeratin, Cd99 and bcl-2 (Figure 2) and negative for Cd31, Cd34 Cd45, Cd20, Cd1a, Cd3, Cd68, Cd45ro, Cd138, muscle specific actin (HHF-35), desmin and myogenin.
Figure 1.
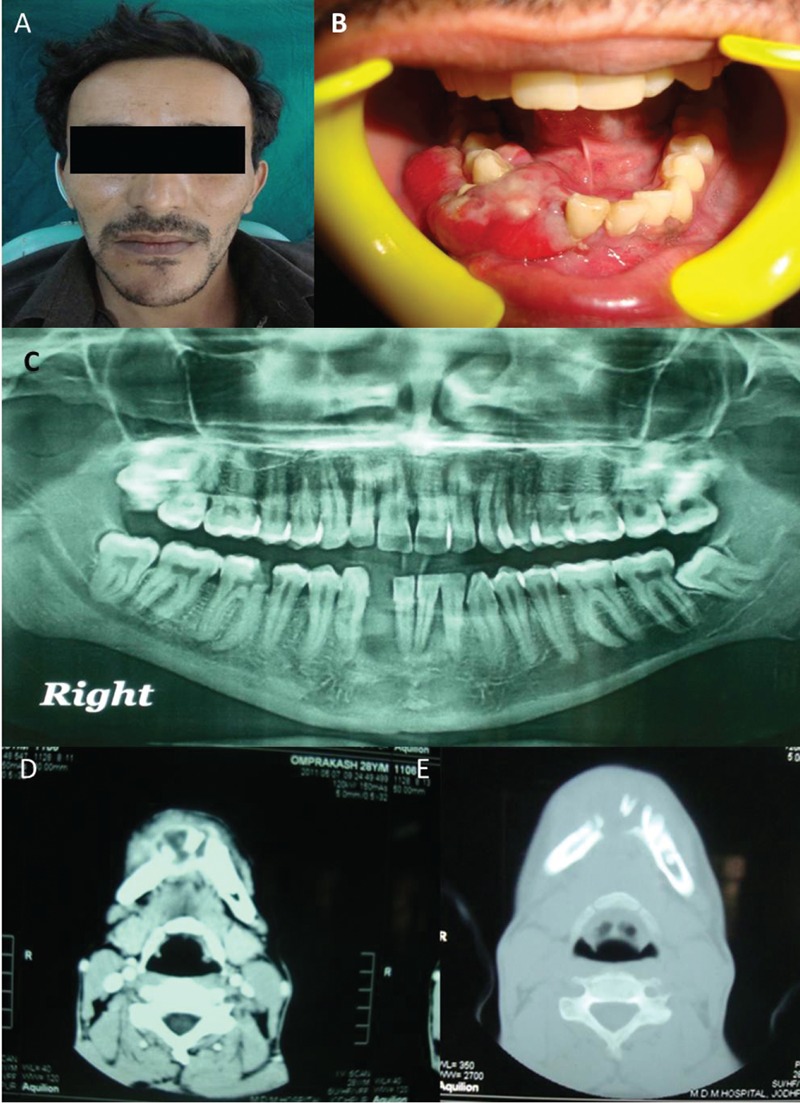
The extra-oral swelling on the lower jaw (A) with an exophytic diffuse and irregular growth on gingiva (B). The imaging features with orthopantomograph (C) show no major findings whereas the computed tomography images (D and E) show an expansile, osteolytic lesion involving the alveolus.
Figure 2.
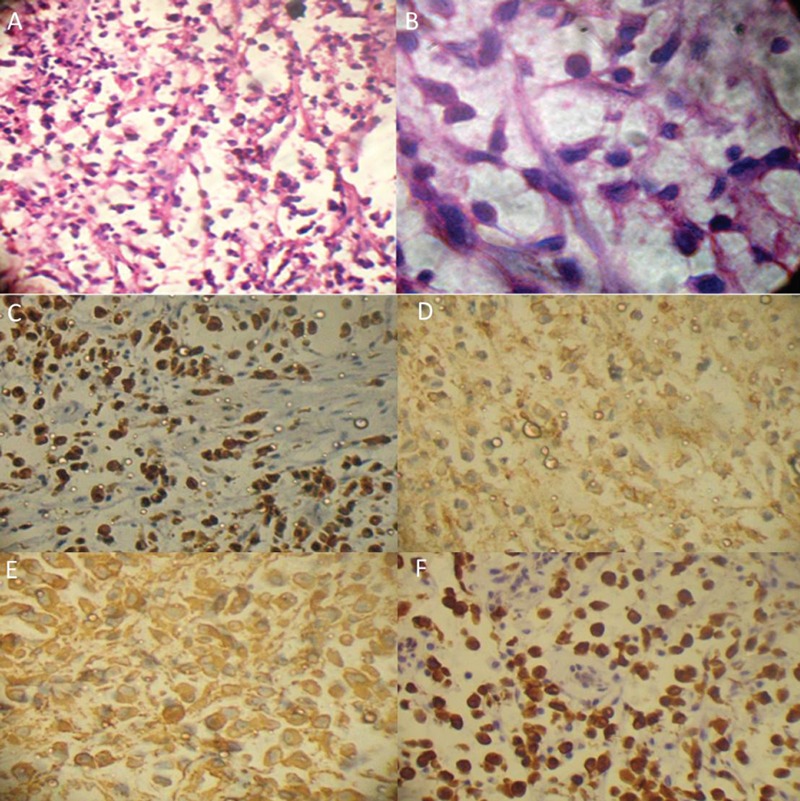
The hematoxylin and eosin stained photomicrographs under low power (A) and high power (B) shows small round to oval tumor cells arranged in an alveolar pattern on a mucinous background with Positive immunohistochemical expression of Pancytokeratin (C), Cd 99 (D), Vimentin (E) and Bcl 2 (F).
Discussion
RMS was first described and defined by Weber in 1854.2 WHO defined rhabdomyosarcoma as a highly malignant tumor of rhabdomyoblasts in varying stages of differentiation with or without cross-striation.5 Stout and Lattes (1967) described adult and juvenile forms of this condition both of which are highly malignant. The adult type usually occurs on the trunk or limbs whereas the juvenile form is more common near mucosal surfaces, especially in the head and neck.6 Described as being the most common soft tissue sarcoma in children, it represents approximately 4–8% of all cases of malignant disease in children younger than 15 years and 40% occur in the head and neck region. Rhabdomyosarcoma is an aggressive malignant tumor. The tumor is composed of neoplastic mesenchymal cells, with varying degrees of striated muscle differentiation with rapidly growing local extensions including bony destruction.1
The main etiology of rhabdomyosarcomas lies in the genetic mutations. Alveolar rhabdomyosarcoma is associated with 2:13 or 1:13 chromosomal translocations, which generate PAX3-FKHR and PAX7-FKHR fusion products, respectively. These translocations result in altered expression, function, and sub cellular localization of the fusion products relative to the wild-type proteins, and ultimately contribute to oncogenic behavior by modifying growth, differentiation, and apoptosis pathways. In contrast to the specific translocations found in alveolar rhabdomyosarcoma, most embryonal rhabdomyosarcoma cases have allelic loss at chromosome 11p15.7,8
A common misconception is that rhabdomyosarcomas arise in skeletal muscle, but in fact many examples arise in viscera such as prostate, urinary bladder, and gall bladder, which are devoid of striated muscle fibers. The exact origin of extramyogenous rhabdomyo -sarcomas is more problematic, because myo-genic transformation can be induced in non muscle cells by genetic manipulation and might be reproduced via tumorigenic influences. Another misconception is that rhab-domyosarcomas represent a single tumor type, whereas there are distinct clinical, pathologic and molecular differences among at least different variants. Such differences suggest that these tumors comprise separate pathobiology phenomena that all share cellular and biologic features with developing muscle. It is important to recognize these distinctions, for they affect clinical outcome and approach to therapy.9 The head and neck RMSs are anatomically divided into 2 categories: parameningeal (including RMS of the nose, nasopharynx, paranasal sinuses, middle ear, mastoid, infratemporal fossa and pterygopalatine fossa) and non-parameningeal (including RMS of the scalp, orbit, parotid gland, oral cavity, oropharynx, and larynx).10
The RMS of oral cavity accounts for 10–12% of all the head and neck RMS cases. The most common site of involvement in the oral cavity is the tongue followed by the soft palate, hard palate and buccal mucosa. It presents typically as a rapidly enlarging, painless sub mucosal mass. Mean age at diagnosis is 20 years and rarely seen after 45 years of age. There is slight male predilection with male:female ratio of 1.5:1. The tumor surface may be smooth or lobulated, sometimes botryoid or grape-cluster in appearance and the tumor becomes fixed to surrounding tissues at an early stage.11 Pain, proptosis, diplopia, strabismus, decreased hearing, nasal obstruction, dysphagia and cervical lymphadenopathy are the other signs and symptoms.5
Staging of rhabdomyosarcoma according to intergroup rhabdomyosarcoma study.5
Localized disease, tumor resected completely, regional lymph nodes not involved.
Localized disease with microscopic residual disease or regional disease with or without microscopic residual disease.
Incomplete resection with gross residual disease.
Metastatic disease.
The histopathological and molecular spectrum manifested by RMS has led to many classification systems. The differing morphological features were recognized in the mid-1900s by Horn and Enterline who divided rhabdomyosarcomas into embryonal, alveolar, botryoid and pleomorphic subtypes (Table 1).3,4,12
Table 1. Different types of rhabdomyosarcoma and their characteristic features.
| Features | Embryonal | Alveolar | Pleomorphic | Botryoid |
|---|---|---|---|---|
| Age | Children < 10 years | 10–25 years | >45 years | Variant of embryonal RMS |
| Site | Extremities | Deep soft tissue of extremities | Deep soft tissue of extremities | |
| Histological features | Comprise of mixture of four types of cells
|
|
|
|
Diagnosis has become easy with the use of immunohistochemistry. In rhabdomyosarcomas, Desmin, Smooth Muscle Actin and Myogenin are considered to be of diagnostic value. Desmin is highly sensitive for all tumors with skeletal differentiation but somewhat nonspecific because it may also stain smooth muscle cells and occasionally even myofibroblasts and Smooth muscle actin positivity is seen in a minority (approximately 10%) of RMS's.2,13 Although myogenin is a sensitive and specific marker for RMS, considered more specific than desmin, muscle-specific actin and myoglobin,2 but in our case desmin, smooth muscle actin and myogenin were negative. So a clinicopathological data, morphology and panel of immunohistochemical markers were more useful in the RMS diagnosis than myogenin alone.
A careful histological examination is required to differentiate such lesions from other more frequent and aggressive lesions affecting the concerned site. The most problematic tumors to differentiate from alveolar rhabdomyosarcoma are neuroblastoma, lymphoma, soft tissue Ewing's sarcoma, langerhans cell histiocytosis and undifferentiated small cell carcinoma. The neuroblastomas have a much more uniform and diffuse distribution of lesional cells and often contain rosettes with neurofilament cores. It also is nonreactive for desmin, myoglobin and skeletal muscle actin, as are the lymphomas, Ewing's sarcoma and the carcinoma.13
In our case, the pleomorphism noted was critical for differentiating rhabdomyosarcoma from Ewing's sarcoma. So a panel of markers like CD31, CD34, CD45, CD20, CD1A, CD3, CD45 and CD138 were used and negativity to this ruled out Ewing's sarcoma and lymphoma. CD68 negativity further ruled out langerhans cell histiocytosis. The presence of an alveolar pattern, pleomorphism, cohesive nature of the cells and the absence of lymphadenopathy further ruled out the diagnosis of lymphoma. Another interesting feature of the tumor observed was the expression of vimentin and absence of desmin expression in the tumor cells. It has been previously reported by Miettinen and colleagues that the two proteins may show extensive coexpression in rhabdomyosarcoma. However, previous results with differentiated rhabdomyosarcomas suggested that the extent of coexpression is limited and the exact nature of the vimentin-positivity incells not readily apparent. It was further proposed that the two may coassemble to form a mixed heteropolymeric intermediate filament.14 Studies on the expression of intermediate filaments during the differentiation of skeletal muscle indicated that, at early stages, vimentin is the predominant component, while desmin is expressed after fusion of myoblasts and gradually exceeds the amount of vimentin within the cells.13 As far as the CD99 expression is concerned, according to Afshin Abdirad and Elham Shafie 27.4% of rhabdomyosarcomas expressed MIC2/CD99.14 So in rare cases CD99 expression may be seen. However the most striking feature in the present case was the negative expression of myogenin, which is considered highly specific for rhabdomyosarcoma. In a recent study by Josefine Heim-Hall and Sophia L. Yohe, it has been emphasized that in MyoD1/myogenin–negative tumors, other features of skeletal muscle differentiation like hematoxylin and eosin morphology and ultrastructure are needed to classify tumor as rhabdomyosarcoma and only 50% of the pleomorphic types are positive for MyoD1 and myogenin.15 Also Soini Y. and Paakko P. in their study showed that bcl-2 expression is activated significantly more often in muscle derived tumours.16 Moreover Stock N. and Chibon F. in their study showed that 20% of RMSs show pancytokeratin expression.17 So on the basis of histological findings and review of literature regarding the immunological expression seen in rhabdomyosarcomas, a final diagnosis of alveolar rhabdomyosarcoma was rendered and it was also concluded that immunohistochemistry is just an auxiliary to the histopathological diagnosis.
In 1994, histological and biological studies resulted in the International Classification of RMS into prognostically significant and diagnostically consistent subgroups based on the level of agreement and power of prognostic prediction. Four broad subtypes of RMS were established: (i) botryoid and spindle cell RMS (both less common variants of embryonal rhabdomyosarcoma); (ii) embryonal rhabdomyosarcoma, generally having a superior prognosis; (iii) alveolar (including the solid-alveolar variant) rhabdomyosarcoma, generally having a poorer prognosis and (iv) undifferentiated sarcoma, also generally having a poorer prognosis. Finally, a category of sarcoma not otherwise specified was created for tumors that could not be classified into a specific subtype. Recently it was added to this classification a subtype of rhabdomyosarcoma with rhabdoid-like features, whose prognosis is not presently valuable. It is now apparent that rhabdomyosarcoma comprises a group of morphologically similar but biologically diverse lesions.3 Prognosis of RMS is excellent in relation to other oral soft tissue malignant lesions. However, it is influenced by clinical staging and anatomic site of the tumor. Within the histological subtypes, according to International Classification of Rhabdomyosarcoma, prognosis was expected as superior prognosis for botryoid and spindle cell types, as intermediate prognosis for embryonal type and as poor prognosis for alveolar & undifferentiated type of sarcomas.2,7 An early and accurate diagnosis of the tumor and a combined therapeutic approach involving surgery, chemotherapy and radiation therapy are known to dramatically improve the survival rates, as seen in cases recorded over the past 20 years. Effective surgical excision is challenging in cases of rhabdomyosarcoma of the head or neck region owing to the involvement of other crucial structures in these locations. Therefore, a multidisciplinary approach with induction chemotherapy and radiotherapy is required to bring about tumor regression in patient.
References
- 1.Davidson LE, Soldani FA, North S. Rhabdomyosarcoma of the mandible in a 6-year-old boy. Int J Paediatr Dent. 2006;16:302–6. doi: 10.1111/j.1365-263X.2006.00732.x. [DOI] [PubMed] [Google Scholar]
- 2.Gabriela VD, Bruno CJ, Mesquita AT, et al. Oral embryonal rhabdomyosarcoma in a child: a case report with immunohistochemical analysis. Oral Oncol Extra. 2006;42:105–8. [Google Scholar]
- 3.França CM, Caran CEM, Alves MT, et al. Rhabdomyosarcoma of the oral tissues - two new cases and literature review. Med Oral Pathol Oral Cir Bucal. 2006;11:E136–40. [PubMed] [Google Scholar]
- 4.Batra R, Gupta DO, Sharma P, Bokariya P. Alveolar rhabdomyosarcoma of oral cavity – A rare case. Al Ameen J Med Sci. 2010;3:255–8. [Google Scholar]
- 5.Konidena A, Kode M. Intraoral rhabdomyosarcoma in a young boy. J Indian Acad Oral Med Radiol. 2010;22:S73–75. [Google Scholar]
- 6.Cove P. An unusual presentation of rhabdomyosarcoma: A case report. Br J Oral Surg. 1974;12:240–3. doi: 10.1016/0007-117x(74)90132-2. [DOI] [PubMed] [Google Scholar]
- 7.Xia SJ, Pressey JG, Barr PF. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002;1:97–104. doi: 10.4161/cbt.51. [DOI] [PubMed] [Google Scholar]
- 8.Perk K, Shachat DA, Moloney JB. Pathogenesis of a rhabdomyosarcoma (undifferentiated type) in rats induced by a murine sarcoma virus (Moloney) Cancer Res. 1968;28:1197–206. [PubMed] [Google Scholar]
- 9.Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children. Arch Pathol Lab Med. 2006;130:1454–65. doi: 10.5858/2006-130-1454-RIAACA. [DOI] [PubMed] [Google Scholar]
- 10.Miloglu O, Altas SS, Buyukkurt MC, et al. Rhabdomyosarcoma of the oral cavity: a case report. Eur J Dent. 2011;5:340–3. [PMC free article] [PubMed] [Google Scholar]
- 11.Dagher R, Helman L. Rhabdomyosarcoma: an overview. Oncologist. 1999;4:34–44. [PubMed] [Google Scholar]
- 12.Rajendran R, Sivapathasundharam B. 6th ed. Noida: Elsevier India; 2009. Shafer's textbook of oral pathology; pp. 192–194. [Google Scholar]
- 13.Geiger S, Czernobilsky B, Marshak G, Geiger B. Embryonal rhabdomyosarcoma: immunohistochemical characterization. Oral Surg Oral Med Oral Pathol. 1985;60:517–23. doi: 10.1016/0030-4220(85)90241-5. [DOI] [PubMed] [Google Scholar]
- 14.Abdirad A, Shafie E. Immunohistochemical differential diagnosis of Ewing's sarcoma/primitive neuroectodermal tumors (ES/PNET) and rhabdomyosarcomas with small round cells: a report of 87 cases. Elect J Pathol Histol. 2003;9:32–4. [Google Scholar]
- 15.Helm-Hall J, Yohe SL. Application of immunohistochemistry to soft tissue neoplasms. Arch Pathol Lab Med. 2008;132:476–89. doi: 10.5858/2008-132-476-AOITST. [DOI] [PubMed] [Google Scholar]
- 16.Soini Y, Paakko P. Bcl-2 is preferentially expressed in tumours of muscle origin but is not related to p53 expression. Histopathology. 1996;28:141–5. doi: 10.1046/j.1365-2559.1996.288334.x. [DOI] [PubMed] [Google Scholar]
- 17.Stock N, Chibon F, Binh MB, et al. Adult-type rhabdomyosarcoma: analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am J Surg Pathol. 2009;33:1850–9. doi: 10.1097/PAS.0b013e3181be6209. [DOI] [PubMed] [Google Scholar]