

Summary
Recent studies that alleles in the hemochromatosis gene may accelerate the onset of Alzheimer's disease (AD) by five years have validated interest in the model in which metals (particularly iron) accelerate disease course. Biochemical and biophysical measurements demonstrated the presence of elevated levels of neurotoxic copper, zinc and iron in the brains of AD patients. Intracellular levels of amyloid precursor protein (APP) holoprotein were shown to be modulated via iron by a mechanism that is similar to the translation control of the ferritin L- and H mRNAs by Iron-responsive Element (IRE) RNA stem loops in their 5′untranslated regions (5′UTRs). Recently, we reported a putative IRE-like sequence to be present in the 5′UTR of the Parkinson's disease (PD) specific alpha synuclein (ASYN) transcript. ASYN encodes the non-Aβ component (NAC) of amyloid plaques. The demonstration of iron-dependent translation of APP mRNA, the involvement of metals in the plaque of AD patients and of increased iron in striatal neurons in the Substantia nigra (SN) of PD patients, have each encouraged the development of metal attenuating agents and iron chelators as a major new therapeutic strategy for the treatment of these neurodegenerative diseases. In the case of AD, metal based therapeutics may ultimately prove more cost effective than the use of an amyloid vaccine as the preferred anti-amyloid therapeutic strategy to ameliorate the cognitive decline of AD patients.
A. METALS AND ALZHEIMER'S DISEASE (AD) AND PARKINSON'S DISEASE (PD) PATHOLOGY
A.1. Introduction
Both extracellular amyloid plaques and intracellular neurofibrillary tangles are the predominant pathological features characterizing the clinical onset of AD (both early onset and late onset AD). A proximal pathological feature of AD is the formation of neurofibrillary tangles by the microtubule associated protein Tau (mutations in Tau cause hereditary frontotemporal dementia (1,2)). However, the amyloid precursor protein (APP) remains central to our understanding AD progression as a devastating neurodegenerative disease. We will discuss the relationship of metals to AD, referring to APP and amyloid formation (3). The chromosome 21 APP gene retains its status as being both the genetic cause of familial AD (FAD) (4) and production of the 40 -42 amino acid amyloid Aβ processing product of APP is the most significant therapeutic target and pathological marker of the AD brain causing amyloid plaque formation (5).
The marked increase in the steady-state levels of metals (Fe, Cu and Zn) in the AD (6) and PD brains (7) induce a gene expression pattern that is associated with deleterious consequences for neuronal survival (6). APP mRNA translational control by iron (8,9) and APP gene transcriptional control by copper (10) each provide new genetic support for the model that APP is a metalloprotein with an integral role in metal metabolism. Recently we identified and published that the alpha synuclein 5′UTR encodes an RNA sequences that closely resembles an active Iron-responsive Element (11).
Genetic and biochemical evidence has linked the biology of metals (Fe, Cu and Zn) to brain aging and major neurodegenerative diseases including AD (12,13). This phenomenon is also associated particularly with elevated iron in the midbrain of PD patients (7). Excess iron, as occurs after Stroke, closely regulates gene expression of the Alzheimer's APP gene at the level of message translation (8,14). This review describes the Fe-regulated control of intracellular levels of APP holoprotein which is modulated by a mechanism that is similar to the pathway by which iron controls the translation of the ferritin L- and H mRNAs by Iron-responsive Element (IRE) RNA stem loops in their 5′untranslated regions (5′UTRs) (8,15). More recently a putative IRE-like sequence was reported to be present in the 5′untranslated region of the Parkinson's disease alpha synuclein (ASYN) transcript (11). APP gene transcription was found to be responsive to copper deficit in a study in which the Cu depleted fibroblasts of a human fibroblasts over-expressing the Menkes protein (MNK) exhibited repressed transcription of APP through metal regulatory and copper regulatory sequences upstream of the 5′ cap site (16).
A.2. APP abundance and mis-processing are a genetic cause of familial Alzheimer's disease (FAD)
Approximately 10% of cases of AD are caused by familial genetic mutations. These cases are associated with autosomal dominant inheritance of APP (17,18) and presenilins PS-1 and PS-2 (19) gene mutations (20). Mutations within the APP gene on chromosome 21 cause altered cleavage of the Aβ-peptide from the APP holoprotein and have been genetically linked with early onset “Familial Alzheimer's Disease”(FAD) (17). APP and PS-1 gene mutations independently enhance cleavage of the precursor into the Aβ peptide [Aβ (1-40) and Aβ (1-42)] that accumulates in the brain as amyloid plaques (21). Trisomy of the APP gene is associated with Down's syndrome (DS) (22). Recently, as in DS, mounting evidence supports that simple APP abundance caused by duplication of the region of chromosome 21 harboring the APP gene, was 100% linked to familial AD in French pedigrees (23,24).
The abundant and ubiquitously expressed APP is a metalloprotein that spans membranes in the endoplasmic reticulum, trans-Golgi and on the cell membrane surface of most cell-types. Originally APP was shown to be a redox active copper-zinc binding protein of unknown function during normal health (25). Our unpublished data suggests that APP, itself, also binds iron. During normal physiology a group of metalloproteases known as the alpha-secretases (26) ADAM-10, ADAM-17 and TACE, cleave the APP to generate the neuroprotective 90 kDa ectodomain of APP (APP(s)) that is released from cells into the cortex, cerebrospinal fluid and the bloodstream; a cytoprotective event that occurs at an increased rate in activated astrocytes during the acute phase response (27).
During the course of AD, APP is the single substrate which is proteolyzed to produce the 40-42 amino acid Aβ peptide that aggregates to form the main component of amyloid plaques (28). The pathogenic Aβ peptide encompasses amino acids 672-714 of APP-770 (longest alternatively spliced isoform of APP), wherein the amino terminus of Aβ includes 28 external amino acids and the remaining carboxyl-terminal half of Aβ is encoded by first residues of the trans-membrane region of nascent APP. A decade of research from multiple laboratories has shown that the γ-secretase multimer (Pen2, presenillin, APH1 and nicastrin) (29) and BACE (30) cleave APP to generate the pathogenic 40 to 42 amino acid Aβ peptide (31).
A.3. Alpha synuclein mutations and abundance are a cause of familial Parkinson's disease
Alpha-synuclein is the ∼15 kd cytosolic protein implicated in pathogenesis of neurodegenerative diseases referred to as α-synucleinopathies (32), including the movement disorders such as PD. The pathological hallmark of PD is the oligomerization of toxic ASYN commonly in Lewy bodies, but also in dystrophic neurites, and glial cytoplasmic inclusions (33). The mechanisms underlying PD have been associated with many cell-based lesions including ubiquitination and LRRK kinase leading to dopaminergic cell death (34). The discovery that mutations in ASYN can cause familial PD(11) (35), and that ASYN accumulates in Lewy bodies suggests that this protein participates in the pathophysiology of PD.
The straightforward protein abundance of ASYN was shown to be the cause of familial PD in families that expressed triplicate copies of the alpha synuclein gene (35), whereas missense mutations have been more frequently quoted in the scientific literature (34). Alpha synuclein knockout mice are fully viable (36), suggesting that the two homologous proteins γ and β synucleins perform a redundant and compensatory function most certainly associated with their its pre-synaptic localization (37). The role of alpha synuclein aggregation mediate neurotoxicity in the SN of Parkinson's disease models was supported by the finding that alpha synuclein knock out mice are resistant to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced neurotoxicity (38).
A.4. Iron and spontaneous Alzheimer's and Parkinson's Disease
Metals (Fe, Cu, Zn) clearly provide one of the ultrastructural requirements needed for polymerization of Aβ peptide (39) in addition to the reported presence of pathological chaperones such as alpha-1 antichymotrypsin (ACT) or Apo-E (40). For example, elemental profiles (S, Fe, Cu, and Zn) were microscopically observed to be physically associated with amyloid plaques using Synchrotron scanning X-ray Fluorescence Microscopy (μ-XRF) (41) (Figure 1). Also Cu, Zn and Fe were demonstrated to be potent natural binding partners of Aβ with a dissociation constant at the attomolar affinity level (42,43). Iron and Cu, were demonstrated to be very important in mediating the neurotoxic action of Aβ protofibrils (44). At the molecular level, the histidine mediated zinc binding site was mapped to a contiguous sequence between positions 6 and 28 of the Aβ sequence (45). Amyloid Aβ is normally secreted from cells grown in culture as a monomer, but also as a covalently linked dimer, in the ratio 55: 30: 15 monomers: dimers: trimers (46). Human amyloid derived from oligomeric Aβ results from a tyrosine cross-lin ked oligomerization that is induced by metal catalyzed oxidation systems.
Figure 1. Elemental profiles (S, Fe, Cu, and Zn) in a typical Alzheimer's Aβ amyloid plaque.
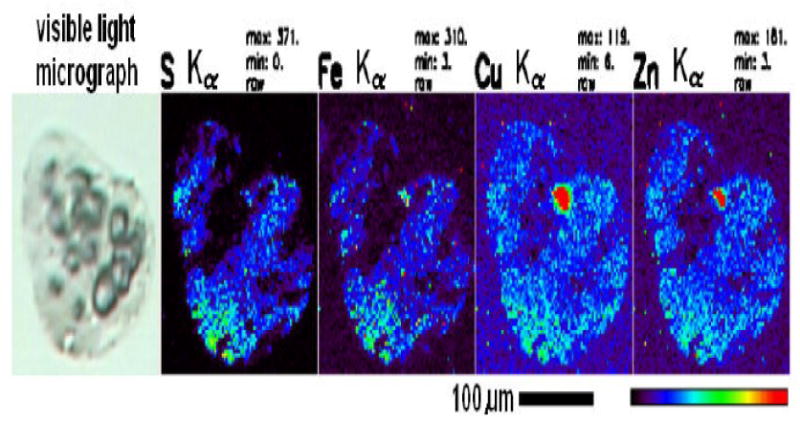
The cryo-sectioned (10 μm thichness) AD brain tissues were stained with 0.1% Thioflavin-T for amyloid plaques. The amyloid plaque-bearing human brain tissues were procured by LCM (Arcturus Pixcell IIE platform) and mounted on Si3N4 membrane grids (2.0 mm × 2.0 mm). Guided by the optical amyloid plaque images, the samples were excited with incident synchrotron X-ray of 10 keV for elemental Kα characteristic emission lines. Elemental profiles (S, Fe, Cu, and Zn) were obtained using Synchrotron scanning X-ray Fluorescence Microscopy (μ-XRF) at the Advanced Photon Source (APS) of the Argonne National Laboratory (ANL).
The current level of understanding of late onset AD has been advanced since the discovery that individuals homozygous for the E4 allele of apolipoprotein E (ApoE) are at increased risk for developing Alzheimer's Disease (47). Other genome scans have identified alpha 2 macroglobin involved in Aβ peptide clearance (48), the insulin degrading enzyme that degrades Aβ (49), alpha-1 antichymotrypsin as a pathological chaperone for profibril formation (40).
A.4.1. Iron and the AD brain
Recently the transferrin C2 allele, relevant to blood iron transport was characterized as a risk factor for AD (50) (51,52). Also several genetic studies demonstrated the clear involvement of the genotype of the hemochomatosis (HFE) gene, additional to the transferrin C2 allele, to increase the risk of AD (53) (54) (50), likely as a consequence of accelerated iron overload and compartmentalization in these individuals. The elegant work of Lee et al., (2006) confirmed the cell biological mechanisms of these AD genetic associations of HFE mutations to their cell based phenotypes in neuroblastoma cells consistent to oxidative stress events causing neurodegeneration (55). The original Italian study, for example, incorporated 107 patients with sporadic late-onset AD and 99 age-matched non-demented controls (56). These authors observed that patients carrying the mutant HFE-H63D allele had a mean age of onset at 71.7 +/- 6.0 years versus 76.6 +/- 5.8 years of those who were homozygous for the wild-type allele (p = 0.001). Further support came from a study using knockout mice deficient in the H-ferritin gene wherein Thompson et al., (2003) reported that null ferritin heterozygous mutants developed oxidative features in the cortex reminiscent of PD and AD (57).
The integral role iron plays in the etiology of AD pathology was directly supported from an MRI imaging study of the brains of Alzheimer's patients that revealed elevated levels of iron, particularly in the neurons of the basal ganglia (58). Confirming these results, elegant spectroscopic and microscopic studies demonstrated that amyloid plaques harbor increased burden of iron, copper and zinc (6) (41) (Figure 1). Iron and zinc levels were measured to be present at concentrations as high as the 1 mM level in the vicinity of amyloid plaques. There has been controversy as to whether the iron-Aβ peptide interaction was neurotrophic or neuroprotective. Aβ-peptide, itself, was found to be neurotrophic under some circumstances (59), and perhaps Aβ toxicity is only evident in the presence of metals. Certainly iron enhanced beta-amyloid action to accelerate Aβ-induced neuroblastoma cell death, providing one direct link between excessive iron and the known loss of neuronal function seen in AD patients (60). Bishop and Robinson (2003) reported that Aβ peptide reduced iron toxicity in rat cerebral cortex (61) although iron remains a candidate pathological mediator of AD (62).
In addition to iron, trace copper in the water supply dramatically accelerated amyloid plaque formation in APP transgenic mice (63). Thus our working model has to account for the fact that the Aβ amyloid precursor protein (APP) of AD is a copper binding metalloprotein (25). From in vitro experiments, copper, zinc and iron accelerated the aggregation the Aβ peptide and enhanced metal catalyzed oxidative stress associated with amyloid plaque formation (44).
Iron imbalance is evident in the AD brain as reflected by the altered expression of several iron proteins in the AD brain, including increased expression of P97 (an iron transporter (64) and potential biomarker (65,66). Ferritin expression is increased in the vicinity of amyloid plaques (67). These data are consistent with the involvement of hemachromatosis and transferrin genes as genetic factors that can increase the risk for late onset sporadic AD.
A.4.2. Iron and the PD brain
The role of iron to cause mis-aggregation has long been a feature of PD research (35), particularly since iron is found in increased abundance in the Substantia nigra, the region of maximal dopaminergic neuronal loss in PD (7). Consistent with this observation, ferritin over-expression, stored and safely sequestered excess iron and offset neuronal loss in mice subjected to the neurotoxic Parkinson's-inducing agent MPTP, suggesting that iron mediated events are important in PD (68). Iron, but also hydrogen peroxide, stimulates the production of intracellular aggregates that contain α-synuclein and ubiquitin as identified by immunocytochemistry, electron microscopy, or the histochemical staining with thioflavine S in striatal regions of the brain (35).
In summary, Metal catalyzed oxidative stress in the vicinity of amyloid remains a central pathological hallmark of AD in the brain cortex region, and metal catalyzed fibrilization of ASYN is proximal to neurotoxic events in the SN of Parkinson's disease patients (35).
B. FERRITIN TRANSLATIONAL CONTROL: LINKS TO APP AND ASYN mRNA EXPRESSION
B.1. Introduction
Iron imbalance, like inflammation, has been recognized genetically in the etiology of neurological disorders. Consistent with these observations, the iron storage protein ferritin is present at increased abundance in neuritic plaques (69) at the same time as the acute phase proteins alpha-1 antichymotrypsin (70). An elucidation of the mechanisms of brain iron homeostasis, as outlined in figure 2, will help our understanding of AD especially since iron binds to Aβ-peptide and enhances beta-amyloid toxicity (45,60,71,72). Excess iron accumulation is a consistent observation in the AD brain. Brain autopsy samples from AD patients have elevated levels of ferritin iron, particularly in the neurons of the basal ganglia (58) and most amyloid plaques contain iron and ferritin-rich cells (12).
Figure 2. Intracellular Iron Homeostasis.

Iron transit across the cell surface membrane is mediated by (i) ferrotransferrin internalization by the transferrin receptor (TfR), (ii) DMT-1, (iii) ferroportin mediated iron efflux from the duodenum into the blood. Ferritin mRNA translation is regulated by the modulated interaction between the IRPs and the IREs in the 5′UTR of ferritin mRNA. MAP kinase signaling events influence ferritin translation and transferrin receptor activity and expression.
This section will outline how iron enhances both the translation of APP mRNA and also cleavage of the Aβ-peptide domain of APP by the metaloprotease alpha secretase (73). Similar to ferritin, the protective effect of the major cleavage product of APP, APP(s), may derive from its capacity to detoxify iron and thus diminish iron/heme aggravated oxidative stress to neuronal cells (25,74). Clinically there is a reported decrease in the rate of decline in AD patients who were treated with the intramuscular iron chelator desferrioxamine (75), and part of this efficacy may be attributable to DFO repression of APP mRNA translation. We need to recognize the “UTRosome’ concept which in essence takes the 5′-UTR from an RNA entity to a RNA-protein complex consisting of proteins (Iron-regulatory proteins)(76) but also note that Smad proteins, and certain transcription factors, bind both proximal promoter region as well as the UTR (77).
Lessons from the genetic control of translation of ferritin mRNAs and transferrin receptor mRNA stability can be applied to the control of APP and also ASYN expression by iron. This information will be relevant to RNA directed drug discovery to offset AD and PD pathology by targeting and inhibiting translation of APP and ASYN gene expression without influencing general translation (78) (79).
B.2. Post-transcriptional Control of Ferritin and Transferrin Receptor (TfR) Expression
Ferritin is the intracellular iron storage protein composed of two subunits (L and H-subunits) that co-assemble to form the 240,000 dalton iron storage shell found in all tissues of the body (80) (Figure 2). Ferritin is cytoprotective (81) because the H-subunit oxidizes Fe 2+, the bioavailable form of iron that causes tissue damage via hydroxyl radicals (Fenton Reaction). Ferric iron then becomes stored as an ferric orthophosphate (Fe3+-P04) crystal inside the internal cavity of the ferritin shell (80,82). Transferrin receptor (TfR) is present at the surface membrane of all cells and tissues. TfRs have a high affinity for binding iron-loaded transferrin (90,000 dalton protein), thereby facilitating the uptake of iron from the bloodstream into tissues (80,82). One of the major markers of any inflammation is lowered serum iron levels accompanied by a reduction in the steady-state levels of blood transferrin levels (83). There is also an increase in serum ferritin levels seen during inflammation (84). In the next sections, we will outline briefly how the iron-regulatory proteins IRP1and IRP2 control intracellular iron homeostasis by modulating ferritin mRNA translation and transferrin receptor mRNA stability (85), and will then compare this to the control of APP and ASYN expression.
B.2. 1. Iron-responsive Element (IRE) RNA stem loops and intracellular iron homeostasis
Transferrin receptors (TfRs) mediate iron uptake into most mammalian cell types wherein iron uptake decreases TfR mRNA stability via five distinct Iron-responsive Element RNA stem loops (IREs) in the 3′UTR of TfR mRNA. In fact, IREs modulate intracellular iron uptake by mediating both TfR mRNA and Divalent Metal Ion Transporter (DMT-1) mRNA stability. In a feedback regulatory circuit, during iron deficit the iron regulatory proteins, IRP1 and IRP2, increase the message stability of TfR mRNA and DMT-1 mRNA by binding to 3′UTR sequences and preventing their degradation (86).
Prior to assembling into the central iron storage protein in all cells, ferritin L- and H subunit translation is controlled by the iron regulatory proteins,IRP1 and IRP2, which bind at high affinity (Kd 40-100 pM) to highly conserved IREs RNA stem loops at the 5′ cap sites of the L- and H-ferritin mRNAs (86,87) (88) (Figures 2 and 3). The IRE/IRP interaction impedes the access of the small 40S ribosome subunit to the 5′ end of the ferritin mRNAs and thereby suppresses L- and H-ferritin mRNA translation (89-91). The potency of the IRE to regulate translation is position dependent (92). Iron influx relieves repression of ferritin translation by removing IRP1 from the ferritin IREs; IRP1 is simultaneously interconverted to a cytoplasmic cis-aconitase (93) (94) with an iron sulfur cluster, and IRP2 is degraded by iron influx (85,95). Removal of IRPs from the ferritin IREs restores recruitment of the 40S ribosome at the 5′ cap sites. Ferritin translation can take place after eIF4E promotes eIF4G binding to eIF3 and hence 40S ribosome recruitment (96) (Figure 3). The 40S ribosome then scans ferritin mRNAs before the eIF2-a dependent 60S ribosome subunit “joining step” occurs, after which protein synthesis begins at the optimal start codon (97). The ratio of IRP1 and IRP2 is tissue specific, but IRP2 is more abundant than IRP1 in pituitary cells (98).
Figure 3. Model for Ferritin mRNA Translational Control.
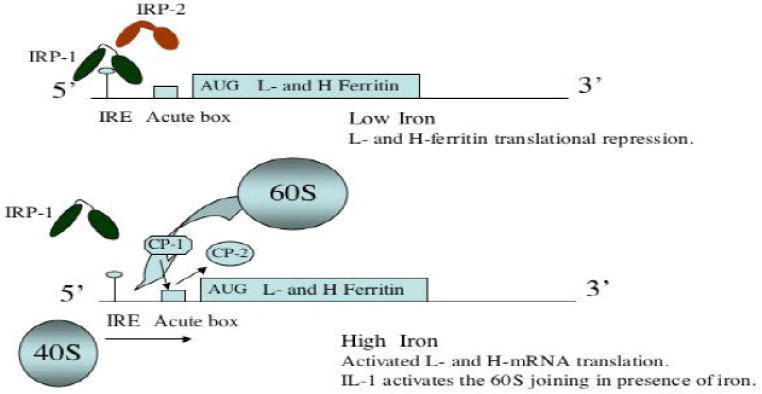
Iron releases IRP1/IRP2 from suppressing ferritin mRNA translation at the Iron responsive Element stem loops (IREs) specific to the Land H- mRNA 5′ cap sites. In the diagram, IRP1 and IRP2 are depicted as two domains separated by a hinge region (line). Our preliminary data suggests that the RNA binding protein (Poly C-binding proteins, CP-1 and CP-2) interact with the ferritin mRNA acute box (AB) domain (box) downstream from the IRE (114).
Hormonal signaling via IRPs certainly modulates intracellular ferritin levels, and appears to be cell line specific. Interleukin-1 (IL-1) does not change steady-state levels of IRP1 and IRP2 but appears to enhance binding to IREs (99). As outlined in the next section, IL-1α and IL-1β also act through the downstream acute box domain in ferritin mRNA 5′UTR. Both Epidermal Growth Factor and Thyroid Releasing Hormone increased the phosphorylation of IRP1 and IRP2 and binding of IRPs to IREs, an event that was shown not always to be correlated with the direction of ferritin translation (98).
Oxidative stress modulates the IRE/IRP regulatory system as an additional control point of ferritin translation. For example, H2O2 as a model for oxidative stress was shown to rapidly activate IRP1 binding to ferritin IRE stem loops (100) (101). These data confirmed that rapid responses in the gene expression of ferritin (and TfR) to oxidative stress was mediated by IRPs (102). Toth et al., (1999) showed that hypoxia altered IRP1 binding to IREs and modulated cellular iron homeostasis in human hepatoma and erythroleukemia cells (103).
The absence of Iron Regulatory Protein-2 (IRP2), which controls iron homeostasis (Figure 1), was associated with a mis-regulated iron metabolism and ferritin translation and TfR mRNA stability in both the gut mucosa and the central nervous system (104). Iwai et al., (1998) previously showed that iron-mediated degradation of IRP2 requires the oxidation of key cysteines that reside within a 73-amino acid region (105) unique to IRP2 and not present in IRP1. However, a mutant IRP2 protein lacking this 73-amino acid region degraded at a rate similar to that of wild-type IRP2 (106). Dycke et al., (2007) showed this sequence is an active non-iron dependent cleavage domain in cultured breast cancer cells, and that it is unlikely that the iron-dependent degradation of IRP2 is mediated by haem binding to the intact 73 aa domain (107). These results are relevant to AD since oxidative stress is important during AD and the APP mRNA has an active IRE in its 5′UTR (Figure 4).
Figure 4.
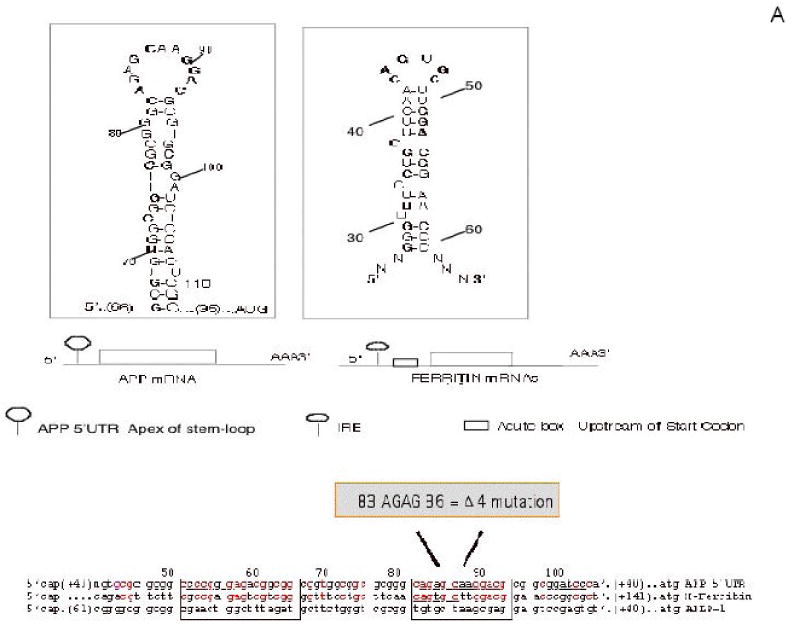
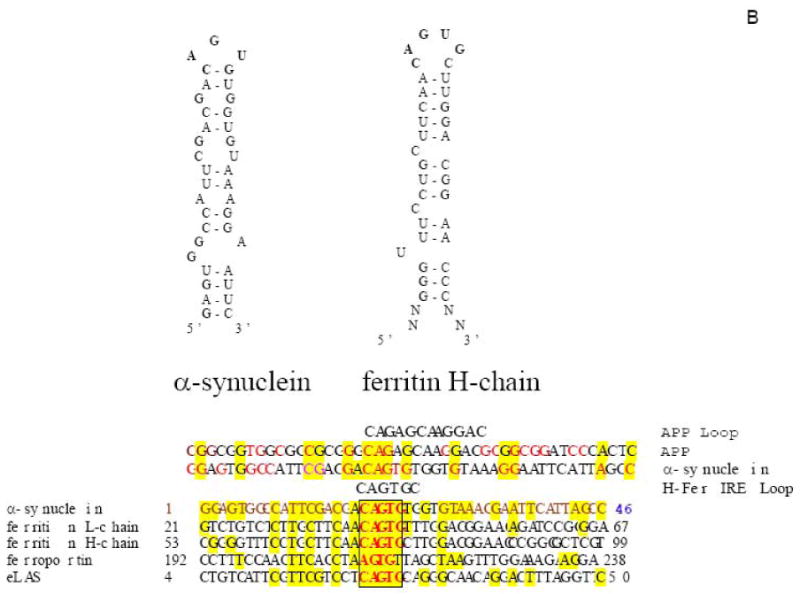
Both APP and alpha synuclein (ASYN) 5′UTRs were predicted to fold into the stable RNA stem loops similar to the 5′UTR-specific IRE in H-ferritin transcript (“APP 5′UTR” DG = -54 kCal/mol) (Zuker et al., 2003 (139)). Panel A The APP mRNA IRE (Type II) was identified after alignment with the ferritin Iron-responsive Element (shown in two clusters of > 70% sequence similarity)(8). The APP IRE is a currently active drug target for translational repression of APP in AD therapeutics (197). Panel B: The ASYN 5′UTR encodes a CAGUGU motif at the exon-1/exon-2 splice junction. This ASYN 5′UTR stem loop (DG =53 kcal/mol) aligns with classical IRE (5′CAGUGN3′ loop motif) that controls L-& H-ferritin translation & transferrin receptor (TfR) mRNA stability. Boxed alignment of the ASYN 5′CAGUGU3′ motif against the IREs of ferritin H- and L- chains (iron storage), ferroportin (iron transport), erythroid eALAS (heme synthesis) mRNAs in addition to an alignment with IRE sequences in APP mRNA.
B. 2.2. Translation of Ferritin RNA by an Interleukin-1 responsive Acute Box Domain
To generate a molecular model for iron sequestration (as observed during anemia of chronic disease), we employed HepG2 cells grown in tissue culture (Figure 2). The anemia of the acute phase response was first observed during inflammation and disease progression by Cartwright in 1946 (108). Here, an enlargement of tissue ferritin pools was seen to be preceded by a marked increase of hepatic and spleen ferritin mRNA translation and subsequent ferritin gene transcription (109). Several groups then showed that enlarged ferritin pools could cause iron sequestration from the bloodstream into rat liver and spleen for tissue storage (84). Like tissue ferritin, serum ferritin may provide cytoprotection both to oxidized low density lipoprotein in vitro and to heme-aggravated damage (81).
For our model system of hepatic ferritin gene expression, interleukin-1 (IL-1) was found to significantly increase ferritin translation by signaling though a novel translation enhancer element, the acute box motif, downstream from the IREs at the 5′ cap site (110-112). This acute box motif is located immediately downstream from the IREs in the 5′UTRs of both the L- and H-ferritin transcripts, and was also found to be present in front of the start codon in the APP transcript (113). This observation was consistent with the pattern of both APP and ferritin expression in response to inflammation during both AD and the anemia associated with chronic disease.
We found that the acute box element in the 5′ leaders of the ferritin transcripts selectively interacted with the Poly C-binding Proteins (CP-1 and CP-2) (114). During erythropoeisis both CP-1 and CP-2 are known to be the mediators of globin mRNA stability (115) and Lipoxygenase mRNA translation (116). In hepatic cells, IL-1 increases ferritin translation at the 60S ribosome subunit “joining step” when protein synthesis begins at the optimal start codon (114,117) after the 40S ribosome has scanned the 5′UTR of ferritin mRNAs. We showed that IL-1 activated iron dependent translation of ferritin H-mRNA by removing the interaction between the CP-1, CP-2 and the H-mRNA acute box (114). Iron influx causes CP-1 to replace CP-2 from acute box residency (114).
The above observations have a biological correlate since serum ferritin levels are markedly increased in the bloodstream after the onset of an inflammation (118). Ferrtin in the serum was shown to be rich in the L-subunit and is known to be immunosuppressive (119). Clinically the average concentration of serum ferritin has been measured over a very broad dynamic range between 2 and 450 μg/liter. Serum ferritin levels are clinically used after trauma as an index of tissue damage and as a predictor of disease (i.e., hepatitis B virus infection was associated with increased serum ferritin levels as a disease biomarker (120). During progression of many chronic conditions the concentration of serum ferritin rises sharply, whereas this is to a level less than that observed during conditions of iron overload such as during hemochromatosis (1000 μg/ml). Inflammation reduces hepatic transferrin gene expression (121), reflecting the levels of transferrin bound iron versus free iron present in the bloodstream during inflammation (the anemia of chronic disease ((76,80)).
B.3. Post-Transcriptional Control of the Alzheimer's APP mRNA
B.3.1. Role an active APP IRE RNA stem-loop
Like ferritin, we characterized the presence of a fully functional iron responsive IRE stem loop in the APP 5′UTR. We originally reported that APP mRNA, but not APLP1 and APLP2 mRNAs, belongs to the family of transcripts that encodes a functional IRE RNA stem loop (8,15). Figure 4A shows the sequence alignment of APP 5′UTR sequences with the known IRE in H-ferritin mRNA. These alignments demonstrated an overall 67% sequence identity between APP 5′UTR sequences (+51 to +94) and the 44 nt IRE in H-ferritin mRNA (+12 to +59) (Figure 4A). Two clusters within this APP 5′UTR IRE-Type II domain showed >70% identity with the ferritin IRE sequences. First an 18 base sequence in APP mRNA (+51 to +66) was found to be 72% similar to 5′ half of the H-mRNA IRE (+12 to +29). Second APP sequences (+82 to +94) (a 13 base cluster) were 76% identical to the loop domain of the H-ferritin IRE (+43 to +55). The IRE alignment shown in figure 4A demonstrated that the IRE-Type II sequence in the APP 5′UTR (+51 to +94) was sited immediately upstream of the IL-1 responsive acute box domain in the APP 5′UTR (+100 to -146). To ensure specificity, the 5′UTR sequences in the APLP-1 transcript only exhibited 25% percent homology with equivalent APP 5′UTR sequences.
This novel Iron-Responsive Element (Type II) in the 5′UTR of APP mRNA was fully functional as assessed by multiple separate transfection assays (8). RNA gel-shift experiments demonstrated that the mutant version of the APP 5′UTR cRNA probe no longer binds to IRP (Figure 4A) (8). Using RNA electrophoretic mobility shift assays (REMSA) we showed that IRP1 specifically binds to this APP IRE stem loop (8). The scientific consensus is that the canonical ferritin type of IRE preferentially binds IRP2 rather than IRP1 as the predominant pathway of IRE mediated control of iron homeostasis (122). Therefore it is of direct relevance to test which of IRP1 or IRP2 preferentially interacts with the APP 5′UTR to control iron dependent translation of APP expression in neural cells.
The fact that an IRP binds strongly to the APP 5′UTR suggests an integral involvement of APP holoprotein in iron metabolism. Comparison of the relative affinity measurement of IRP1/IRP2 as binding partners to the APP 5′UTR will provide useful information for screening novel lead compounds that limit APP translation, particularly if IRP1 and/or IRP2 expression can be functionally linked to APP expression.
Lahiri and his co-workers elegantly aligned the 5′UTR sequences of APP, APLP-1 and APLP-2 throughout the animal kingdom reporting, the presence of a “CAGA box” in the APP gene unique to amyloid plaque-forming species and absent in all APLP-1/2 genes with implications for AD (123). Here, the “CAGA” sequence was identified proximal to the “ATG” start codon and was unique to APP genes of amyloid plaque-forming genes surveyed. This CAGA box is immediately upstream of an interleukin-1-responsive element (the “acute box”, see next section). Interestingly, this proximal CAGA box is present in the 11 base apex of the CAGAGN stem-loop structure in both human and guinea pig APP mRNA IRE (see Figure 4A). We also note that the amyloid species specific CAGA box is indeed the same sequence responsible for binding to IRP1 in the core IRE sequence in the APP transcript (8,15). The convergence of these findings implies that IRP1 regulation of APP gene expression may yet be found to be associated with an intrinsic capacity of the encoded APP sequence to form brain amyloid and ultimately amyloid plaque in vivo.
The role of 5′-UTR can be considered in a broader context. We propose that APP 5′-UTR in concert with proximal promoter region (PPR) functions to both down-regulate and up-regulate APP gene expression (124). Notably, the proximal promoter (PPR) and the APP 5′UTR participate at both transcriptional and posttranscriptional levels. Based on our studies, multi-protein DNA- and RNA protein complexes in a gene's 5′-UTR and PPR may interact and cooperate as a unit that can regulate gene expression at both the transcriptional and post-transcriptional levels (82,124,125).
B.3.2. Translation of APP mRNA by an IL-1 Responsive Acute Box Domain
As a Cu-Zn metalloprotein (25), we first reported that the APP gene is acutely regulated at the translational level by changes in cellular iron status in neuronal cells but also by IL-1 in astrocytes (113). Furthermore, IL-1-responsive sequences were characterized as active RNA regulatory elements in the 146 APP 5′UTR analogous to the IL-1 responsive acute-box RNA enhancer immediately in front of the start codons of the ferritin L- and H-chain mRNAs (113).
Since poly(C)-binding protein affects H-ferritin translation via its acute box domain, it is worth speculating that poly(C)-binding protein isoforms may also have a role in determining the expression of APP in response to IL-1 signaling. Mini-strokes associated with AD may generate an ischemic inflammation with the same genetic consequences (126). Zhu et al., (2002) used immunostaining to show that poly(C)-binding protein 1, but not poly(C)-binding protein 2, expression was increased in the ischemic boundary zone (penumbra) of the frontal cortex after 90 min of ischemia, and persisted for at least 72 h after reperfusion (127). These results demonstrated that poly(C)-binding protein 1 and poly(C)-binding protein 2 in cortical neurons are differentially affected by hypoxic/ischemic insults, suggesting that there are functional differences between poly(C)-binding protein isoforms. We are currently investigating the role of poly(C)-binding proteins relevant to the regulation of APP mRNA translational control.
B.3.3. Mechanisms for APP and Ferritin mRNA Translational Control
The 5′UTR specific translational control circuits that regulate ferritin L- and H-mRNAs in response to iron and IL-1 are summarized in the model shown in figure 3. The mRNA for FMR1 (Fragile X) presents as a typical eukaryotic mRNA in which RNA secondary structure suppresses protein synthesis (128). However, like the ferritin mRNAs, the 146 base APP 5′UTR is unique to the precursor transcript that encodes stable RNA secondary structure, the acute box, that regulates 40S ribosome scanning and facilitate the onset of APP synthesis (Figure 4A).
In sum, our data supports the hypothesis that IL-1 actively stimulates the translation of the APP transcript by a pathway that is similar to the translational regulation of the mRNAs encoding the L- and H-subunits of ferritin (112). We also reported that APP 5′UTR sequences encode functional sequences that are 75% similar to the iron-responsive element (IRE) in the mRNAs for the ferritin L- and H-subunits (Ferritin IREs) (Figure 3). There are common features of ferritin and APP translation by iron and Interleukin-1 (8,113). These two sets of mRNAs appear each to encode very different metalloproteins whose regulation is controlled by common RNA binding protein interactions but by radically different mechanisms.
B. 4. IREs in the Transcripts of Other Proteins of Iron Metabolism
A consistent finding is that IREs are involved in the post-transcriptional regulation of several genes that control intracellular iron homeostasis. The AD brain shows dysregulated binding of IRPs to IREs (129), which might have implications for the expression of APP and other iron-related proteins. Like ferritin, the other transcripts that encode active IREs in their 5′UTR include:- (i) the mRNA for erythroid aminoluvenyl synthase (eALAS). eALAS is the rate limiting enzyme that controls heme biosynthesis in the mitochondria of developing reticulocytes (130,131); (ii) transferrin (Tf) transports iron throughout the bloodstream to tissues, and translation of Tf is activated by binding of IRPs to the 5′UTR of the transferrin transcript (132); (iii) Thomson et al., (1999) reviewed the mechanism of action of 5′UTR specific IREs that control the translation of IREG-1 (ferroportin), which is responsible for iron efflux from the duodenum into the bloodstream and macrophage iron efflux; (iii) Like the transferrin receptor, the divalent metal ion transporter (DMT-1) is known to be regulated by an IRE in its 3′UTR as one of the alternatively spliced DMT-1 transcripts (86). DMT-1 is responsible for uptake of iron and other divalent cations from the gut into the bloodstream into (133,134).
B. 5. An IRE in the Parkinson's Disease ASYN Transcript
The biology of iron-regulatory proteins is highly relevant to that of movement disorders. IRP1 knockout mice, although viable, show an enhanced PD-like neurological phenotype in a gene-dose manner. Here heterozygotes for the IRP1 gene knockout, that already have a double deletion in the IRP2 gene, showed an accelerated movement disorder with ataxia, bradykinesia and tremor (135,136) relative to counterparts with two allelic copies of the IRP1 gene.
ASYN mRNA translational control may be significantly de-regulated in Lewy Body Dementia brains (137,138). In this context, we reported sequences in the ASYN mRNA 5′untranslated region (5′UTR) to be homologous to the Iron-responsive Element (IRE) RNA stem-loops encoded in the 5′UTRs of transcripts for the L- and H-ferritin chains (iron-storage), erythroid amino-levulynyl synthase (eALAS) (heme-biosynthesis) (11), and the Alzheimer's APP (Cu/Zn-metalloprotein (8). Five 3′UTR IRE stem-loops control iron-dependent transferrin-receptor (TfR) mRNA stability and hence cellular iron transport (see the model for intracellular iron homeostasis in figure 2). The loop domain, CAGUG in the novel ASYN IRE is precisely juxtaposed to the splice junction of exons-1 and -2 (Fig. 4B) (RNA folding program of Zucker and colleagues (139)). The presence of this putative IRE in ASYN mRNA is of added significance since individual neurons in the Substantia nigral neurons of PD patients harbor twofold increased iron relative to aged matched controls (7). Therefore the ASYN IRE is a potential regulatory element through which iron influx may increase ASYN expression at the translational level. In neuronal cells this event would be predicted to contribute to disease pathology since ASYN over expression. [Trisomy of the chromosome 4 alpha synuclein locus has already been genetically linked to familial Parkinson's disease (140)]. It remains to be determined whether the 5′UTR specific IRE in ASYN mRNA is functional. Scherzer et al., (2008) demonstrated a GATA motif in intron-2 of the alpha synuclein gene (141). This study is consistent with possibility that the IRE in the ASYN 5′UTR may be fully functional and provides substantial further evidence for a role of alpha synuclein in iron metabolism.
B.6. APP mRNA Stability Coupled to Translation, Role of 5′UTR and 3′UTR sequences
Cytokines, which are physiologically relevant to AD, change the efficiency of APP mRNA translation and APP mRNA stability by signaling through 3′UTR RNA sequences (142) additional to the APP 5′UTR (113). By this route APP gene expression is increased at the post transcriptional level to generate more template for the deposition of more amyloid Aβ. Two cis-acting elements in the APP 3′UTR regulate the stability of the precursor transcript where TGFβ induces a 68 kDa protein to bind to an 81nt motif and thus stabilize the APP mRNA (143). Addition of serum growth factors overrides the action of another 3′UTR element, a 29-base sequence (2285-2313), that normally destabilizes APP mRNA in endothelial cells (and peripheral blood lymphocytes) (144).
In astrocytes, we found that APP gene expression was up-regulated by IL-1 at the translational level via 5′UTR sequences by a regulatory pattern similar to that for the universal iron storage protein, ferritin (112) (113). de Sauvage et al., (1991) reported that polyadenylation site selection in the APP mRNA 3′UTR was critical for its efficient translation in xenopus oocytes (145). The longer 3.3 Kb APP mRNA was translated 3-fold more efficiently than the shorter 3.042 Kb transcript. Mbella et al (2000) then observed that these 3′UTR sequences enhanced APP mRNA translation in mammalian (CHO) cells, and more closely mapped two guanosine residues that are crucial for this action (146). These authors used RNA gel-shift assays to determine that a translational repressor protein interacts with the shorter transcript (146). Thus in addition to the APP 5′UTR system, the presence of the 3′UTR specific 258 nucleotide poly(A) regulatory region (PAR) (nt +3042 to +3300) removes binding of this APP mRNA repressor and facilitates longer APP mRNA translation (146).
In sum, APP 3′ untranslated region sequences operate in conjunction with the APP 5′UTR to establish the post-transcriptional control of APP gene expression (and Aβ production) (146). Future studies will rigorously test how APP mRNA 5′UTR sequences and 3′UTR interact to effectively regulate the translation of the Aβ-precursor protein. We will continue to characterize how the anticholinesterase phenserine, as well as the trivalent iron chelator desferrioxamine, generate anti-amyloid efficacy in vivo perhaps by inhibition of APP mRNA translation via 3′UTR sequences acting in concert with APP 5′UTR sequences (147).
When mRNAs compete for ribosome binding, neither the cap structure nor the poly(A) tail alone are sufficient for optimal translation, but together they may synergize and direct ribosome entry to the 5′ end (148-150). However, the elegant studies of Chen et al., (1995) and others demonstrated that initiation of protein synthesis by the eukaryotic translational apparatus includes the formation of circular RNAs where the 3′ end interacts with the 5′ end of any given message (151). Using this model, an APP mRNA circularization step and internal ribosome entry may have to be invoked to explain the mechanism by which APP mRNA is translated. Consistent with an internal 40S ribosome entry mechanism to mediate APP translation, metal chelators inhibited precursor translation acting through an APP IRE stem loop that is positioned > 50 bases from the 5′ cap site of the APP transcript (15). The canonical IRE had previously been shown to no longer confer 5′ cap dependent translation when situated this distance from the 5′ cap site (92).
There is also notable RNA binding protein interaction between the coding region of the APP transcript and the fragile X mental retardation protein (FMRP), which is a cytoplasmic mRNA binding protein whose expression is absent in fragile X syndrome. Westmark and Malter (2007) showed that FMRP binds to the coding region of APP mRNA at a guanine-rich, G-quartet-like sequence (152). Stimulation of cortical synaptoneurosomes or primary neuronal cells with the metabotropic glutamate receptor agonist, DHPG, increased APP translation in wild-type but not fmr-1 knockout samples. This interaction may affect APP levels in vivo. Indeed, this mechanism may explain a recent human study by Sokol and colleagues who detected high levels of plasma APP in children with severely autistic behavior and aggression (153). Finally, a predicted IRE-like stem loop was found in the Aβ region of APP mRNA, and IRP1 and IRP2 may interact at this RNA sequence (154). However, mutations silent for familial AD disrupt this predicted IRE, thus down playing its pathogenic importance (155). We propose this coding sequence IRE-like stem loop present in Aβ may represent a binding site of an, as yet uncharacterized, disease associated RNA binding proteins other than IRP1 and IRP2.
B.7. Disease relevance of IRE dependent APP translational control
The brains of AD patients may exhibit disrupted iron distribution with a changed brain pattern of iron regulatory proteins (156). As discussed, ferritin is the universal iron storage protein, and the transferrin receptor (TfR) is the universal receptor responsible for transferrin-mediated iron transport. Ferritin and TfR are respectively regulated by modulated binding of IRPs to the 5′ cap site-and 3′UTR specific IRE RNA stem loops in each transcript (85). It has been demonstrated that 30% of AD brain samples displayed a stronger interaction between the IRP1 and IRP2 and the canonical IRE stem loop in the 5UTRs of the transcripts coding for H-ferritin and the 3′ untranslated region of the transferrin receptor mRNA (TfR-mRNA) (129). Since IRP binding to the IREs controls intracellular iron homeostasis, a change in this RNA binding affinity would be predicted to enhance iron transport into neuronal cells during AD, but decrease ferritin levels, and thus diminish the cellular iron storage capacity. In these circumstances, neuronal cells in the AD brain would harbor an enlarged cellular pool of dangerous unstored iron (129). This model of reduced ferritin synthesis may be accurate at earlier stages in AD progression, but the hypothesis has to account for the fact that most amyloid plaques contain ferritin-rich cells. To account for this discrepancy, ferritin appears to be deposited into plaques at later stages in disease progression when iron homeostasis in neurons has been reset by the enhanced preceding increase in transferrin receptor activity. Certainly ferritin synthesis of microglial, rather than neuronal origin, may also account for most plaque associated ferritin in the AD brain (157).
Our report that the 5′ untranslated region of APP mRNA is related to the 5′UTRs of L- and H-ferritin mRNAs (113) provided a further direct link between iron metabolism and AD pathogenesis. IRP1/IRP2 appear to be the specific trans-activators of the APP 5′UTR translational control, and thereby central regulators of APP translation (8). The post-transcriptional regulatory domains in the APP transcript include the APP 5′ untranslated region comprising both IL-1 and iron dependent enhancers of APP translation. The presence of a functional IRE in the APP 5′UTR is a further indication for the essential requirements of metal metabolism for the regular hitherto unknown function of APP. This view is supported by the finding that the APP cytoplasmic tail can be interchanged with that of the transferrin receptor and maintain 50% endocytosis of transferrin bound iron (158).
C. CHELATION THERAPY FOR ALZHEIMER'S DISEASE AND PARKINSON'S DISEASE
C.1. Introduction (Overview of iron and copper chelators as therapeutic agents for AD treatment)
The presence of metals in the plaque of AD patients (Fig 1), and of increased iron in striatal neurons in the Substantia nigra (SN) of PD patients (7), together with the demonstration of iron dependent translation of APP mRNA (8), have each encouraged the development of metal attenuating agents and iron chelators as a major new therapeutic strategy for the treatment of these neurodegenerative diseases (159,160). In the case of AD, metal based therapeutics complements the use of anti Aβ passive immunity and the use of an amyloid vaccine (161) as part of the trend to combination therapeutic strategies to ameliorate the cognitive decline of AD patients. In keeping with a metal regulatory element (IRE-Type II) in the APP 5′untranslated region, we identified both iron and copper chelators to be a major class of APP 5′UTR-directed leads from our screen of a library of 1,200 FDA pre-approved drugs (162). Drug screens targeted to the APP 5′ untranslated region identified dimercaptopropanol (Hg and Pb chelators), and tetrathiolmobdylate (Cu chelator) as FDA pre-approved leads that limited APP holoprotein expression and (Aβ-peptide output) (163).
Iron and copper chelators appear to operate at two levels as therapeutic agents for the treatment of AD. A clinical trial that was run in Toronto (Canada) showed that desferrioxamine provided clear and effective therapeutic relief to Alzheimer's patients as registered by measurement of cognitive performance (75). Notably desferrioxamine, which is a highly specific iron chelator (Kd Fe3+ =10-31 M), suppressed intracellular levels of APP holoprotein by inhibiting translation of APP mRNA from it's 5′ untranslated region 5′UTR) (164). An alternative mechanism for the anti-amyloid efficacy of chelators comes from the reported broad specificity of the copper-zinc-iron chelator, clioquinol, that dissolved extra cellular fibrillar Aβ and thereby diminished plaque burden (165).
Clioquinol has been tested in a Phase II clinical trial in Melbourne Australia, wherein the metal binding drug improved cognition in severe AD patients (166) but was withdrawn from trials because of impurities during preparation. More recently a replacement small molecule metal-protein attenuating compound, of the 8-hydroxyquinoline class PBT-2 has shown promise in later trials (13). PBT-2 was deemed more brain penetrable, and was designed to be safer and more efficacious than parent compound clioquinol.
C.2. Iron chelators (inhibitors of APP mRNA translation)
Although APP transcription is up-regulated by copper (167), Fe chelation with DFO consistently suppressed APP expression (and Aβ levels) without any change to APP mRNA levels in SH-SY5Y cells (8). A major mechanism for the observed anti-amyloid efficacy of Fe chelators is that these agents inhibit APP translation through the active iron responsive IRE RNA stem-loop in the APP 5′UTR (76). In support, EGCG in Chinese green tea exhibited potent iron-chelating activity comparable to that of the prototypic iron chelator desferrioxamine, and was found to dose dependently (1-10 μM) inhibit both the immature and full-length cellular holo-APP, as shown by two-dimensional gel electrophoresis, without altering APP mRNA levels. EGCG operated via the APP 5′UTR (9).
C.2.1. Intramuscular Injection of Desferrioxamine
Daily intramuscular injection of the intracellular Fe3+ chelator, desferrioxamine, was shown to decrease the cognitive decline in a large cohort of AD patients (75). Desferrioxamine (DFO) was at first thought to chelate aluminium, which was considered a risk factor for AD (168). However desferrioxamine is commonly used as an iron chelator in the treatment of Sickle Cell disease (169), and for treatment of patients suffering from poisoning during acute iron overload (170-172). Thus the mechanism of action of desferrioxamine in the treatment of AD is likely associated with iron chelation rather than aluminum chelation. Desferrioxamine appears to specifically suppresses APP mRNA translation (Figure 4) (164).
Desferrioxamine (Df) has a dissociation constant for binding to Fe 3+ at 10-31M which provides the very high specificity for the chelation of iron required for the treatment of patients with transfusion iron overload (170-172). As such DFO has been very closely monitored as clinical agent for 20 years (171). The main clinical drawback of desferrioxamine is that the chelator can only be administered by intramuscular injection (usually to Sickle Cell Disease patients in crisis). There has been an active program to introduce new orally active iron chelators (170-172).
C.2.2. Iron Chelators to Supercede desferioxamine (Deferiprone and Ferralex)
Deferiprone is the orally active iron chelator that has been most often used for the treatment of iron overload disorders (173). It will be of interest to test whether deferiprone provides efficacy for AD based on the experimental capacity of chelators to limit APP translation, as has been observed for both desferrioxamine (Fe3+ chelator) and dimercaptopropanol (Hg2+, Cu2+ chelator)(174). There is controversy concerning the use of deferiprone as an agent to replace desferrioxamine since the chelator has caused hepatic fibrosis in cases of transfusion iron overload (172).
C.2.3. Iron Chelation and Tau Phosphorylation
Aside from the amyloid based model for AD, iron enhanced phosphorylation of Tau and the formation of the neurofibrillary tangles associated with the AD brain (160). In this report a novel trivalent cationic chelator, Feralex, effectively dissociated binding of aluminum and iron to hyperphosphorylated Tau (neurofibrillary tangle) pertinent to its use as a therapeutic agent for AD ((159)).
C.2.4. Future Directions for Iron Chelators
New oral iron chelators are rapidly being developed to combat disorders of iron overload such as sickle cell anemia, transfusional iron overload, including (a) the hexadentatephenolicaminocarboxylate HBED [n,N'bis(2-hydroxybenzyl)ethylenediamine-N,N-diacetic acid], (b) the tridentate desferrithiocin derivative 4′-OH-dadmDFT[4′dihydroxy-(S)-deszaddesmethyl-desferrithiocin, (S)-4,5-dihydro-2-(2,4-dihydroxylphenyl)-4-thiazolecarboxylic acid], (c) the tridentate triazole ICL670A[CGP72 670A; 4-[3,5-bis-(hydroxylphenyl)-1,2,4triazol-1-yl]-benzoic acid], and (d), the bidentate hydroxypyridin-4-one deferoprone [L1,CP20: 1,2-dimethyl-3-hydroxypyridin-4-one] (170). The successful use of new oral iron chelators for blood-associated diseases will predict their therapeutic capacity for the treatment of AD. Their efficacy will be measured using the same both histochemical and nuclear magnetic resonance imaging techniques that originally identified that the brain cortex of Alzheimer's disease patients displayed an abnormal iron distribution of (58). New Fe chelators such as PBT2 (175) and Ferelex are under investigation (PBT2 in Phase II clinical trials for improvement in cognitive performance (159)). Some of their therapeutic impact may be mediated by mechanisms of translational repression of APP gene expression to subsequently generate anti-amyloid efficacy.
C.3. Antioxidants Retard Metal-dependent Amyloid Induced Oxidative Stress and Neurotoxicity
For a long time oxidative stress has been strongly associated with the neurotoxicity in Alzheimer's disease (176-178). It has been recently shown that over-expression of superoxide dismutase -1 protected neurons against Abeta-amyloid peptide toxicity (179). The antioxidant, Co-enzyme Q, has already been tested for efficacy in animal models receiving increased oxidative stress, and is currently being tested for efficacy to AD (180).
One important mechanism to link Aβ peptide induced neurotoxicity and oxidative damage came from the finding that Aβ itself, is a generator of metal-catalyzed oxidative stress (Aβ (1-42) > Aβ (1-40) (71,181). Electron spin trap measurements have shown that Aβ expressed in C-elegans does generate superoxides in solution (182). However, Fe, Cu and Zn bind strongly with Aβ peptide (42). Huang et al., (1999) demonstrated that Cu and Fe are reduced by Aβ peptides, and that this catalytic reaction transfers electrons to molecular oxygen thereby generating neurotoxic H2O2 and superoxide ions (44). Using in-vitro assays, the presence of iron or copper (100 nM) with Aβ (10 μM) was found to catalyze H2O2 production (71). Redox interactions between Aβ and Cu(II) and Fe(III) engender production of reduced metal ions, Cu(I) and Fe(II), and consequential generation of ROS- , H2O2 and OH-. Like Cu(II), Zn(II) precipitates Aβ in-vitro (Aβ 1-42 > Aβ 1-40 > rat Aβ (1-40).
Amyloid induced neuronal loss in AD (44) can be explained by the binding of Aβ-peptides to copper and iron to catalyze the generation of toxic H2O2. Equation 1 shows the standard Fenton reaction by which iron or copper react with hydrogen peroxide and superoxides to generate the toxic and deadly hydroxyl radicals that mediate neuronal loss during AD (183). The presence of reduced copper (Cu1+) and iron (Fe2+) also implied that even more damaging hydroxyl radical would be formed by the Fenton and Haber-Weiss chemical reactions (39,184) (Equation 1).
Equation 1.
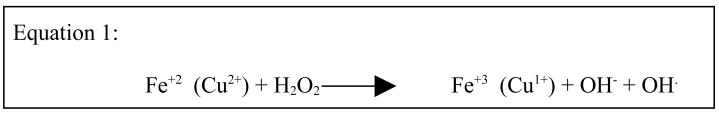
Metal-catalyzed neuronal oxidative damage via hydroxyl radicals.
The pathologically damaging production of hydrogen peroxide generated by Aβ may be the result of a corruption of a beneficial superoxide dismutase activity associated with Aβ. In fact, Aβ dimer may form a copper - zinc protein (8 kDa) protein that has intrinsic superoxide dismutase activity that is capable of converting O2- into H2O2. (185). This hydrogen peroxide can then be converted into two water molecules by catalase to complete an antioxidant function. Pathogenic corruption of Aβ function may occur when oxygen is consumed as a substrate instead of O2- radicals, thereby changing a potentially cytoprotective peptide into a pro-oxidant which has deleterious activity.
Trace amounts of copper in water induce Aβ-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease (63). At a therapeutic level, it is worth remembering that Zn2+ is redox-inert and appears to play an inhibitory role in H2O2-mediated Aβ toxicity by copper. After co-incubation of zinc with Cu(II), Zn(II) was found to rescue primary cortical and human embryonic kidney 293 cells that were exposed to Aβ 1-42, correlating with the effect of Zn(II) in suppressing Cu(II)-dependent H2O2 formation from Aβ 1-42 (186). Other antioxidants will also prove beneficial to counteract the toxicity of the redox interaction between Fe and Cu and Aβ peptide. For example, coenzyme coenzyme Q10 is a well known nutraceutical that may serve this important antioxidant purpose for AD therapeutics (187).
C.4. Chelators (Cu Zn) that dissolve amyloid plaques and reduce oxidative burden
It is clear that the strategy of using chelators to eliminate both Aβ fibrilization and Aβ-dependent metal oxidative stress neurotoxicity could well provide a major new therapeutic impact on AD progression. Certainly the copper chelator, clioquinol, has been successfully used as an inhibitor of copper-dependent aggregation of Aβ peptide. Also clioquinol suppressed metal catalyzed H2O2 production by copper interacting with Aβ peptide (181). Thus clioquinol both chelated metals and dissolved amyloid plaques, and continuous diet with the chelator to transgenic mice over-expressing APP have improved amyloid burden, and slowed the rate of cognitive decline (165). Clioquinol underwent phase II clinical trails for its therapeutic impact to Alzheimer's Disease patients (166). As discussed, PBT-II, as developed by Prana, Ltd., is the current top replacement for clioquinol as an active agent that may dissolve amyloid plaques (188).
There remains considerable controversy as to whether copper actually accelerates the onset of AD. Two studies demonstrated that copper appeared to lower the amyloid burden in the AD brain of APP transgenic mice. In the first study Phinney et al., 2003 demonstrated an in vivo reduction of amyloid-Aβ by a mutant copper transporter (189). They used a well-known spontaneous mutation of a special strain of mutant toxic-milk mice that accumulate too much copper. Eventually those animals get a liver disease which is a facsimile of a human disorder called Wilson's disease. In direct contrast to the results of Sparks and Scheur (2003) (63), this study demonstrated that an excess of copper in the brain actually reduced of amyloid burden. Superoxide dismutase-1 activity was found to be stabilized by excess dietary copper and reduced amyloid Aβ production in APP23 transgenic mice (190). These data suggested that copper may not be is a causative risk factor in AD since excess of dietary copper can be beneficial. On the other hand copper deficiency associated with the Menkes disease protein (copper pump) reduced APP mRNA levels and APP proximal promoter activity in fibroblasts (10). This latter finding was consistent with the model that copper may enhance APP production, be amyloidogenic and act as a promoter of Alzheimer's Disease. To accommodate these controversies at the therapeutic level it may be possible to screen for drugs that alter copper transport and distribution in patients. These experimental lines will help us understand the interface between copper biology and APP biology. Interestingly, copper and iron metabolism are closely linked (133).
It is intriguing to speculate that iron-specific chelators (porphyrins) may be considered a viable therapeutic option in aging and AD (191). These findings are in keeping with an a earlier therapeutic strategy for AD that was designed to inhibit Aβ fibrilization, and thus block amyloid plaque formation in the pathogenesis of AD (192). To monitor drug efficacy in reducing the extent of Aβ fibrilization, scientists at Smith Kline and Beecham developed a monoclonal antibody-based assay (DELFIA) that detected fibril formation over the presence of monomer (dimer Aβ). Using this assay, hemin and related porphyrins were found to effectively inhibit Aβ -amyloid aggregation (191). The rank order of fibril inhibition in-vitro was hemin > hematin > zinc > protoporphyrin IX in the absence of cytotoxic action of the compounds (191).
Porphyrins and phthalocyannines also inhibited protease resistant prion protein formation relevant to Bovine Spongiform Encephalopathy (BSE) and the appearance of Creutzfeldt-Jakob disease in humans (193,194). Scrapie is transmitted by intra-peritoneal injection of protease sensitive prion (PrP -sens), and porphyrin-specific suppression of PrP conversion to a pathogenic protease resistant PRP (PrP-res) occurs in the periphery before crossing the blood brain barrier to cause encephalopathy (193). By contrast, during the progression of AD amyloid plaques form in the brain cortex whereas heme and proteoporphyrin do not cross the blood-brain barrier, and thus it is uncertain that the use of porphyrines will have therapeutic impact in this case. Also shorter protofibrils of prefibrillar, diffusible assemblies of Aβ peptide are deleterious agents during the progression of Alzheimer's disease. These facts might argue against identifying drugs, like heme, targeted to inhibit the formation of larger fibrils as a treatment for AD (195).
Conclusions
In summary, brain levels of Zn, Cu, and Fe, and their binding proteins are dysregulated in the brain cortex of AD patients and dopaminergic neurons of PD patients (67,157). Levels of Zn and Fe are increased to concentrations as high as 1 mM in cerebral amyloid plaques (Cu is at 400 μM) (6). Normally Zn, Cu, and Fe are concentrated in the temporal cortex at relatively lower levels [eg: 7.94 μM Fe / mg protein, in the temporal cortex and 20 μM Fe / mg in the motor cortex (67)]. The distributions of both the brain iron storage proteins, transferrin and ferritin, are also altered in the white matter versus the gray matter in the brains of AD patients compared to age matched controls (69,196). Key to this review we discuss the implications of our discovery of fully functional IRE RNA stem loops in the 5′UTRs of the AD specific APP mRNA and the PD specific ASYN mRNA, which each encode two key proteins and causative agents for these neurodegenerative diseases. Iron also causes neural damage, behavioral changes and microgliosis in mouse behavioral models of each disease and we have not ruled out a role for the APP and ASYN IREs to contribute to the pathology of AD and PD. Thus, an alternative mechanism for the action of Fe chelators (such as DFO and EGGC) in their therapeutic efficacy against neurodegenerative diseases such as AD and PD, is their role as translation inhibitors of APP (or indeed alpha synuclein) transcripts. By contrast metal attenuating agents such as clioquinol and PBT-2 may well preferentially operate at the protein levels to dissolve amyloid plaques.
Abbreviations
- APP
Amyloid Precursor Protein
- P97
mellanotransferrin
- PS
Presenillin
- ADAM
A Disintegrin and Metalloprotease Domain-10
- ADAM-17
A Disintegrin and Metalloprotease Domain
- TACE-1
Tumor Necrosis Factor alpha Converting Enzyme
- ORF
Open Reading Frame
- ASYN
Alpha synuclein
- IRE
Iron-responsive Element
- IRP
Iron-regulatory Protein
- Cu
Copper
- Fe
Iron
- Zn
Zinc
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kosik K, Ahn J, Stein R, Yeh L. Discovery of compounds that will prevent tau pathology. J Mol Neurosci. 2002;19:261–266. doi: 10.1385/JMN:19:3:261. [DOI] [PubMed] [Google Scholar]
- 2.Lu M, Kosik K. Competition for microtubule-binding with dual expression of Tau missense and splice isoforms. Mol Biol Cell. 2001;12:171–184. doi: 10.1091/mbc.12.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J. Framing β-amyloid. Nature Genetics. 1992;1:233–234. doi: 10.1038/ng0792-233. [DOI] [PubMed] [Google Scholar]
- 5.Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008;283:22529–22540. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 7.Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA, Elstner M, Morris CM. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology. 2007;68:1820–1825. doi: 10.1212/01.wnl.0000262033.01945.9a. [DOI] [PubMed] [Google Scholar]
- 8.Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 9.Reznichenko L, Amit T, Zheng H, Avramovich-Tirosh Y, Youdim MB, Weinreb O, Mandel S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer's disease. J Neurochem. 2006;97:527–536. doi: 10.1111/j.1471-4159.2006.03770.x. [DOI] [PubMed] [Google Scholar]
- 10.Bellingham SA, Lahiri DK, Maloney B, La Fontaine S, Multhaup G, Camakaris J. Copper depletion down-regulates expression of Alzheimer's disease Amyloid-beta precursor protein gene. J Biol Chem. 2004 doi: 10.1074/jbc.M400805200. [DOI] [PubMed] [Google Scholar]
- 11.Friedlich AL, Tanzi RE, Rogers JT. The 5′-untranslated region of Parkinson's disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol Psychiatry. 2007;12:222–223. doi: 10.1038/sj.mp.4001937. [DOI] [PubMed] [Google Scholar]
- 12.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 13.Barnham KJ, Bush AI. Metals and neuroscience. Curr Opin Chem Biol. 2008 doi: 10.1016/s1367-5931(99)00073-3. [DOI] [PubMed] [Google Scholar]
- 14.Komori N, Kittel A, Kang D, Shackelford D, Masliah E, Zivin JA, Saitoh T. Reversible ischemia increases levels of Alzheimer amyloid protein precursor without increasing levels of mRNA in the rabbit spinal cord. Brain Res Mol Brain Res. 1997;49:103–112. doi: 10.1016/s0169-328x(97)00133-2. [DOI] [PubMed] [Google Scholar]
- 15.Bandyopadhyay S, Huang X, Cho H, Greig NH, Youdim MB, Rogers JT. Metal specificity of an iron-responsive element in Alzheimer's APP mRNA 5′untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J Neural Transm. 2006:237–247. doi: 10.1007/978-3-211-33328-0_25. [DOI] [PubMed] [Google Scholar]
- 16.Bellingham SA, Ciccotosto GD, Needham BE, Fodero LR, White AR, Masters CL, Cappai R, Camakaris J. Gene knockout of amyloid precursor protein and amyloid precursorlike protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J Neurochem. 2004;91:423–428. doi: 10.1111/j.1471-4159.2004.02731.x. [DOI] [PubMed] [Google Scholar]
- 17.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. see comments. [DOI] [PubMed] [Google Scholar]
- 18.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of β-amyloid. Nature Genetics. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 19.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 20.Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 21.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 22.Wisniewski KE, Dalton AJ, McLachlan C, Wen GY, Wisniewski HM. Alzheimer's disease in Down's syndrome: clinicopathologic studies. Neurology. 1985;35:957–961. doi: 10.1212/wnl.35.7.957. [DOI] [PubMed] [Google Scholar]
- 23.Rovelet-Lecrux A, Hannequin D, Raux G, Meur NL, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 24.Rovelet-Lecrux A, Frebourg T, Tuominen H, Majamaa K, Campion D, Remes AM. APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 2007 doi: 10.1136/jnnp.2006.113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Multhaup G, Schlicksupp A, Hesse L, Beher D, Ruppert T, masters C, Beyreuther K. The Amyloid Precursor protein of Alzheimer's disease in the reduction of copper(II) to copper(I) Science. 1996;271:1406–1409. doi: 10.1126/science.271.5254.1406. [DOI] [PubMed] [Google Scholar]
- 26.Bandyopadhyay S, Goldstein LE, Lahiri DK, Rogers JT. Role of the APP Non-Amyloidogenic Signaling Pathway and Targeting alpha-Secretase as an Alternative Drug Target for Treatment of Alzheimer's Disease. Curr Med Chem. 2007;14:2848–2864. doi: 10.2174/092986707782360060. [DOI] [PubMed] [Google Scholar]
- 27.Bandyopadhyay S, Hartley DM, Cahill CM, Lahiri DK, Chattopadhyay N, Rogers JT. Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J Neurosci Res. 2006;84:106–118. doi: 10.1002/jnr.20864. [DOI] [PubMed] [Google Scholar]
- 28.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 29.LaVoie M, Fraering P, Ostaszewski B, Ye W, Kimberly W, Wolfe M, Selkow D. Assembly of the gamma-secretase complex involves early formation of an intermediate of APh-1 and nicastrin. J Biol Chem. 2003;278:37213–37222. doi: 10.1074/jbc.M303941200. [DOI] [PubMed] [Google Scholar]
- 30.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. see comments. [DOI] [PubMed] [Google Scholar]
- 31.De Strooper B. Aph-1, Pen, and Nicastrin with presenilin generate an active gamma secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 32.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee VM, Giasson BI, Trojanowski JQ. More than just two peas in a pod: common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci. 2004;27:129–134. doi: 10.1016/j.tins.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci. 2000;20:6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, et al. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 38.Duka T, Rusnak M, Drolet RE, Duka V, Wersinger C, Goudreau JL, Sidhu A. Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. Faseb J. 2006;20:2302–2312. doi: 10.1096/fj.06-6092com. [DOI] [PubMed] [Google Scholar]
- 39.Bush AI. The Metallobiology of Alzheimer's Disease. Trends in Neurosciences. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 40.Nilsson LN, B K, DiCarlo G, Gordon MN, Morgan D, Paul SM, Potter H. Alpha-1-antichymotrypsin promotes beta-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:1444–1451. doi: 10.1523/JNEUROSCI.21-05-01444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu G, Huang W, Moir RD, Vanderburg CR, Lai B, Peng Z, Tanzi RE, Rogers JT, Huang X. Metal exposure and Alzheimer's pathogenesis. J Struct Biol. 2006;155:45–51. doi: 10.1016/j.jsb.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 42.Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, Romano DM, Hartshorn MA, Tanzi RE, Bush AI. Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 43.Atwood C, Huang X, Khatri A, Scarpa R, Kim YS, Moir RD, Tanzi RE, Roher AE, Bush AI. Copper catalyzed oxidation of Alzheimer Aβ. Cellular and Molecular Biology. 2000;46:777–783. [PubMed] [Google Scholar]
- 44.Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, Stokes KC, Leopold M, Multhaup G, et al. Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 45.Bush A, Pettingell WH, Multhaup G, ParadisD, Vonsattel, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Rapid induction of Azheimer A-beta amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 46.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corder E, Saunders AM, Strittmatter MD, Schmechel DE, Gaskell PC, Rimmler MS, Locke BA, Conneally PM, Schmader KE, Tanzi RE, Gusella J, Small GW, Roses AD, Pericak-Vance MA, Haines JL. Apolipoprotein E survival in Alzheimer's disease patients, and the competing risks of death and Alzheimer's disease. Neurology. 1995;45:1323–1328. doi: 10.1212/wnl.45.7.1323. [DOI] [PubMed] [Google Scholar]
- 48.Qiu Z, Strickland DK, Hyman BT, Rebeck GW. Alpha2-macroglobulin enhances the clearance of endogenous soluble beta- amyloid peptide via low-density lipoprotein receptor-related protein in cortical neurons. J Neurochem. 1999;73:1393–1398. doi: 10.1046/j.1471-4159.1999.0731393.x. [DOI] [PubMed] [Google Scholar]
- 49.Farris W, Mansurian S, Chang Y, Lindsey L, Eckman E, Frosch M, Eckman C, Tanzi R, Selkoe D, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta protein, and the beta amyloid precursor protein intracellular domain in vivo. PNAS. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 51.Zatta P, Messori L, Mauri P, van Rensburg SJ, van Zyl J, Gabrielli S, Gabbiani C. The C2 variant of human serum transferrin retains the iron binding properties of the native protein. Biochim Biophys Acta. 2005;1741:264–270. doi: 10.1016/j.bbadis.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 52.Zambenedetti P, De Bellis G, Biunno I, Musicco M, Zatta P. Transferrin C2 variant does confer a risk for Alzheimer's disease in caucasians. J Alzheimers Dis. 2003;5:423–427. doi: 10.3233/jad-2003-5601. [DOI] [PubMed] [Google Scholar]
- 53.Robson KJ, Lehmann DJ, Wimhurst VL, Livesey KJ, Combrinck M, Merryweather-Clarke AT, Warden DR, Smith AD. Synergy between the C2 allele of transferrin and the C282Y allele of the haemochromatosis gene (HFE) as risk factors for developing Alzheimer's disease. J Med Genet. 2004;41:261–265. doi: 10.1136/jmg.2003.015552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Connor JR, Lee SY. HFE mutations and Alzheimer's disease. J Alzheimers Dis. 2006;10:267–276. doi: 10.3233/jad-2006-102-311. [DOI] [PubMed] [Google Scholar]
- 55.Lee SY, Patton SM, Henderson RJ, Connor JR. Consequences of expressing mutants of the hemochromatosis gene (HFE) into a human neuronal cell line lacking endogenous HFE. Faseb J. 2007;21:564–576. doi: 10.1096/fj.06-6397com. [DOI] [PubMed] [Google Scholar]
- 56.Sampietro M, Caputo L, Casatta A, Meregalli M, Pellagatti A, Tagliabue J, Annoni G, Vergani C. The hemochromatosis gene affects the age of onset of sporadic Alzheimer's disease. Neurobiol Aging. 2001;22:563–568. doi: 10.1016/s0197-4580(01)00219-6. [DOI] [PubMed] [Google Scholar]
- 57.Thompson K, Menzies S, Muckenthaler M, Torti F, Wood T, Torti S, Nentze M, Beard M, Connor J. Mouse brains deficient in H-ferritin have normal iron nconcentration but protein profile of iron deficiency and increased oxidative stress. J Neuroscie Res. 2003;71:46–63. doi: 10.1002/jnr.10463. [DOI] [PubMed] [Google Scholar]
- 58.Bartzokis G, Sultzer D, Cummings J, Holt LE, Hance DB, Henderson VW, Mintz J. In vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Arch Gen Psychiatry. 2000;57:47–53. doi: 10.1001/archpsyc.57.1.47. [DOI] [PubMed] [Google Scholar]
- 59.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 60.Schubert D, Chevion M. The role of iron in beta amyloid toxicity. Biochem Biophys Res Commun. 1995;216:702–707. doi: 10.1006/bbrc.1995.2678. [DOI] [PubMed] [Google Scholar]
- 61.Bishop G, Robinson S. Human Abeta1-42 reduces iron-induced toxicity in rat cerbral cortex. J Neuroscie Res. 2003;73:316–323. doi: 10.1002/jnr.10661. [DOI] [PubMed] [Google Scholar]
- 62.Bishop G, Robinson S, Liu Q, Perry G, Atwood C, Smith M. Iron: a pathological mediator of Alzheimer's disease? Dev Neurosci. 2002;24:184–187. doi: 10.1159/000065696. [DOI] [PubMed] [Google Scholar]
- 63.Sparks D, Schreurs B. Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer's disease. PNAS. 2003;100:11065–11069. doi: 10.1073/pnas.1832769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kennard M, Richardson D, Gabathuler R, Ponka P, Jeffries W. A novel iron uptakemechanism mediated by GPI-anchored human p97. EMBO J. 1995;14:4178–4186. doi: 10.1002/j.1460-2075.1995.tb00091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jeffries W, Dickstein D, Ujiie M. Assessing p97 as an Alzheimer's disease serum biomarker. J Alzheimers Dis. 2001;3:339–344. doi: 10.3233/jad-2001-3309. [DOI] [PubMed] [Google Scholar]
- 66.Ujiie M, Dickstein D, Jeffries W. p97 as a biomarker for Alzheimer's disease. Front Biosci. 2002;7:42–47. doi: 10.2741/ujiie. [DOI] [PubMed] [Google Scholar]
- 67.Connor JR, Snyder BS, Arosio P, Loeffler DA, LeWitt P. A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer's diseased brains. J Neurochem. 1995;65:717–724. doi: 10.1046/j.1471-4159.1995.65020717.x. [DOI] [PubMed] [Google Scholar]
- 68.Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- 69.Robinson S, Noone DF, Kril J, Haliday GM. Most amyloid plaques contain ferritin rich cells. Alzheimer's Research. 1995;1:191–193. [Google Scholar]
- 70.Abraham CR, Selkoe DJ, Potter H, Price DL, Cork LC. Alpha 1-antichymotrypsin is present together with the beta-protein in monkey brain amyloid deposits. Neuroscience. 1989;32:715–720. doi: 10.1016/0306-4522(89)90292-3. [DOI] [PubMed] [Google Scholar]
- 71.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, et al. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 72.Mantyh PW, Ghilardi JR, Rogers S, DeMaster E, Allen CJ, Stimson ER, Maggio JE. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J Neurochem. 1993;61:1171–1174. doi: 10.1111/j.1471-4159.1993.tb03639.x. [DOI] [PubMed] [Google Scholar]
- 73.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 74.Bush AI, Atwood C, Goldstein L, Huang X, Rogers JT. Could Aβ and βPP be antioxidants? J Alzheimer's Disease. 2000;2:83–84. doi: 10.3233/jad-2000-2203. [DOI] [PubMed] [Google Scholar]
- 75.Crapper McLachlan D, Dalton AJ, Kruck TPA, Bell MY, Smith WL, Kalow W, Andrews DF. Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 76.Rogers J. APP and Ferritin Translation, Metals and Alzheimer's Disease, in Metals and Medicine. In: Sessler J, editor. J Am Chem Soc. Vol. 13 2006. [Google Scholar]
- 77.Lesne S, Docagne F, Gabriel C, Liot G, Lahiri DK, Buee L, Plawinski L, Delacourte A, MacKenzie ET, et al. Transforming growth factor-beta 1 potentiates amyloid-beta generation in astrocytes and in transgenic mice. Journal of Biological Chemistry. 2003;278:18408–18418. doi: 10.1074/jbc.M300819200. [DOI] [PubMed] [Google Scholar]
- 78.Payton S, Cahill CM, Randall JD, Gullans SR, Rogers JT. Drug discovery targeted to the Alzheimer's APP mRNA 5'-untranslated region: the action of paroxetine and dimercaptopropanol. J Mol Neurosci. 2003;20:267–275. doi: 10.1385/JMN:20:3:267. [DOI] [PubMed] [Google Scholar]
- 79.Tucker S, Ahl M, Cho HH, Bandyopadhyay S, Cuny GD, Bush AI, Goldstein LE, Westaway D, Huang X, Rogers JT. RNA Therapeutics Directed to the Non Coding Regions of APP mRNA, In Vivo Anti-Amyloid Efficacy of Paroxetine, Erythromycin, and N-acetyl cysteine. Curr Alzheimer Res. 2006;3:221–227. doi: 10.2174/156720506777632835. [DOI] [PubMed] [Google Scholar]
- 80.Rogers J. Genetic regulation of the iron transport and storage genes: Links with the acute phase response. In: Lauffer R, editor. Iron and Human diseases. CRC Press; Boca Raton: 1992. [Google Scholar]
- 81.Balla G, J H, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM. Ferritin: A cytoprotective antioxident strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- 82.Rogers JT, Lahiri DK. Metal and inflammatory targets for Alzheimer's disease. Curr Drug Targets. 2004;5:535–551. doi: 10.2174/1389450043345272. [DOI] [PubMed] [Google Scholar]
- 83.Gordeuk V, Prithviraj P, Dolinar T, Brittenham GM. Interleukin-1 administration in mice produces hypoferremia despite neutropenia. J Clin Invest. 1988;82:1934–1938. doi: 10.1172/JCI113812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tran TN, Eubanks SK, Schaffer KJ, Zhou CY, Linder MC. Secretion of ferritin by rat hepatoma cells and its regulation by inflammatory cytokines and iron. Blood. 1997;90:4979–4986. [PubMed] [Google Scholar]
- 85.Thomson AM, Rogers JT, Leedman PJ. Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation. Int J Biochem Cell Biol. 1999;31:1139–1152. doi: 10.1016/s1357-2725(99)00080-1. [DOI] [PubMed] [Google Scholar]
- 86.Gunshin H, Allerson CR, Polycarpou-Schwarz M, Rofts A, Rogers JT, Kishi F, Hentze MW, Rouault TA, Andrews NC, Hediger MA. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. 2001;509:309–316. doi: 10.1016/s0014-5793(01)03189-1. [DOI] [PubMed] [Google Scholar]
- 87.Kim HY, Klausner RD, Rouault TA. Translational repressor activity is equivalent and is quantitatively predicted by in vitro RNA binding for two iron-responsive element-binding proteins, IRP1 and IRP2. J Biol Chem. 1995;270:4983–4986. doi: 10.1074/jbc.270.10.4983. [DOI] [PubMed] [Google Scholar]
- 88.Kaldy P, Menotti E, Moret R, Kuhn LC. Identification of RNA-binding surfaces in iron regulatory protein-1. Embo J. 1999;18:6073–6083. doi: 10.1093/emboj/18.21.6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eisenstein R, Theil E. Combinatorial mRNA Regulation; Iron regulatory Protein and Iso Iron-responsive elements. J Biol Chem. 2000;275:40659–40662. doi: 10.1074/jbc.R000019200. [DOI] [PubMed] [Google Scholar]
- 90.Leibold E, Munro HN. Cytoplasmic protein binds in-vitro to a highly conserved sequence in the 5′ untranslated region of ferritin heavy- and light-subunit mRNAs. PNAS. 1988;85:2171–2175. doi: 10.1073/pnas.85.7.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leedman P, Stein AR, Chin WW, Rogers JT. Thyroid hormone modulates the interaction between the iron regulatory protein and ferritin mRNA Iron-responsive elements. J Biol Chem. 1996;271:12017–12023. doi: 10.1074/jbc.271.20.12017. [DOI] [PubMed] [Google Scholar]
- 92.Goossen B, Hentze MW. Position is the critical determinant for function of iron-responsive elements as translational regulators. Mol Cell Biol. 1992;12:1959–1966. doi: 10.1128/mcb.12.5.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Walden WE, Selezneva AI, Dupuy J, Volbeda A, Fontecilla-Camps JC, Theil EC, Volz K. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science. 2006;314:1903–1908. doi: 10.1126/science.1133116. [DOI] [PubMed] [Google Scholar]
- 94.Dupuy J, Volbeda A, Carpentier P, Darnault C, Moulis JM, Fontecilla-Camps JC. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure. 2006;14:129–139. doi: 10.1016/j.str.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 95.Martelli A, Salin B, Dycke C, Louwagie M, Andrieu JP, Richaud P, Moulis JM. Folding and turnover of human iron regulatory protein 1 depend on its subcellular localization. Febs J. 2007;274:1083–1092. doi: 10.1111/j.1742-4658.2007.05657.x. [DOI] [PubMed] [Google Scholar]
- 96.Muckenthaler M, Gray NK, Hentze MW. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol Cell. 1998;2:383–388. doi: 10.1016/s1097-2765(00)80282-8. [DOI] [PubMed] [Google Scholar]
- 97.Wu S, Kaufman RJ. Double-stranded (ds) RNA binding and not dimerization correlates with the activation of the dsRNA-dependent protein kinase (PKR) J Biol Chem. 1996;271:1756–1763. doi: 10.1074/jbc.271.3.1756. [DOI] [PubMed] [Google Scholar]
- 98.Thomson AM, Rogers JT, Leedman PJ. Thyrotropin-releasing hormone and epidermal growth factor regulate iron- regulatory protein binding in pituitary cells via protein kinase C-dependent and -independent signaling pathways. J Biol Chem. 2000;275:31609–31615. doi: 10.1074/jbc.M002354200. In Process Citation. [DOI] [PubMed] [Google Scholar]
- 99.Pinero DJ, Hu J, Cook BM, Scaduto RC, Connor JR. Interleukin-1beta increases binding of the iron regulatory protein and the synthesis of ferritin by increasing the labile iron pool. Biochim Biophys Acta. 2000;1497:279–288. doi: 10.1016/s0167-4889(00)00066-5. In Process Citation. [DOI] [PubMed] [Google Scholar]
- 100.Caltagirone A, Weiss G, Pantopoulos K. Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J Biol Chem. 2001;276:19738–19745. doi: 10.1074/jbc.M100245200. [DOI] [PubMed] [Google Scholar]
- 101.Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pantopolulos K, Hentze MW. Rapid responses to oxidative sress mediated by iron regulatory protein. EMBO. 1995;14:2917–2924. doi: 10.1002/j.1460-2075.1995.tb07291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Toth I, Yuan L, Rogers JT, Boyce H, Bridges KR. Hypoxia alters iron-regulatory protein-1 binding capacity and modulates cellular iron homeostasis in human hepatoma and erythroleukemia cells. J Biol Chem. 1999;274:4467–4473. doi: 10.1074/jbc.274.7.4467. [DOI] [PubMed] [Google Scholar]
- 104.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 105.Iwai K, Drake SK, Wehr NB, Weissman AM, LaVaute T, Minato N, Klausner RD, Levine RL, Rouault TA. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: implications for degradation of oxidized proteins. Proc Natl Acad Sci U S A. 1998;95:4924–4928. doi: 10.1073/pnas.95.9.4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hanson ES, Rawlins ML, Leibold EA. Oxygen and iron regulation of iron regulatory protein 2. J Biol Chem. 2003;278:40337–40342. doi: 10.1074/jbc.M302798200. [DOI] [PubMed] [Google Scholar]
- 107.Dycke C, Bougault C, Gaillard J, Andrieu JP, Pantopoulos K, Moulis JM. Human iron regulatory protein 2 is easily cleaved in its specific domain: consequences for the haem binding properties of the protein. Biochem J. 2007;408:429–439. doi: 10.1042/BJ20070983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cartwright G, Lauritsen MA, Humphreys S, Jones PJ, Merrill IM, Wintrobe MM. Anemia associated with chronic infection. Science. 1946;103:72. doi: 10.1126/science.103.2664.72. [DOI] [PubMed] [Google Scholar]
- 109.Konijn AM, Carmel N, Levy R, Hershko C. Ferritin synthesis in inflammation. II. Mechanism of increased ferritin synthesis. Br J Haematol. 1981;49:361–370. doi: 10.1111/j.1365-2141.1981.tb07238.x. [DOI] [PubMed] [Google Scholar]
- 110.Rogers JT, Bridges KR, Durmowicz GP, Glass J, Auron PE, Munro HN. Translational control during the acute phase response. Ferritin synthesis in response to interleukin-1. J Biol Chem. 1990;265:14572–14578. [PubMed] [Google Scholar]
- 111.Rogers JT, A J, Lacroix L, Kasschau KD, Durmowicz GP, Bridges KR. Translational regulation of H-ferritin mRNA by Il-1β acts through 5′ leader sequences distinct from the Iron Responsive Element. Nucl Acids Research. 1994;22:2678–2686. doi: 10.1093/nar/22.13.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rogers JT. Ferritin translation by interleukin-1: the role of sequences upstream of the start codons of the heavy and light subunit genes. Blood. 1996;87:2525–2537. [PubMed] [Google Scholar]
- 113.Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN. Translation of the alzheimer amyloid precursor protein mRNA is up- regulated by interleukin-1 through 5′-untranslated region sequences. J Biol Chem. 1999;274:6421–6431. doi: 10.1074/jbc.274.10.6421. [DOI] [PubMed] [Google Scholar]
- 114.Thomson AM, Cahill CM, Cho HH, Kassachau KD, Epis MR, Bridges KR, Leedman PJ, Rogers JT. The Acute Box cis-Element in Human Heavy Ferritin mRNA 5′-Untranslated Region Is a Unique Translation Enhancer That Binds Poly(C)-binding Proteins. J Biol Chem. 2005;280:30032–30045. doi: 10.1074/jbc.M502951200. [DOI] [PubMed] [Google Scholar]
- 115.Chkheidze AN, Lyakhov DL, Makeyev AV, Moraqles J, Kong J, Liebhaber SA. Assembly of the α-Globin mRNA Stability Complex Reflects Binary Interaqxtion between thre Pyrimidine-Rich 3' Untranslated Region Determinant and Poly(C) Binding Protein αCP. Mol Cell Biol. 1999;19:4572–4681. doi: 10.1128/mcb.19.7.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ostareck DH, Ostareck-Lederer A, Wilm M, Thiele BJ, Mann M, Hentze MW. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3′ end. Cell. 1997;89:597–606. doi: 10.1016/s0092-8674(00)80241-x. [DOI] [PubMed] [Google Scholar]
- 117.Hentze M, Kuhn LC. Molecular control of vertebrate iron mertabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. PNAS. 1996;83:8175–8182. doi: 10.1073/pnas.93.16.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jacobs A, Woodworth M. Ferritin in Serum. N Engl J Med. 1975;292:951–956. doi: 10.1056/NEJM197505012921805. [DOI] [PubMed] [Google Scholar]
- 119.Broxmeyer H, Cooper S, Arosio P. Mutated recombinant human heavy-chain ferrittins and myelosuppression in vitro and in-vivo: A link between ferritin ferroxidase activity and biological function. PNAS. 1991;88:770–774. doi: 10.1073/pnas.88.3.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lustlader E, Hann EWL, Blumberg BS. Serum Ferritin as a predictor of host response to hepatitis Bvirus infection. Science. 1983;220:423–425. doi: 10.1126/science.6301008. [DOI] [PubMed] [Google Scholar]
- 121.Ramadori G, Sipe JD, Dinarello CA, Mizel B, Colten HR. Pretranslational modulation of acute phase hepatic protein synthesis by murine recombinant interleukin I (IL-1) and purified human IL-1. J Exp Med. 1985;162:930–942. doi: 10.1084/jem.162.3.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang W, Di X, D'Agostino RB, Jr, Torti SV, Torti FM. Excess capacity of the iron regulatory protein system. J Biol Chem. 2007;282:24650–24659. doi: 10.1074/jbc.M703167200. [DOI] [PubMed] [Google Scholar]
- 123.Maloney B, Ge YW, Greig N, Lahiri DK. Presence of a “CAGA box” in the APP gene unique to amyloid plaque-forming species and absent in all APLP-1/2 genes: implications in Alzheimer's disease. Faseb J. 2004;18:1288–1290. doi: 10.1096/fj.03-1703fje. [DOI] [PubMed] [Google Scholar]
- 124.Lahiri D, Chen D, vivien D, Ge YW, Greig N, Rogers J. the role of cytokines in the gene expression of amyloid b-protein precursor: Identification of a 5′UTR Binding nuclear factor and its implications for Alzheimer's Disease. JAD. 2003;5:81–90. doi: 10.3233/jad-2003-5203. [DOI] [PubMed] [Google Scholar]
- 125.Lahiri DK, Ge YW, Maloney B. Characterization of the APP proximal promoter and 5′-untranslated regions: identification of cell type-specific domains and implications in APP gene expression and Alzheimer's disease. Faseb J. 2005;19:653–655. doi: 10.1096/fj.04-2900fje. [DOI] [PubMed] [Google Scholar]
- 126.Fujioka M, Taoka T, Matsuo Y, Mishima K, Ogoshi K, Kondo Y, Tsuda M, Fujiwara M, Asano T, et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann Neurol. 2003;54:732–747. doi: 10.1002/ana.10751. [DOI] [PubMed] [Google Scholar]
- 127.Zhu Y, Sun Y, Mao XO, Jin KL, Greenberg DA. Expression of poly(C)-binding proteins is differentially regulated by hypoxia and ischemia in cortical neurons. Neuroscience. 2002;110:191–198. doi: 10.1016/s0306-4522(01)00522-x. [DOI] [PubMed] [Google Scholar]
- 128.Feng Y, Zhang F, Lokey LK, Chastain JL, Lakkis L, Eberhart D, Warren ST. Translational suppression by trinucleotide repeat expansion at FMR1. Science. 1995;268:731–734. doi: 10.1126/science.7732383. [DOI] [PubMed] [Google Scholar]
- 129.Pinero DJ, Hu J, Connor JR. Alterations in the interaction between iron regulatory proteins and their iron responsive element in normal and Alzheimer's diseased brains. Cell Mol Biol (Noisy-Le-Grand) 2000;46:761–776. In Process Citation. [PubMed] [Google Scholar]
- 130.Melofors O, Goossen B, Johansson B, Stripecke R, Gray NK, Hentze MW. Translational control of 5-aminolevulinate synthase mRNA by Iron-responsive Elements in erythroid cells. J Biol Chem. 1993;268:5974–5978. [PubMed] [Google Scholar]
- 131.Cox TC, Bawden MJ, Martin A, May BK. Human erythroid 5-aminolevulinate synthase: promoter analysis and identification of an iron-responsive element in the mRNA. Embo J. 1991;10:1891–1902. doi: 10.1002/j.1460-2075.1991.tb07715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cox LA, Adrian GS. Posttranscriptional regulation of chimeric human transferrin genes by iron. Biochemistry. 1993;32:4738–4745. doi: 10.1021/bi00069a007. [DOI] [PubMed] [Google Scholar]
- 133.Chung J, Haile DJ, Wessling-Resnick M. Copper-induced ferroportin-1 expression in J774 macrophages is associated with increased iron efflux. Proc Natl Acad Sci U S A. 2004;101:2700–2705. doi: 10.1073/pnas.0306622101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Knutson MD, Vafa MR, Haile DJ, Wessling-Resnick M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood. 2003;102:4191–4197. doi: 10.1182/blood-2003-04-1250. [DOI] [PubMed] [Google Scholar]
- 135.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 136.Smith SR, Cooperman S, Lavaute T, Tresser N, Ghosh M, Meyron-Holtz E, Land W, Ollivierre H, Jortner B, et al. Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann N Y Acad Sci. 2004;1012:65–83. doi: 10.1196/annals.1306.006. [DOI] [PubMed] [Google Scholar]
- 137.Jellinger KA. Alpha-synuclein pathology in Parkinson's and Alzheimer's disease brain: incidence and topographic distribution--a pilot study. Acta Neuropathol (Berl) 2003;106:191–201. doi: 10.1007/s00401-003-0725-y. [DOI] [PubMed] [Google Scholar]
- 138.Cantuti-Castelvetri I, Klucken J, Ingelsson M, Ramasamy K, McLean PJ, Frosch MP, Hyman BT, Standaert DG. Alpha-synuclein and chaperones in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2005;64:1058–1066. doi: 10.1097/01.jnen.0000190063.90440.69. [DOI] [PubMed] [Google Scholar]
- 139.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 141.Scherzer CR, Grass JA, Liao Z, Pepivani I, Zheng B, Eklund AC, Ney PA, Ng J, McGoldrick M, et al. GATA transcription factors directly regulate the Parkinson's disease-linked gene {alpha}-synuclein. Proc Natl Acad Sci U S A. 2008;105:10907–10912. doi: 10.1073/pnas.0802437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rajagopalan LE, Malter JS. Growth factor-mediated stabilization of amyloid precursor protein mRNA is mediated by a conserved 29-nucleotide sequence in the 3′- untranslated region. J Neurochem. 2000;74:52–59. doi: 10.1046/j.1471-4159.2000.0740052.x. [DOI] [PubMed] [Google Scholar]
- 143.Amara FM, Junaid A, Clough RR, Liang B. TGF-beta(1), regulation of alzheimer amyloid precursor protein mRNA expression in a normal human astrocyte cell line: mRNA stabilization. Brain Res Mol Brain Res. 1999;71:42–49. doi: 10.1016/s0169-328x(99)00158-8. [DOI] [PubMed] [Google Scholar]
- 144.Zaidi SH, Malter JS. Amyloid precursor protein mRNA stability is controlled by a 29-base element in the 3′-untranslated region. J Biol Chem. 1994;269:24007–24013. [PubMed] [Google Scholar]
- 145.de Sauvage F, Kruys V, Marinx O, Huez O, Octave JN. Alternative polyadenylation of the amyloid protein precursor mRNA. EMBO. 1992;11:3099–3103. doi: 10.1002/j.1460-2075.1992.tb05382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mbella EG, Bertrand S, Huez G, Octave JN. A GG nucleotide sequence of the 3′ untranslated region of amyloid precursor protein mRNA plays a key role in the regulation of translation and the binding of proteins. Mol Cell Biol. 2000;20:4572–4579. doi: 10.1128/mcb.20.13.4572-4579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Venti A, Giordano T, Eder P, Lahiri DK, Greig NH, Rogers JT. The Integrated Role of Desferrioxamine and Phenserine Targeted to an Iron-Responsive Element in the APP-mRNA 5′-Untranslated Region. Ann N Y Acad Sci. 2004;1035:34–48. doi: 10.1196/annals.1332.003. [DOI] [PubMed] [Google Scholar]
- 148.Sachs AB, Sarnow P, Hentze MW. Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell. 1997;89:831–838. doi: 10.1016/s0092-8674(00)80268-8. [DOI] [PubMed] [Google Scholar]
- 149.Preiss T, Muckenthaler M, Hentze MW. Poly(A)-tail-promoted translation in yeast: implications for translational control. Rna. 1998;4:1321–1331. doi: 10.1017/s1355838298980669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Preiss T, Hentze MW. Dual function of the messenger RNA cap structure in poly(A)-tail- promoted translation in yeast. Nature. 1998;392:516–520. doi: 10.1038/33192. [DOI] [PubMed] [Google Scholar]
- 151.Chen CY, Sarnow P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science. 1995;268:415–417. doi: 10.1126/science.7536344. [DOI] [PubMed] [Google Scholar]
- 152.Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007;5:e52. doi: 10.1371/journal.pbio.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sokol DK, Chen D, Farlow MR, Dunn DW, Maloney B, Zimmer JA, Lahiri DK. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J Child Neurol. 2006;21:444–449. doi: 10.1177/08830738060210062201. [DOI] [PubMed] [Google Scholar]
- 154.Tanzi RE, Hyman BT. Alzheimer's mutation. Nature. 1991;350:564. doi: 10.1038/350564a0. [DOI] [PubMed] [Google Scholar]
- 155.Zubenko G, Farr J, Stiffler S, Hughes HB, Kaplan BB. Clinically-silent mutation in the putative Iron-Responsive Element in exon 17 of the beta-amyloid precursor protein gene. J Neuropath Expt Neurol. 1992;51:459–463. doi: 10.1097/00005072-199207000-00008. [DOI] [PubMed] [Google Scholar]
- 156.Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer's disease. J Neurosci Res. 1992;31:327–335. doi: 10.1002/jnr.490310214. [DOI] [PubMed] [Google Scholar]
- 157.Grundke-Iqbal I, Fleming J, Tung YC, Lassmann H, Iqbal K, Joshi JG. Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta Neuropathol. 1990;81:105–110. doi: 10.1007/BF00334497. [DOI] [PubMed] [Google Scholar]
- 158.Lai A, Sisodia SS, Trowbridge IS. Characterization of the sorting signals in the beta-amyloid precursor protein cytoplasmic domain. J Biol Chem. 1995;270:3565–3573. [PubMed] [Google Scholar]
- 159.Kruck TP, Percy ME, Lukiw WJ. Metal sulfate-mediated induction of pathogenic genes and repression by phenyl butyl nitrone and Feralex-G. Neuroreport. 2008;19:245–249. doi: 10.1097/WNR.0b013e3282f4cb7e. [DOI] [PubMed] [Google Scholar]
- 160.Shin R, Kruck T, Murayama H, Kitamoto T. A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated tau of Alzheimer's disease. Brain Res. 2003;961:139–146. doi: 10.1016/s0006-8993(02)03893-3. [DOI] [PubMed] [Google Scholar]
- 161.Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF. Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 162.Rogers JT, R J, Eder PS, Huang X, Bush AI, Tanzi RE, Venti A, Payton SM, Giordano T, Nagano S, Cahill CM, Moir R, Lahiri DK, Greig N, Sarang SS, Gullans SR. Alzheimer's disease drug discovery targeted to the APP mRNA 5′untranslated region. J Mol Neurosci. 2002;19:77–82. doi: 10.1007/s12031-002-0014-6. [DOI] [PubMed] [Google Scholar]
- 163.Payton S, Cahill C, Randall J, Gullans S, Rogers J. Alzheimer's Disease Drug Discovery Targeted to the APP mRNA 5′Untranslated region; paroxetine and dimercaptopropanol are drug hits. Journal of molecular Neuroscience. 2003;20:267–275. doi: 10.1385/JMN:20:3:267. [DOI] [PubMed] [Google Scholar]
- 164.Rogers JT, R J, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig N, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans S. An iron-responsive element type II in the 5′ untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 165.Cherny R, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:641–642. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 166.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny RA, Li QX, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer's disease: a pilot phase 2 clinical trial. Archives of Neurology. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 167.Bellingham SA, Lahiri DK, Maloney B, La Fontaine S, Multhaup G, Camakaris J. Copper depletion down-regulates expression of the Alzheimer's disease amyloid-beta precursor protein gene. J Biol Chem. 2004;279:20378–20386. doi: 10.1074/jbc.M400805200. [DOI] [PubMed] [Google Scholar]
- 168.Campbell A, Bondy SC. Aluminum induced oxidative events and its relation to inflammation: a role for the metal in Alzheimer's disease. Cell Mol Biol (Noisy-Le-Grand) 2000;46:721–730. In Process Citation. [PubMed] [Google Scholar]
- 169.Halliwell & Hedlund. Therapeutic Strategies to inhibit iron-catalyzed tissue damage. Iron and Human Disease. 1992:477–500. [Google Scholar]
- 170.Brittenham G. Iron chelators and iron toxicity. Alcohol. 2003;30:151–158. doi: 10.1016/s0741-8329(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 171.Brittenham G, Nathan D, Oliveieri N, Pippard M, Weatherall D. Deferiprone versus desferrioxamine in thalassaemia, and T2 validation and utility. The Lancet. 2003;361 doi: 10.1016/s0140-6736(03)12223-4. [DOI] [PubMed] [Google Scholar]
- 172.Brittenham G, Nathan D, Olivieri N, Porter J, Pippard M, Vichinsky E, Weatherall D. Deferiprone and hepatic fibrosis. Blood. 2003;101:5089–5091. doi: 10.1182/blood-2002-10-3173. [DOI] [PubMed] [Google Scholar]
- 173.Konoghiorghes G, Neocleous K, Kolnagou A. Benefits and risks of deferiprone in iron overload in Thalassaemia and other conditions: comparison of epidemiological and therapeutic aspects with deferoxamine. Drug Safety. 2003;26:553–584. doi: 10.2165/00002018-200326080-00003. [DOI] [PubMed] [Google Scholar]
- 174.Morse LJ, Payton SM, Cuny GD, Rogers JT. FDA-Preapproved Drugs Targeted to the Translational Regulation and Processing of the Amyloid Precursor Protein. J Mol Neurosci. 2004;24:129–136. doi: 10.1385/JMN:24:1:129. [DOI] [PubMed] [Google Scholar]
- 175.Doraiswamy PM, Xiong GL. Pharmacological strategies for the prevention of Alzheimer's disease. Expert Opin Pharmacother. 2006;7:1–10. doi: 10.1517/14656566.7.1.1. [DOI] [PubMed] [Google Scholar]
- 176.Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, Wiggert B, Petersen RB, Perry G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer's disease. Am J Pathol. 1994;145:42–47. [PMC free article] [PubMed] [Google Scholar]
- 178.Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Celsi F, Ferri A, D'Amrosi N, Rotliiio G, Costa A, Volonte C, Carri M. Overexpression of superoxide dismutase 1 protects against beta=amyloid peptide toxicity: effect of estrogen and copper chelators. Neurochem Int. 2004;44:25–33. doi: 10.1016/s0197-0186(03)00101-3. [DOI] [PubMed] [Google Scholar]
- 180.Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Ann N Y Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- 181.Opazo C, Huang X, Cherny RA, Moir RD, Roher AE, White AR, Cappai R, Masters CL, Tanzi RE, et al. Metalloenzyme-like activity of Alzheimer's disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2) J Biol Chem. 2002;277:40302–40308. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 182.Yatin SM, Aksenova M, Aksenov M, Markesbery WR, Aulick T, Butterfield DA. Temporal relations among amyloid beta-peptide-induced free-radical oxidative stress, neuronal toxicity, and neuronal defensive responses. J Mol Neurosci. 1998;11:183–197. doi: 10.1385/JMN:11:3:183. [DOI] [PubMed] [Google Scholar]
- 183.Halliwell B, G JMC. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gutteridge JM. Hydroxyl radicals, iron, oxidative stress, and neurodegeneration. Ann N Y Acad Sci. 1994;738:201–213. doi: 10.1111/j.1749-6632.1994.tb21805.x. [DOI] [PubMed] [Google Scholar]
- 185.Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem. 2001;276:20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 186.Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI. Evidence that the beta-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem. 2000;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- 187.Beal MF, Shults CW. Effects of Coenzyme Q_{10} in Huntington's disease and early Parkinson's disease. Biofactors. 2003;18:153–161. doi: 10.1002/biof.5520180218. [DOI] [PubMed] [Google Scholar]
- 188.Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, et al. Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 189.Phinney A, Drisaldi B, Schmidt S, Lugowski S, Coronado V, Liang Y, Horne P, Yang J, Sekoulidis J, et al. In vivo reduction of amyloid-β by a mutant copper transporter. PNAS. 2003;100:14193–14198. doi: 10.1073/pnas.2332851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bayer T, Schafer S, Simons A, Kemmling A, Kamer T, Tepests R, Eckert A, Schussel K, Eikenberg O, et al. Dietry Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Aneta production in APP23 transgenic mice. PNAS. 2003;100:14187–14192. doi: 10.1073/pnas.2332818100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Howlett D, Cutler P, Heales S, Camilleri P. Hemin and related porphyrins inhibit beta-amyloid aggregation. FEBS Lett. 1997;417:249–251. doi: 10.1016/s0014-5793(97)01290-8. [DOI] [PubMed] [Google Scholar]
- 192.Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 193.Priola SA, Raines A, Caughey WS. Porphyrin and phthalocyanine antiscrapie compounds. Science. 2000;287:1503–1506. doi: 10.1126/science.287.5457.1503. see comments. [DOI] [PubMed] [Google Scholar]
- 194.Caughey WS, Raymond LD, Horiuchi M, Caughey B. Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc Natl Acad Sci U S A. 1998;95:12117–12122. doi: 10.1073/pnas.95.21.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 196.Connor JR, Menzies SL, St Martin SM, Mufson EJ. A histochemical study of iron, transferrin, and ferritin in Alzheimer's diseased brains. J Neurosci Res. 1992;31:75–83. doi: 10.1002/jnr.490310111. [DOI] [PubMed] [Google Scholar]
- 197.Bandyopadhyay S, Ni J, Ruggiero A, Walshe K, Rogers MS, Chattopadhyay N, Glicksman MA, Rogers JT. A high-throughput drug screen targeted to the 5′untranslated region of Alzheimer amyloid precursor protein mRNA. J Biomol Screen. 2006;11:469–480. doi: 10.1177/1087057106287271. [DOI] [PubMed] [Google Scholar]