

Abstract
Objectives
Pompe disease is a progressive neuromuscular disorder due to acid alpha glucosidase (GAA) deficiency. Cross-reacting immunologic material (CRIM)-negative infants with null GAA mutations have the most severe phenotype and develop anti-GAA antibodies following exposure to enzyme replacement therapy (ERT). Antibodies influence bio-distribution, attenuate the beneficial effects of ERT, and are believed to influence infusion-associated reactions (IARs), which occur in nearly all early-onset patients. We evaluated the potential that B-cell depletion prior to ERT initiation would block GAA antibody responses and improve clinical outcome.
Study Design
Six Pompe subjects (four CRIM-negative) between 2–8 months of age received rituximab and sirolimus or myophenolate prior to ERT. Four subjects continued to receive sirolimus, every 12-week rituximab and monthly intravenous immunoglobulin for the duration of ERT. Sirolimus trough levels, IgG levels, CD3, CD4, CD8, CD19, CD20, NTproBNP, CK, CK-MB, CRP, platelet, alkaline phosphatase, AST, ALT were measured regularly.
Results
Immunomodulation achieved B-cell depletion without adverse effects. After 17–36 months of rituximab, sirolimus and ERT, all subjects lacked antibodies against GAA, four continued to gain motor milestones, yet two progressed to require invasive ventilation. Absence of infusion associated reactions allowed accelerated infusion rates. No IARs were observed at standard or accelerated infusion rates.
Conclusions
B-cell depletion and T-cell immunomodulation in infants naïve to ERT was accomplished safely, eliminated immune responses against GAA, thereby optimizing clinical outcome, however this approach did not necessarily influence sustained independent ventilation. Importantly, study outcomes support the concept of initiating immunomodulation prior to beginning ERT since the study regimen allowed for prompt initiation of treatment.
Keywords: immunomodulation, rituximab, sirolimus, ERT, acid alpha glucosidase (GAA), antibodies
Introduction
Immune responses to therapeutic proteins in patients with enzyme deficiencies secondary to severe mutations have been shown to limit long-term efficacy of enzyme replacement therapy (ERT) and have been particularly well-described in Pompe disease [1]. Infantile onset Pompe disease is the most severe phenotype of this recessive disorder and results in death from cardiorespiratory failure within 12–24 months in the absence of treatment. Milder forms of the disease presenting in childhood or in adults are the result of less deleterious mutations. Generally, acid alpha-glucosidase (GAA) activity less than 1% of wild type level correlates with presentation in infancy, while 2–20% GAA activity is seen in later-onset disease [1]. Approximately 25% of infants with less than 1% GAA activity have no detectable GAA protein by western blot and are considered cross-reactive immunological material (CRIM) negative [2].
In CRIM-positive patients, the presence of residual GAA protein usually correlates with a lack of antibody (Ab) formation against GAA after initiation of ERT. In contrast, CRIM-negative patients lack tolerance to GAA protein and mount robust humoral immune responses to ERT, attenuating the efficacy to treatment. In a related adult study, 40% of administered recombinant alglucosidase alfa (Myozyme®, Genzyme) was captured by circulating anti-GAA Ab [3]. Hence, CRIM-negative patients on ERT have a poor prognosis and diminished survival unless some form of immunosuppression is administered [4]. Immunosuppressive drugs that have been used with some success include daily oral cyclophosphamide (cyclophosphamide, Roxane Laboratories-Boehringer Ingelheim), methotrexate (methotrexate, Mylan), omalizumab (omalizumab, Genentech/Novartis) and rituximab (rituximab, Genentech and Biogen) as well as plasmapheresis [5, 6]. In this 5-year study, we address the immunologic and clinical consequences of B-cell depletion and T-cell immunomodulation prior to initiation of ERT in infants with early onset Pompe Disease.
METHODS
Subjects
Five infants with the diagnosis of Pompe disease, GAA enzyme activity of less than 1% and confirmed GAA mutations were enrolled into an observational study of Pompe disease at the University of Florida. Parents consented to pre-ERT immunosuppression between February 2007 and November 2010. Data from an additional CRIM-positive patient with infantile-onset Pompe disease enrolled into the observational study who did not receive pre-ERT immunosuppression is included as a reference subject. The end date for analysis of results was March 15, 2012.
The protocol was approved by the University of Florida Institutional Review Board. The patients’ parents were informed that standard of care treatment for this disease was initiation of ERT as soon as the diagnosis was confirmed by GAA activity assay and was available as alternative therapy to the proposed treatment. Stated risks of the immunomodulatory regimen included risk of infection, anaphylaxis, malignancy and death. Written informed consent was obtained from the parents prior to initiation of immunosuppression.
Study Design
Inclusion criteria for the study included diagnosis of Pompe disease before 12 months of age, cardiac hypertrophy as defined by 2D Left Ventricular Mass Index (LVMI) of greater than 2 z-scores, GAA activity less than 1% in peripheral blood mononuclear cells (PBMC) or dried blood spot, absence of infection or complication that could be worsened by systemic immunosuppression, and no prior exposure to ERT.
After consent, all subjects received methylprednisolone (methylprednisolone, Prizer) 10 mg/kg intravenously (IV) and induction rituximab, which was dosed one of two ways depending on the infant’s clinical status and ability to tolerate IV fluids. Subjects (A, E) received two 750 mg/M2 doses of rituximab, 10–14 days apart. Remaining subjects received a loading dose of rituximab 375 mg/M2 per week for three weeks, to lessen the fluid load with each administration. After rituximab induction doses, each subject was placed on daily oral immunosuppression and received sirolimus (sirolimus, Wyeth) at a dose of 0.6–1 mg/M2 per day adjusted to maintain a goal trough serum sirolimus level of 3–7 ng/m; one subject received mycophenolate (mycophenolic acid, Roche) 300 mg/M2 per day, which was used at the beginning of this study protocol. After induction rituximab followed by oral immunosuppression, all patients began recombinant human alglucosidase alfa (20 mg/kg IV every 7–10 days), infused over six hours initially. ERT dosing interval was subsequently increased to every 10–14 days if clinical improvement was demonstrated as measured by discontinuation of ventilatory assistance (invasive or non-invasive) and attainment of feeding goals as well as discharge from the inpatient setting. ERT infusion rates were also increased stepwise over time to achieve a goal of two-hour infusions periods as long as infusion reactions were not observed and no anti-GAA antibodies were detected.
Once the induction doses of rituximab were completed, all subjects began monthly IVIG (Gamunex, Talecris Biotherapeutics or Privigen, CSL Behring AG) at a dose of 500–1000 mg/kg, adjusted to maintain a trough serum IgG level of 700–1000 mg/dL. IVIG was given to provide passive immunity since subjects were not permitted to receive well-child vaccines other than the seasonal inactivated influenza vaccine for the duration of B-cell depletion. After initiation of ERT, maintenance rituximab at a dose of 375 mg/M2 every 12 weeks was continued in four of five infants.
Mutation Analysis
Archive quality DNA was isolated from PBMC using Puregene Blood Core Kits (Quiagen, USA) and then purified (MinElute, Quiagen, USA) before PCR amplification and sequencing. PCR amplified products containing all 20 exons and the flanking intronic sequences of the GAA gene were generated using published flanking primers (Integrated DNA Technologies), sequenced at the Interdisciplinary Centre for Biotechnology Research (ICBR, University of Florida, FL) using the Applied Biosystems Model 3130 Genetic Analyzer, and then compared to published sequences (GenBank Accession: NT_024871.11) using Clone Manager Professional Suite version 8.
CRIM assay
CRIM status was determined at the Powell Gene Therapy Center (Gainesville, FL) by Western blot analysis of skin fibroblasts. Twenty micrograms (μg) of cell lysate was subjected to SDS-PAGE, run on 4–12% gradient Tris-glycine gels, and transferred to nitrocellulose membranes. Blots were probed with 1:10,000 dilution of a protein-A purified rabbit anti-GAA polyclonal Ab allowing detection of unprocessed and processed forms of GAA, followed by incubation with goat anti-rabbit IRDye 800CW Ab at 1:20,000 dilution (LI-COR Biosciences, NE). The membranes were scanned using infrared imaging (Odyssey, LI-COR Biosciences, NE) (Figure 1A), and quantification of GAA was reported as integrated intensity, proportional to the amount of membrane-bound Ab (Figures 1B and 1C).
Figure 1.
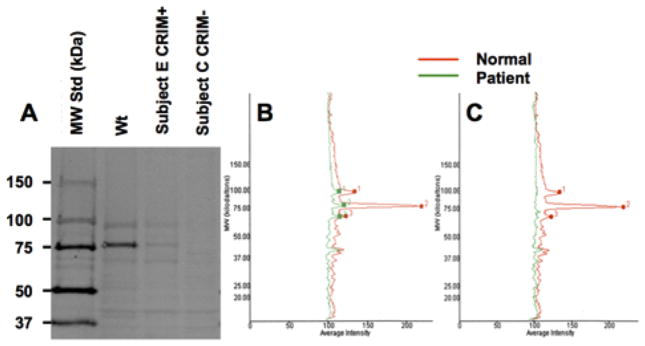
A. Representative CRIM Western Blot for normal control, CRIM-positive (Subject E) and CRIM-negative (Subject C) patients with Pompe disease. B. Integrated Intensity of CRIM-positive Subject E vs. normal control C. Integrated Intensity of CRIM negative Subject C vs. normal control.
Anti-GAA Antibody ELISA
Plasma anti-GAA titers from patients and controls were measured by enzyme linked immunosorbent assay (ELISA) at the Powell Gene Therapy Center (Gainesville, FL). Briefly, microtiter plates (Fisher, PA) were coated with 5μg/ml rhGAA protein overnight at 4 C, blocked with 10% FBS for 2 hours at room temperature (RT) and then washed with 0.05% Tween-20 in PBS. Plasma samples diluted in blocking buffer were added to wells overnight at 4°C, washed with PBS-Tween, and then incubated with 1:10,000 dilution of secondary sheep anti-human IgG-HRP Ab (Amersham PLC, UK) at RT for 2-hours. 3,3′,5,5′-tetramethylbenzidine (TMB; Invitrogen, CA) was used for color development and reactions were stopped with 0.5M sulphuric acid. Plates were read at 450nm wavelength using a μQuant analyzer (Bio-Tek Instruments, VT) and anti-GAA Ab titers were determined using standard curves generated with positive and negative controls.
Immune Markers
Enumeration of B and T lymphocyte subpopulations in peripheral blood was done by flow cytometry using monoclonal Ab to CD19 or CD20 for B cells and CD3, CD4 and CD8 for T cells in the UF Shands Hospital Hematopathology Laboratory (Gainesville, FL).
RESULTS
Subjects
Subjects with infantile Pompe disease (three females, three males) were from Florida and included three Hispanic, two Caucasian and one African American infant. Key features of the initial symptoms included difficulty feeding, failure to thrive and cardiomegaly detected between the ages of 3 weeks and 5 months. All subjects received pre-ERT immunosuppression and were diagnosed with Pompe disease by measurement of GAA activity (below 1% of normal between 6 weeks and 7 months of age) and severe cardiac hypertrophy (LVMI greater than 2 z-scores; Table 1). The reference subject is a CRIM-positive Caucasian patient (Subject F), who received standard ERT without immunosuppression, presented with cardiac hypertrophy and was diagnosed with infantile Pompe disease by enzyme assay in skin fibroblasts at 8 months of age. Subject F was included in an earlier study [7] and is included here as a reference patient and for historical comparison. Subject E presented with severe symptoms and received the same inductive immunosuppression but was later determined to be CRIM+. CRIM− historical subjects serve as controls as all subjects anticipated to benefit from immune modulation and ERT were assigned to the study regimen. Given the established practice of initiating ERT upon establishing a diagnosis of Pompe we began immune modulation prior to GAA exposure to avoid any delay in initiating ERT.
Table 1.
Characteristics of infantile Pompe patients relative to CRIM status.
| Subject A | Subject B | Subject C | Subject D | Subject E | Subject F | |
|---|---|---|---|---|---|---|
| Sex | F | F | M | F | M | M |
| Age at Diagnosis (month) | 4.5 | 7 | 1.5 | 5.5 | 2.5 | 6 |
| Ethnicity | African American | Hispanic | Hispanic | Caucasian | Hispanic | Caucasian |
| GAA mutation allele 1 | Ex.18 c.2560C>T | Ex. 9 c.1396delG | Ex.5 c.925G>A | Ex.10 c.1548G>A | Ex. 14 c.1933G>A | Ex.3 c.655G>A |
| Nucleotide position | p.Arg854X | p.Val466Phefs X11 | p.Gly309Arg | p.Try516X | p.Asp645Asn | p.Gly219Arg |
| Type of mutation | Nonsense | Deletion, Frame shift, early stop | Missense | Nonsense | Missense | Missense |
| GAA mutation allele 2 | Ex. 18 c.2560C>T | Ex. 12 c.1705dupT | Ex.5 c.925G>A | Not found | Ex. 18 c.2501_2502 | Ex.12 c.1735G>A |
| Nucleotide position | p.Arg854X | p. Tyr569LeufsX67 | p.Gly309Arg | Not found | p.Thr834Arg X49 | p.Glu579Lys |
| Type of Mutation | Nonsense | Insertion, Frame shift, early stop – new mutation | Missense | Not found | Deletion | Missense |
| CRIM Status | Negative | Negative | Negative | Negative | Positive | Positive |
| GAA Activity | 4nmol/hr/mg (WBC) <1% | 1.3pmol/punch/hour (Dried Blood Spot) | 1.2 nmol/mg protein/hr (Leukocytes) <1% | 7–9 pmol/hr/spot (Dried Blood Spot) <1% | 9 pmol/hr/spot (Dried Blood Spot) <1% | 1.3 nmol/hr/mg (Skin Fibroblasts) <1% |
| Start Immune Suppression (month) | 8 | 8 | 2 | 5.5 | 3 | None |
| Start ERT (month) | 8 | 8 | 2.75 | 6 | 3 | ~9 |
| Age at end of study (month) | 19 | 44 | 33 | 30 | 19 | 116 |
Mutation Analysis of Early Onset Pompe Patients
Patients with Pompe disease inherit two abnormal alleles of the GAA gene located on human chromosome 17. A comparison of the identified mutations in our patients with the Pompe Center Erasmus MC Rotterdam Database, which includes mutation severity ratings, revealed that two CRIM-negative infants (Subjects A and C) were homozygous for distinct mutations known to correlate with severe and less severe disease phenotypes, respectively (Table 1). Subject A was of African American descent and was homozygous for the very severe Ex.18c2560C>T mutation common in African American patients. Subject C was homozygous for a missense mutation (Table 1). Subject B was CRIM-negative and found to be a compound heterozygote for a very severe deletion and a previously unknown insertion that introduces a translational frameshift and a premature stop codon into the protein (Ex.12c.1705dupT; Table 1). Subject D was CRIM negative; we identified one mutant allele, a putative severe nonsense mutation (Table 1). Subject E was initially identified to be CRIM-negative, but was later determined to be weakly CRIM positive by Western blot of cultured fibroblasts. The mutations in subject E includes one very severe deletion and one potentially less severe missense mutation (Table 1). Subject F was CRIM-positive by Western blot and was compound heterozygous for two less severe GAA mutations (Table 1).
Treatment Outcomes
B-cell Depletion with Induction Rituximab
At study enrollment and prior to immunosuppression, all infants had normal B-cell and T-cell numbers and subset distribution for age except for Subject C who had persistent T-lymphocytopenia from study initiation that did not improve with interruption of daily sirolimus therapy (Figure 2). All subjects had normal serum immunoglobulins (IgG, IgA, IgM) and lymphocyte proliferative responses to pokeweed mitogen, phytohemagglutinin and concanavalin A (data not shown). Induction rituximab given either as two doses of 750 mg/M2, 10–14 days apart, or at 375 mg/M2 per dose weekly for three weeks in those who were suspected to be sensitive to larger fluid volume infusion. All subjects were effectively depleted of all circulating CD19/CD20 positive B cells by flow cytometry. B-cell repopulation was not observed in subjects continuing to receive maintenance rituximab every 12 weeks while on ERT.
Figure 2.

Enumeration of Peripheral Blood CD3-positive T cells and CD19/CD20-positive B cells by Flow Cytometry in Subjects with Infantile Pompe Disease on Immunosuppression Over Time.
Subject A received daily mycophenolate 300 mg/M2 and subjects B, C, D, and E received daily sirolimus at doses adjusted to maintain a serum trough levels between 3–7 ng/ml. All infants tolerated immunosuppression well and (with the exception of Subject C), their T-cell numbers and subset distribution remained within normal limits for age for the duration of the study.
Lack of Antibody Formation Against GAA after B-cell Depletion
During the time period that subjects had undetectable peripheral CD19/CD20 positive B cells following rituximab, total serum immunoglobulins (IgA, IgM, IgG) were low and anti-GAA-specific Ab was undetectable (Figure 3 and data not shown). Subject A received only induction rituximab and without maintenance rituximab infusions, B-cells were observed after four months. He continued to receive myocphenolate during this period and anti-GAA antibody titer was associated with return of CD20 positive B-cells. Three months after B-cell recovery, the serum immunoglobulins were normalized (data not shown) and no further IVIG was given. The return of B-cell function was associated with rapid production of high titer Ab against GAA, subsequent clinical deterioration and death five months later.
Figure 3.

Anti-GAA Antibody Titers in Subjects with Infantile Pompe Disease receiving pre-ERT Immunomodulation. A) CRIM-negative Subject A treated with induction rituximab only, followed by ERT and daily mycophenolate. B) CRIM-negative subjects B, C, D, E treated with induction and maintenance rituximab, daily sirolimus and ERT. Rituximab, Rapamycin and Myozyme were discontinued for Subject B at ~13 months post initiation of treatment. C) Sequence of treatment events for immunomodulation and ERT.
Ongoing B-cell depletion with every 12-week rituximab infusions and daily sirolimus prevented anti-GAA antibody was used in the remaining infants (Subjects B, C, D, E). In subject C, discontinuation of rituximab following 10 months of ERT resulted in re-emergence of peripheral blood B cells within four months despite daily sirolimus. However, anti-GAA Ab was not detected at the time of B-cell recovery. Sixteen months after discontinuation of rituximab and 14 months after discontinuation of sirolimus, his serum anti-GAA titers remained undetectable (Figure 3). Subject C was unique in that his clinical response was the least favorable of the of the study cohort.
Lack of Infusion Reactions to Enzyme Replacement Therapy After B-cell Depletion
There was no anti-GAA Ab formation in subjects A, B, C, D and E. Accelerated infusion schedules were achieved in subjects A–E as well as subject F who had remained Ab negative because he was strongly CRIM +. ERT was administered at the standard dose of 20 mg/kg every 7–14 days; however the infusion rate was accelerated to deliver the total GAA dose over two hours. This was accomplished by giving 10% of the total dose the first 30 minutes as an initial test dose and the remaining 90% of the dose was administered in the remaining 90 minutes. There were no infusion-associated reactions observed with this schedule. Notably, more frequent infusions of ERT were achieved without adverse events. Subject F, who did not produce anti-GAA Ab, also received ERT 20 mg/kg every 14 days over two hours without complications. The accelerated infusion schedule was pursued to improve the theoretical peak concentration of GAA during infusion and to maximize delivery by receptor mediated uptake.
Adverse Effects of Immunosuppression
No subjects developed opportunistic or serious bacterial infections that could be directly attributed to immunomodulation. Subject B experienced chronic gram-negative urinary tract infections related to uncorrected grade III vesicourethral reflux despite prophylactic antibiotics as well as an episode of Clostridium difficile colitis. Subject C required biPAP and then tracheostomy and chronic invasive ventilation at 10 months of age after four months of ERT. He had multiple viral respiratory illnesses that did not resolve or improve with interruption of daily sirolimus therapy for 2–8 weeks at a time over six months of immunosuppression. After tracheostomy, Subject C restarted daily sirolimus and no longer experienced upper respiratory infections, although he continued to have decreased peripheral blood CD3 cells. However, rituximab was discontinued after 10 months and sirolimus was discontinued two months later following a serious adverse event related to complications of mechanical ventilation. The patient suffered a respiratory arrest and neurological injury following mechanical failure of assisted ventilation. He continued to receive IVIG and ERT until his transfer to a hospice facility where the decision was made to discontinue all treatments. He remained alive on assisted ventilation 16 months later at conclusion of the study. There were no adverse events in the other subjects.
Cardiac Functional Measurement
Subject A’s LVMI on echocardiogram was not significantly changed after 10 months of ERT, although there was a significant decrease in her serum level of N-terminal pro-brain natriuretic peptide (NT Pro-BNP), which is a biomarker for left ventricular function (Table 2). In contrast, Subjects B, C, D, and E who continued rituximab every 12 weeks while on ERT demonstrated reduction in LVMI as shown in Table 2. The rate of change in LVMI or degree of improvement was not significantly different than previously reported.
Table 2.
Improved cardiac markers and increased ventilator-free survival in early onset Pompe subjects receiving immunomodulation.
| Subject A | Subject B | Subject C | Subject D | Subject E | Subject F | |
|---|---|---|---|---|---|---|
| Anti-rhGAA Titer at End of Study
|
Very High | None | None | None | None | None |
| Duration of ERT (Month)
|
10 | 36 | 31 | 34 | 16 | 107 |
| LVMI before ERT (g/m2)
|
191.22 | 199.97 | 93.49 | 61.05 | 61.72 | 35.57 |
| LVMI at End of Study (g/m2)
|
162.42 | 118.15 | 44.11 | 69.05 | 61.72 | 35.57 |
| NTproBNP at Beginning of Study
|
22328 | 9110 | 8987 | >54000 | 34975 | <50 |
| NTproBNP at End of Study
|
292 | 3578 | <50 | <50 | 160 | - |
| Chronic Ventilator Support During Study (Age at ventilation in months) | No (died) | No | Yes (17) | No | No | Yes (47) |
Subsequent to this study Subject B developed the need for ventilator support at 43 months.
Impact on Ventilator-free Survival
Subject A, who received only induction rituximab, had progressive cardiopulmonary failure, and the family elected not to pursue invasive ventilation. Subject B was managed without a tracheostomy until nearly four years of age when she developed slowly progressive increased work of breathing, especially at night. She was managed for a brief period with biPAP, but eventually required invasive ventilation subsequent to end of data collection for this study. Subject C remained marginally compensated at baseline and during any illness or low grade fever, also required non-invasive ventilatory assistance and intensive care, possibly related to his baseline T-lymphocytopenia. Ultimately, he required placement of a tracheostomy and invasive ventilation at 10 months of age, despite absence of an anti-GAA antibodies. Subject D required assisted ventilation at the time of diagnosis of infantile Pompe disease for two months, but was subsequently ventilator-independent through 22 month of ERT. In contrast, subject E never required invasive ventilation and had no evidence of respiratory deterioration at the end of the study after 17 months of ERT. Subject F, who did not receive immunosuppression, developed progressive muscle weakness necessitating assisted ventilation for the last eight years.
Clinical Outcomes
Subject B developed severe mitral regurgitation as a result of cardiac dilation, which contributed to congestive heart failure and subsequent need for tracheostomy and invasive ventilatory support during sleep after 36 months of ERT. Subjects A, C, and F, developed significant sleep disturbance observed by polysomnography at the completion of the study. These findings further highlight the difficulty in managing ventilatory deficits in this patient population.
Subjects B, D and E continued to make developmental strides and gain muscle strength globally. All subjects continued to have markedly elevated creatine kinase (CK) and CK-MB levels at the end of study (data not shown). Aided by intensive physical rehabilitation, these infants were able to achieve independent sitting at the end of the study, and Subjects B and D were able to pull to standing and bear weight, however, no subject achieved independent ambulation.
All subjects showed mild cognitive delay, more in speech than receptive language, but less so than suggested by registry data [8](data not shown). Brain MRIs of Subjects B and D demonstrated abnormal myelination without atrophy after 32 months and 12 months of ERT, respectively. Subject B had mild sensorineural hearing loss bilaterally by ear-specific visual reinforcement audiometry after 32 months of ERT. Brain MRI of Subject E was normal after 11 months of ERT. Gastrostomy tubes were required in all subjects for adequate caloric intake.
Discussion
Immune response to protein replacement therapy has long been recognized as a limitation to the use of recombinant proteins, especially in recessive conditions such as Pompe disease. The first study of ERT in three infants with Pompe disease documented the clinical decline of two participants who developed high and sustained Ab titers to GAA [9]. In the pivotal study leading to the approval of alglucosidase alfa, the high rates of infusion-associated reactions (95%) and seroconversion (88%) prompted the consideration of immune modulation in Subject A, who was begun on rituximab and mycophenolate in February, 2007, immediately after the commercial release of Myozyme.
While initial studies focused on CRIM-negative patients with null GAA mutations and no residual GAA protein, recently it has become clear that some patients who are characterized as CRIM-positive by current assay methods will also develop antibody to GAA [3]. Immunomodulation of such high-risk patients using rituximab, as well as anti-IgE antibody omalizumab, have been used previously to control anti-GAA Ab production during ERT [10, 11]. However, elimination of existing Ab-producing plasma cells is a substantial challenge since these cells do not express CD20 and are resistant to rituximab, which is a humanized anti CD20 Ab and depletes by targeting the B cell surface CD20 antigen [12]. The side effects of increasingly toxic immunotherapy to inhibit pre-established anti-GAA immune responses may in fact outweigh gains achieved by ERT.
Our study indicates that immunomodulation of high-risk Pompe infants (both CRIM-negative and CRIM-positive) is safe, effectively controls anti-GAA responses, and eliminates the need for more intensive immunosuppression given after ERT initiation with the development of anti-GAA Ab and infusion associated reactions. Sirolimus, an inhibitor of the mammalian target of the rapamycin (mTOR) pathway, was used in this study rather than cytotoxic drugs or signal blockers to modulate T-cell responses to recombinant GAA. We hypothesized that sirolimus was advantageous since other evidence supports that sirolimus selectively promotes the survival and expansion of regulatory T (Treg) cells while allowing for programmed cell death of activated effector T lymphocytes [13–16]. Clinical studies support improved immune regulation using sirolimus [17]. Therefore, we believe the optimal immune modulatory regimen in this population benefits from the properties of both rituximab and sirolimus.
Pretreatment with only an induction dose of rituximab and maintenance mycophenolate proved insufficient to control anti-GAA Ab formation in Subject A. B-cell recovery and detection of low-level anti-GAA Ab after four months of ERT were the initial clues that a resurgent immune response with considerably increased anti-GAA Ab had developed in Subject A by seven months of ERT. However, rituximab pre-treatment followed by sirolimus and ERT within two weeks of the initial rituximab dosing and maintenance every 12-week rituximab proved to be a straightforward and successful protocol to avoid anti-GAA immune responses. This protocol successfully depleted B cells in the four subjects on this immunomodulatory regimen. They did not develop immune responses to GAA, including absence of anti-GAA Ab, for the duration of the study (17–36 months).
Passive immunity was provided with monthly IVIG infusions, and subjects did not have increased bacterial or viral infections other than recurrent UTIs in Subject B with uncorrected vesicourethral reflux and increased viral respiratory illnesses in Subject C until tracheostomy placement. The study cohort, which included four CRIM-negative infants, had the same rate of respiratory failure as subjects in the pivotal ERT studies that were subsequently evaluated in the Pompe registry. Study of additional patients will be necessary to substantiate this point [7, 8]. All subjects except Subject C received ERT at home, and the absence of anti-GAA allowed for accelerated infusion rates with no infusion-associated reactions (total of 433 infusions among all subjects). Subject E received immunomodulation due to the severity of his phenotype, although he was subsequently demonstrated to be weakly CRIM-positive based on Western blot of skin fibroblasts. The development of a defined CRIM assay is required to differentiate weakly CRIM-positive patients, with a high probability of developing anti-GAA immune responses, from patients who are non-responsive to alglucosidase alfa. Subject F did not develop ERT infusion reactions or related immune responses, highlighting the wide range of reactions observed among early onset Pompe disease patients and the difficulty in predicting risk of subsequent anti-GAA immune responses without an early and detailed CRIM assay. The decision to initiate immune modulation was based on the age of onset and severity of symptoms. The need for continuation of immune modulation is in our opinion requires an individualized approach since overall clinical condition and response to treatment influences this decision. Further experience in this limited patient population will certainly help establish more definitive guidelines for management of this challenging patient population.
In addition to preventing Ab formation against GAA, we determined that subjects managed as described had favorable reduction in surrogate markers of heart failure and expected reductions in left ventricular mass. The density of mannose-6-phosphate receptor and blood flow in the heart favor therapeutic protein delivery which may explain a more consistent response to ERT in the heart. Importantly, despite maximal delivery of alglucosidase alfa with early and weekly ERT dosing (up to 40 mg/kg), there was no further augmentation of effectiveness in cardiac mass reduction over that observed in the first pivotal alglucosidase alfa study[7]. We followed the same dosing regimen as in the initial pivotal study where, GAA dose was increased in subjects with declining clinical condition. This finding suggests that the limitation of current therapy is not solely due to Ab-mediated change in biodistribution of GAA. Several possible explanations include the limited phosphorylation on mannose residues of the currently available product or that trafficking of GAA from the cell surface in vivo is not as efficient as initially observed in in vitro studies. A direct impact on glycogen synthase may be another potential benefit for the use of sirolimus in this setting, given the recent findings in an animal model of Pompe disease [18]. The high dose cohort in the initial pivotal ERT study had a higher rate or infusion associated reactions whereas the subjects in this study had no infusion associated reactions, reinforcing the relationship between anti-GAA antibodies and infusion associated reactions.
Another critical outcome measure for studies in early onset Pompe disease is the management of respiratory insufficiency. Early after clinical presentation, the severe cardiac hypertrophy leads to reduction in intrathoracic volume that results in loss of left lung volume. Our subjects were all initially managed with high-flow nasal cannula using room air to avoid mechanical ventilation. The strategy may have facilitated the avoidance of assisted ventilation during the first four months of ERT in Subjects A, B, C, E. During the follow-up period, subjects were closely monitored for evidence of ventilatory insufficiency and three (Subjects A, B, C) developed progressive respiratory insufficiency to a variable degree, especially when challenged with intercurrent usually mild viral illnesses that led to increased ventilatory demand.
The proportion of children in this small cohort who required mechanical ventilation is the same as for the 15 of 38 patients in the pivotal alglucosidase alfa studies [7, 19] and in registry data [19] [20]. These findings are not unexpected because of the recent findings from preclinical studies that confirm a neural component of the respiratory dysfunction in the mouse model of Pompe disease [21]. ERT is unable to cross the blood brain barrier and has no influence on the lower motor neuron dysfunction that contributes to respiratory failure [21–23].
Evaluation of developmental outcomes in this patient cohort demonstrates improvement over that observed in other reports [7, 19] particularly in acquisition of motor skills, although all subjects remained significantly delayed compared to unaffected age-matched controls. All infants were able to sit independently and two were able to bear weight on their lower extremities with no evidence of regression in motor skills among those who were gaining function. In contrast, CRIM-negative subjects in the initial pivotal studies did not survive past 36 months of age. Similarly, receptive and expressive language delays were noted in our subjects.
In conclusion, prevention of Ab formation against GAA by B-cell depletion and T-cell immunomodulation was safe, eliminated serious ERT infusion reactions, and facilitated some improvement in clinical outcomes of affected infants. However, lack of anti-GAA did not significantly augment recovery of nerve and muscle function, or moderate residual glycogen storage leading to cardiac hypertrophy. These findings suggest that reduced efficacy of ERT is not due solely to anti-GAA effects, rather that currently available alglucosidase alfa has limited efficiency in muscle transduction and no effect on lower motor neuron function. Since immunomodulation did not significantly delay initiation of ERT and lead to overall clinical improvement for the duration of this study, we suggest immunomodulation be initiated before ERT in all cases of early onset Pompe. Moreover, this immunosuppressive strategy may prove effective for control of immune responses to therapeutic enzyme replacement in other autosomal recessive disorders as well.
Acknowledgments
This work was supported by NIH grants HL59412 (support for Sushrusha Nayak), HL107904 (to Roland Herzog and to Barry Byrne) and DK58327 (to Barry Byrne).
The faculty of the Division of Pediatric Immunology, Rheumatology, and Infectious Diseases, and the Pediatric Hospitalist Group at the University of Florida were key to the care of the study subjects. We are grateful for the assistance of Vini Vijayan, MD; Sukesh Sukumaran MD, Pamela Russell, ARNP, Jenna Lammers, PT. Lindsay Falk, RN and Sarah K Black, BS, for the coordination of this project.
Footnotes
The authors have no disclosures and no competing financial interests.
References
- 1.Byrne BJ, Kishnani PS, Case LE, Merlini L, Muller-Felber W, Prasad S, et al. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab. 2011;103:1–11. doi: 10.1016/j.ymgme.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Bali DS, Goldstein JL, Banugaria S, Dai J, Mackey J, Rehder C, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. 2012;160:40–9. doi: 10.1002/ajmg.c.31319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Vries JM, van der Beek NA, Kroos MA, Ozkan L, van Doorn PA, Richards SM, et al. High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol Genet Metab. 101:338–45. doi: 10.1016/j.ymgme.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 14:135–42. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banugaria SG, Patel TT, Mackey J, Das S, Amalfitano A, Rosenberg AS, et al. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: Need for agents to target antibody-secreting plasma cells. Mol Genet Metab. 2012 doi: 10.1016/j.ymgme.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genetics in medicine: official journal of the American College of Medical Genetics. 2009;11:210–9. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 8.Byrne BJ, Falk DJ, Pacak CA, Nayak S, Herzog RW, Elder ME, et al. Pompe disease gene therapy. Hum Mol Genet. 2011;20:R61–8. doi: 10.1093/hmg/ddr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–8. [PubMed] [Google Scholar]
- 10.Rohrbach M, Klein A, Kohli-Wiesner A, Veraguth D, Scheer I, Balmer C, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis. 2010;33:751–7. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]
- 11.Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Engl J Med. 2009;360:194–5. doi: 10.1056/NEJMc0806809. [DOI] [PubMed] [Google Scholar]
- 12.Banugaria S, Nampoothiri S, Feldman J, Kabori J, MsGann J, Koeberl D, et al. Bortezomib: a solution to the challange of antibodies in diseases treated with therapeutic proteins? Mol Genet Metab. 2011;102:S6–S7. [Google Scholar]
- 13.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–8. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 14.Bestard O, Cassis L, Cruzado JM, Torras J, Franquesa M, Gil-Vernet S, et al. Costimulatory blockade with mTor inhibition abrogates effector T-cell responses allowing regulatory T-cell survival in renal transplantation. Transpl Int. 24:451–60. doi: 10.1111/j.1432-2277.2011.01223.x. [DOI] [PubMed] [Google Scholar]
- 15.Nayak S, Cao O, Hoffman BE, Cooper M, Zhou S, Atkinson MA, et al. Prophylactic immune tolerance induced by changing the ratio of antigen-specific effector to regulatory T cells. J Thromb Haemost. 2009;7:1523–32. doi: 10.1111/j.1538-7836.2009.03548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moghimi B, Sack BK, Nayak S, Markusic DM, Mah CS, Herzog RW. Induction of tolerance to factor VIII by transient co-administration with rapamycin. J Thromb Haemost. 9:1524–33. doi: 10.1111/j.1538-7836.2011.04351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levitsky J, Mathew JM, Abecassis M, Tambur A, Leventhal J, Chandrasekaran D, et al. Systemic immunoregulatory and proteogenomic effects of tacrolimus to sirolimus conversion in liver transplant recipients. Hepatology. 2012 doi: 10.1002/hep.25579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashe KM, Taylor KM, Chu Q, Meyers E, Ellis A, Jingozyan V, et al. Inhibition of glycogen biosynthesis via mTORC1 suppression as an adjunct therapy for Pompe disease. Mol Genet Metab. 2010;100:309–15. doi: 10.1016/j.ymgme.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 21.DeRuisseau LR, Fuller DD, Qiu K, DeRuisseau KC, Donnelly WH, Jr, Mah C, et al. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci U S A. 2009;106:9419–24. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee KZ, Qiu K, Sandhu MS, Elmallah MK, Falk DJ, Lane MA, et al. Hypoglossal neuropathology and respiratory activity in pompe mice. Front Physiol. 2011;2:31. doi: 10.3389/fphys.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu K, Falk DJ, Reier PJ, Byrne BJ, Fuller DD. Spinal delivery of AAV vector restores enzyme activity and increases ventilation in Pompe mice. Mol Ther. 2012;20:21–7. doi: 10.1038/mt.2011.214. [DOI] [PMC free article] [PubMed] [Google Scholar]