

The mechanisms underlying imatinib resistance in chronic myelogenous leukemia (CML) include BCR-ABL gene rearrangement, overexpression of Bcl-Abl tyrosine kinase fusion protein, and activation of Bcr-Abl-independent pathways such as those of Src kinases.1,2 Although impressive therapeutic responses have been achieved with imatinib treatment, many patients fail to respond optimally due to innate or acquired resistance.1,2 The available treatments for these patients are limited and improved treatment options are urgently needed. In patients with Philadelphia chromosome positive CML, tyrosyl phosphorylation of the Bcr-Abl oncoprotein enables binding to the Src homolog-2 (SH2) domain of the adapter protein growth factor receptor-bound 2 (Grb2), thereby activating downstream signaling pathways, including the Ras/MAPK pathway, controlling cell proliferation.3 Grb2 mutants missing one of its two SH3 domains impair cell transformation by Bcr-Abl, highlighting the importance of Grb2 interactors, such as SOS1, in oncogenic signaling.4 These findings also suggest that Grb2 binding antagonists may provide an effective alternative or adjunct therapeutic strategy for CML patients resistant to imatinib.
Small molecule Grb2 SH2 domain binding antagonists have been developed that are selective, potent and phosphatase resistant.5 These have been shown to block the growth of tumor-derived cells in culture as well as tumor angiogenesis and metastasis in mice.3,5,6 We report here that TB03 (Supplementary Figure 1), a member of this compound class, synergistically enhanced inhibition of K562 leukemia cell proliferation by imatinib (Figure 1). Treatment with TB03 or imatinib alone resulted in dose-dependent growth inhibition (Figure 1a, b); when combined with TB03, imatinib activity was significantly enhanced (Figure 1b). Combining imatinib with TB03 at a ratio of 1:100 was synergistic and achieved an ED90 with a combination index (CI) of 0.774. At ratios of 1:50, 1:25, 1:12.5, and 1:6.25, all combinations were synergistic as indicated by combination indices at ED50, ED75, or ED90 values (Supplementary Table 1 and Figure 1c).
Figure 1. Inhibition of K562 cell growth by imatinib and TB03.

a. K562 cells were treated with varying concentrations of TB03 alone for 48 h, stained with Cell Counting Kit-8 reagent and optical densities were read at 450 nm; absorbance values obtained from cells treated with vehicle alone were regarded as 100% growth. Values represent mean +/− SD of triplicate wells; an IC50 value of 39 µM was obtained by curve fitting using GraphPad Prism software. Please see Supplementary Materials and Methods for further details related to all panels.
b. Cells treated with imatinib alone (squares; IC50 = 0.21 µM) or imatinib in combination with TB03 at the indicated concentrations and cell proliferation was determined as described for panel a.
c. The Fa-CI plot of the ratios of 1:6.25 (red “+”) and 1:12.5 (black “x”) imatinib:TB03 obtained from cell growth assays performed as described in b but with varying ratios of imatinib to TB03. Data were analyzed using CalcuSyn 2.1; values below the dotted horizontal line indicate synergy.
d. Flow cytometric analysis of K562 cell apoptosis induced by treatment with imatinib alone (0.25 µM), TB03 alone (20 µM), or the combination of these two compounds for 48 h. Cells were harvested, stained with Annexin-V-FITC and propidium iodide followed by flow cytometry; values represent the mean +/− SD or triplicate samples (*p<0.05, n=3).
e. Percentage of K562 cells reaching the 8th round of division after 72 h treatment with vehicle alone (unfilled bar) or the indicated concentrations of imatinib (yellow bars), TB03 alone (blue bar), or the combination of these two agents (green bars).
f. Percentage of K562 cells in cell cycle phases listed on the x-axis following 48 treatment with vehicle (unfilled bars), imatinib alone (0.5 µM, yellow bars), TB03 alone (blue bars), or imatinib + TB03 (green bars). Flow cytometric analysis using Vybrant DyeCycle Green was performed as described in Supplementary Materials and Methods. Data from one representative experiment of three independent experiments is shown.
Flow cytometry experiments were performed to further characterize synergistic inhibition of K562 cell proliferation by the combination of imatinib and TB03. Analysis using annexin V-FITC/propidium iodide double staining showed that treatment with either imatinib or TB03 alone did not affect apoptosis relative to the vehicle control group, but the combination of these two agents increased apoptosis by >50% (20 vs 36%; Figure 1d), indicating functional complementation of distinct pro-apoptotic effects. Flow cytometry analysis using CellTrace Violet indicated that vehicle-treated K562 cells divided eight times in 72 h. Treatment with 0.25 or 2.5 µM imatinib alone resulted in 0.04% and 0.03% of cells reaching the eighth cell division, respectively, and treatment with 2 µM TB03 alone resulted in 0.08% of cells reaching the eighth cell division (Figure 1e). Combined imatinib/TB03 treatments further reduced the number of cells reaching eight divisions: 0.02% for 2 µM TB03 plus 0.25 µM imatinib and 0.01% for 2 µM TB03 plus 2.5 µM imatinib; the latter combination represents an eight-fold reduction in cell survival (Figure 1e). Short-term analysis of cell cycle progression using Vybrant DyeCycle Green showed that imatinib treatment increased the G0/G1 population while decreasing the S and G2/M phase populations, consistent with cell-cycle suppression (Figure 1f). Treatment with TB03 alone had little effect, but combined TB03/imatinib treatment increased the number of cells in G0/G1 in a dose-dependent manner, with proportionately fewer cells reaching S and G2/M. The increased number of cells in sub-G1 phase with imatinib treatment relative to control (1.5% vs 4.7%) may reflect pro-apoptotic effects. Consistent with the complementation of apoptosis observed by annexin staining, TB03 plus imatinib further increased the number of cells in this phase in a dose-dependent manner, from 4.7% to 7% for imatinib:TB03 at a ratio 0.5:6.25 µM, and from 4.7% to 11% for imatinib:TB03 at 0.5:12.5 µM (Figure 1f). These results reinforce the hypothesis that TB03 alone inhibits only cell cycle progression, but enhances imatinib-induced apoptosis and cell cycle suppression, resulting in synergistic inhibition of K562 proliferation.
Cancer stem cells (CSCs) have become prime suspects for mediating resistance to therapy, disease relapse and progression.7 High-levels of aldehyde dehydrogenase (ALDH) activity have been recognized as a CSC maker in many cancers, including leukemia.8 The effects of imatinib, TB03 and combined imatinib/TB03 treatment on a putative leukemia stem cell subpopulation of K562 was investigated by flow cytometry using ALDEFLUOR, which provides a fluorescent ALDH reaction product to identify stem and progenitor cells by ALDH activity. Among vehicle-treated K562 cells 11.4% were ALDH+, this was unaltered by TB03 treatment but decreased to 8.9% and 2.7% with imatinib treatment at 0.25 µM and 25 µM, respectively (Supplementary Figure 2). When combined with TB03 (20 µM), imatinib at 0.25 and 25 µM concentrations further reduced the ALDH+ K562 population to 5.0% and 1.8%, respectively (Supplementary Figure 2). To our knowledge this is the first evidence of leukemia stem cell suppression by imatinib or by imatinib combined with a Grb2 SH2 domain binding antagonist.
Imatinib competitively inhibits Abl kinase activation, a critical transforming event in CML cases displaying the BCR-ABL gene rearrangement, but the activation of other tyrosine kinase-mediated signaling pathways can sustain important SH2 domain-mediated signaling events, including those of Grb2. We therefore investigated imatinib/TB03 synergy at the level of protein tyrosyl phosphorylation and Grb2 recruitment. Immunoblot analysis of K562 cell lysates after 1 h treatment with imatinib alone showed dose-dependent inhibition of multiple protein tyrosyl phosphorylation events relative to vehicle control, including phospho-Bcr-Abl (Figure 2a, arrows). TB03 alone had a modest effect, but the effects of imatinib were clearly enhanced when the two agents were combined (Figure 2a). Small molecule Grb2 SH2 domain binding antagonists block the binding of Grb2 to the activated growth factor receptor tyrosine kinases and effectors containing the Grb2 SH2 domain recognition motif.3,6 As anticipated, immunoprecipitation of Grb2 from intact K562 cells treated with imatinib or TB03 alone, followed by immunoblotting with anti-phosphotyrosine antibody, showed reduced capture of multiple phosphoproteins ranging from 150 kDa to 250 kDa (Figure 2b, left). The combination of TB03 and imatinib further reduced the association of tyrosyl phosphoproteins with Grb2, to the point where almost all bands were undetectable by immunoblotting (Figure 2b, right). These biochemical effects appear to be the most immediate and direct manifestations of synergy between TB03 and imatinib.
Figure 2. Inhibition of protein tyrosine phosphorylation and binding of tyrosyl phosphoproteins to Grb2.

a. Upper panel: immunoblot analysis of tyrosyl protein phosphorylation in K562 cells were treated with the indicated concentrations of imatinib, TB03 alone, or imatinib in combination with TB03 for 48 h. Cells were then harvested, extracted in detergent, and 50 µg of cell lysates were subjected to immunoblot analyses using phosphotyrosine antibody as described in Supplementary Materials and Methods. Changes in protein tyrosyl phosphorylation, including that of phospho-Bcr-Abl (labeled at right), are indicated by arrows at right. Lower panel: immunoblot for GADH as a control for sample loading. Data from one representative experiment of three independent experiments is shown.
b. Upper panel: immunoblot analysis of tyrosyl phosphoproteins captured by immunoprecipitation with anti-Grb2 antibody in K562 cells were treated with the indicated concentrations of imatinib, TB03 alone, or imatinib in combination with TB03 for 48 h as described in Supplementary Materials and Methods. Changes in the recovery tyrosyl phosphorylated proteins, including that of phospho-Bcr-Abl (labeled at right), are indicated by arrows at right. Lower panel: immunoblot for GADH as a control for sample loading. Data from one representative experiment of three independent experiments is shown.
c. Densitometric quantitation of bFGF (unfilled bars) and EGF (gray bars) content in media conditioned by K562 cells treated with imatinib (0.25 µM), TB03 (20 µM), or imatinib + TB03, relative to vehicle treated cells, for 48h (additional details can be found in Supplementary Materials and Methods). Values are the mean density in arbitrary units +/− SD. Data from one representative experiment of three independent experiments is shown.
The suppression of Bcr-Abl-driven intracellular protein tyrosyl phosphorylation and adapter protein recruitment by imatinib and TB03 was also accompanied by reduced secretion of epidermal (EGF) and basic fibroblast growth factors (bFGF; Figure 2c). Media conditioned by K562 cells for 48 h treated with vehicle or TB03, imatinib, or a combination of these two drugs, was collected and analyzed by immunoblotting a growth and angiogenesis factor array. Imatinib and TB03 each reduced bFGF secretion relative to the vehicle control by >50%, and by 66% when combined. EGF secretion was unaffected by either agent alone, but was reduced 40% by the combined treatment (Figure 2c). Although leukemia cells do not express EGF receptors,9 EGF promotes tumor angiogenesis in a variety of malignancies,10 and several angiogenic regulators, including bFGF, appear to contribute to the survival of chronic lymphocytic leukemia (CLL) cells through as yet undefined mechanisms.11
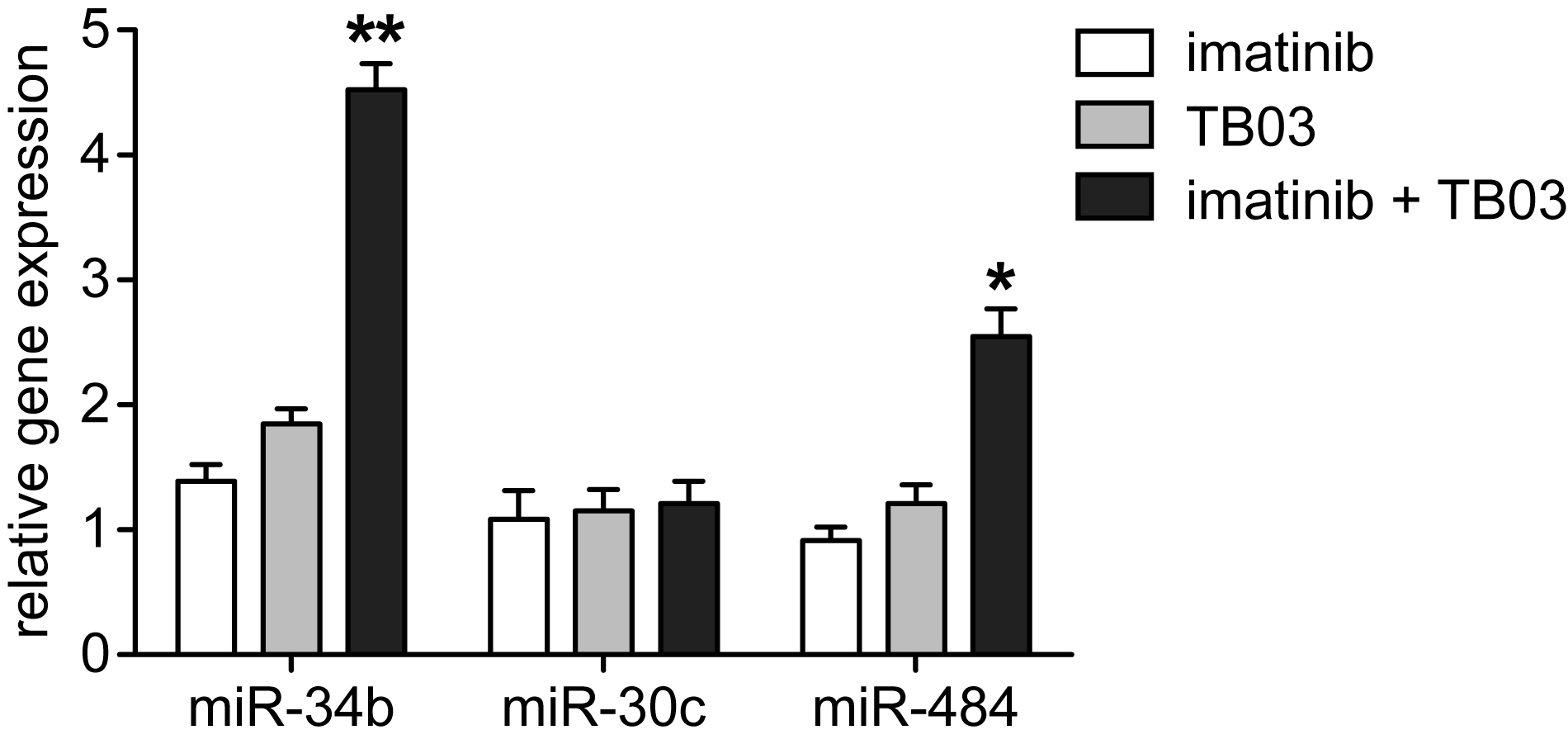
The emerging importance of microRNAs as gene regulators, their dysfunction in cancer, and their potential utility as pharmacodynamic markers of imatinib/TB03 synergy prompted us to screen a microRNA array12 to identify those significantly affected by treatment with TB03, imatinib, or the two combined (Supplementary Table 2). Treatment with imatinib for 48 h modulated the expression of 13 microRNAs, three of which were also modulated by TB03. Of the 17 microRNAs modulated by imatinib plus TB03, six also responsive to either agent alone were modulated substantially more when the drugs were combined. Ten of 17 were unique to the combination, suggestive of distinct but complementary effects (Supplementary Table 2). The microRNAs affected most by these treatments have no known roles in cancer; hsa-miR-34b13 and hsa-miR-30c14 are tumor suppressors and miR-484 expression was found to be low in CLL patients developing autoimmune anemia (AHA),15 so these results were validated by qPCR. Drug effects on hsa-miR-30c were modest, consistent with microarray profiling, whereas the imatinib/TB03 combination significantly increased expression of hsa-miR-34b and hsa-miR-484 relative to either agent alone or vehicle controls (p<0.05, n=3; Supplementary Figure 3). If the upregulation of miR-484 by imatinib/TB03 in K562 cells can be extended to CLL cell models, further studies of its impact on AHA may be warranted. Hypermethylation of the miR-34b promoter in leukemia allows overexpression of the CREB proto-oncogene, thereby enhancing cell proliferation and altering normal differentiation.13 Whether increased miR-34b expression associated with the combination of imatinib and TB03 in K562 was related to affects on promoter methylation is not yet clear, but such an effect in vivo would be anticipated to enhance treatment efficacy and also warrants further study.
In summary, synergistic inhibition of K562 leukemia cell proliferation by combined treatment with imatinib and a small molecule Grb2 SH2 domain binding antagonist was associated with cell cycle arrest, induction of apoptosis, reduced protein tyrosyl phosphorylation and Grb2 recruitment, suppression of a progenitor/stem cell subpopulation and increased expression of relevant tumor suppressor microRNAs. Further characterization of these effects could lead to improved diagnosis and more effective treatment of CML.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by The Children’s Leukemia Research Association, Inc. (a.k.a. National Leukemia Research Association) and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Supplementary Information accompanies this paper on the Leukemia website
Conflict of Interest Statement: The authors disclose that there are no conflicts of interest.
References
- 1.Maru Y. Molecular biology of chronic myeloid leukemia. Cancer Sci. 2012;103(9):1601–1610. doi: 10.1111/j.1349-7006.2012.02346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yeung DT, Hughes TP. Therapeutic targeting of Bcr-Abl: prognostic markers of response and resistance mechanism in chronic myeloid leukaemia. Crit Rev Oncog. 2012;17(1):17–30. doi: 10.1615/critrevoncog.v17.i1.30. [DOI] [PubMed] [Google Scholar]
- 3.Giubellino A, Burke TR, Jr, Bottaro DP. Grb2 signaling in cell motility and cancer. Expert Opin. Ther. Targets. 2008;12(8):1021–1033. doi: 10.1517/14728222.12.8.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gishizky ML, Cortez D, Pendergast AM. Mutant forms of growth factor-binding protein-2 reverse Bcr-Abl-induced transformation. Proc. Natl. Acad. Sci. U.S.A. 1995;92(24):10889–10893. doi: 10.1073/pnas.92.24.10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soriano JV, Liu N, Gao Y, Yao ZJ, Ishibashi T, Underhill C, et al. Inhibition of angiogenesis by growth factor receptor bound protein 2-src homology 2 domain bound antagonists. Mol. Cancer Ther. 2004;3(10):1289–1299. [PubMed] [Google Scholar]
- 6.Giubellino A, Gao Y, Lee S, Lee MJ, Vasselli JR, Medepalli S, et al. Inhibition of tumor metastasis by a growth factor receptor bound protein 2 src homology 2 domain-binding antagonist. Cancer Res. 2007;67(13):6012–6016. doi: 10.1158/0008-5472.CAN-07-0022. [DOI] [PubMed] [Google Scholar]
- 7.Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011;7(2):292–306. doi: 10.1007/s12015-010-9208-4. [DOI] [PubMed] [Google Scholar]
- 8.Fleischman AG. Aldh marks leukemia stem cell. Blood. 2012;119(15):3376–3377. doi: 10.1182/blood-2012-02-406751. [DOI] [PubMed] [Google Scholar]
- 9.Boehrer S, Adès L, Braun T, Galluzzi L, Grosjean J, Fabre C, et al. Erlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical study. Blood. 2008;111(4):2170–2180. doi: 10.1182/blood-2007-07-100362. [DOI] [PubMed] [Google Scholar]
- 10.Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77(6):400–410. doi: 10.1159/000279388. [DOI] [PubMed] [Google Scholar]
- 11.Xia Y, Lu R-N, Li J. Angiogenic factors in chronic lymphocytic leukemia. Leuk. Res. 2012;36(10):1211–1217. doi: 10.1016/j.leukres.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 12.Liu C-G, Calin GA, Volinia S, Croce CM. MicroRNA expression profiling using microarrays. Nat Protoc. 2008;3(4):563–578. doi: 10.1038/nprot.2008.14. [DOI] [PubMed] [Google Scholar]
- 13.Pigazzi M, Manara E, Bresolin S, Tregnago C, Beghin A, Baron E, et al. Mir-34b promoter hypermethylation induces CREB overexpression and contributes to myeloid transformation. Haematologica. 2013;98(4):602–610. doi: 10.3324/haematol.2012.070664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russ AC, Sander S, Lück SC, Lang KM, Bauer M, Rücker FG, et al. Integrative nucleophosmin mutation-associated microRNA and gene expression pattern analysis identifies novel microRNA - target gene interactions in acute myeloid leukemia. Haematologica. 2011;96(12):1783–1791. doi: 10.3324/haematol.2011.046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrer G, Navarro A, Hodgson K, Aymerich M, Pereira A, Baumann T, et al. MicroRNA expression in chronic lymphocytic leukemia developing autoimmune hemolytic anemia. Leuk. Lymphoma. 2013;54(9):2016–2022. doi: 10.3109/10428194.2012.763123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.