

T-cell malignancies, including T-cell acute lymphoblastic leukemia (T-ALL) and T-cell non-Hodgkin lymphoma (T-NHL), are a biologically and clinically heterogeneous group of rare and aggressive neoplastic disorders that result from clonal expansion of lymphocytes committed to the T-cell lineage 1. Although the prognoses for T-cell malignancies have improved in recent years due to intensified combination chemotherapies, the outcome of patients with resistant or relapsed disease remains poor 2. Therefore, current research efforts are focused on more effective and less toxic targeted therapies, which will require an improved understanding of the molecular events underlying these malignancies.
Janus kinase 3 (JAK3) is constitutively activated in a number of T-cell malignancies and has emerged as a molecular target for cancer therapy 3,4,5. JAK3 belongs to a family of cytoplasmic non-receptor tyrosine kinases (JAK1, JAK2, JAK3 and TYK2) that mediate signals initiated by cytokine and growth factor receptors 6. While JAK1, JAK2 and TYK2 are ubiquitously expressed, JAK3 is primarily confined to the hematopoietic lineage where it plays a vital role in lymphoid cell development and homeostasis7. Its restricted expression and critical function in lymphoid cells has attracted significant attention in recent years as a therapeutic target for immune-mediated diseases 8, however achieving JAK3 selectivity has remained a significant challenge 9. The first-in-class JAK3 inhibitor, tofacitinib, has been hindered by undesirable pan-JAK inhibitory side-effects that include anemia and neutropenia, presumably related to JAK2 inhibition and interference with cytokines such as erythropoietin and colony stimulating factors 10,11. Thus, although efficacy of a next-generation JAK3 inhibitor is paramount, drug selectivity to JAK3 over JAK2 may determine its clinical usefulness.
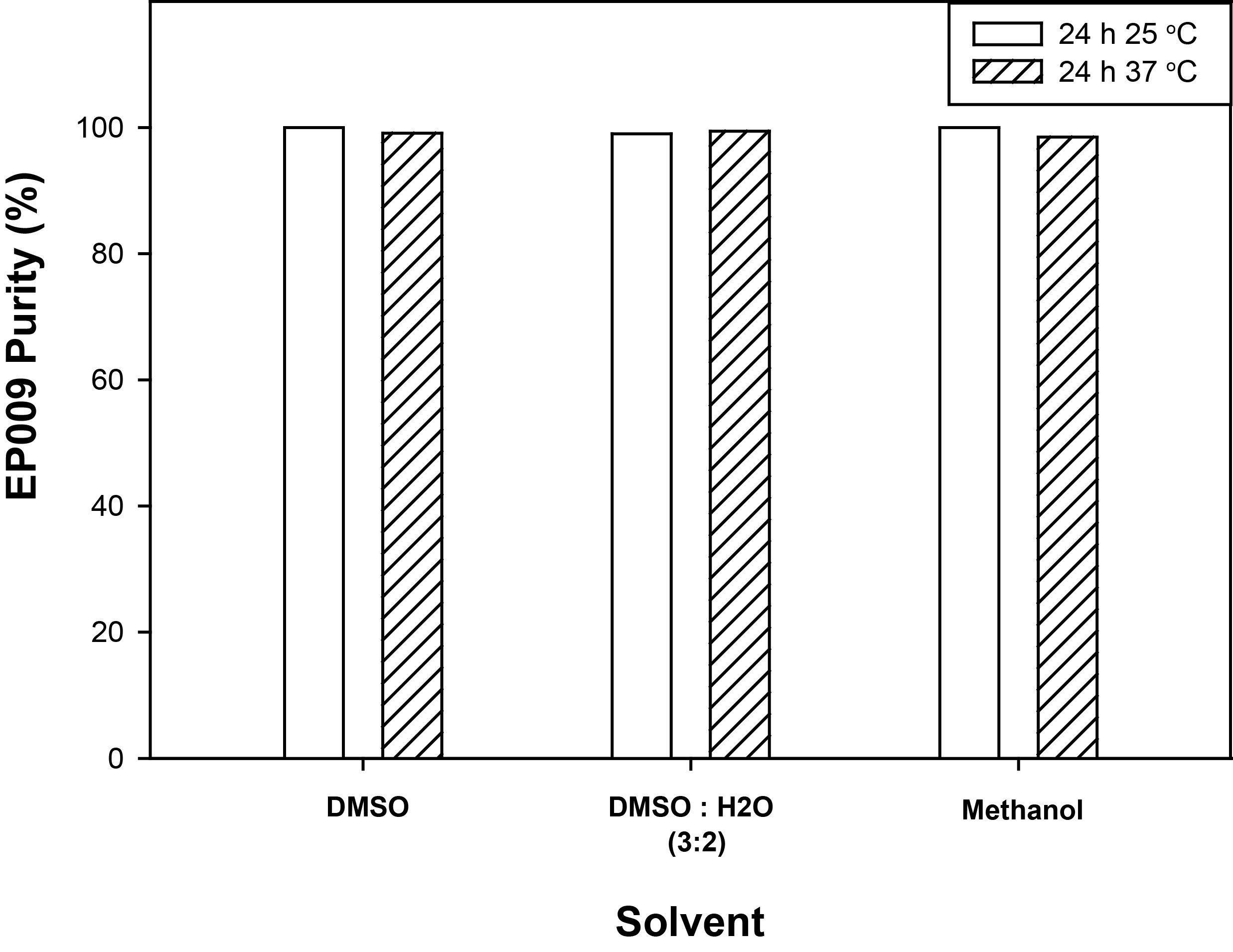
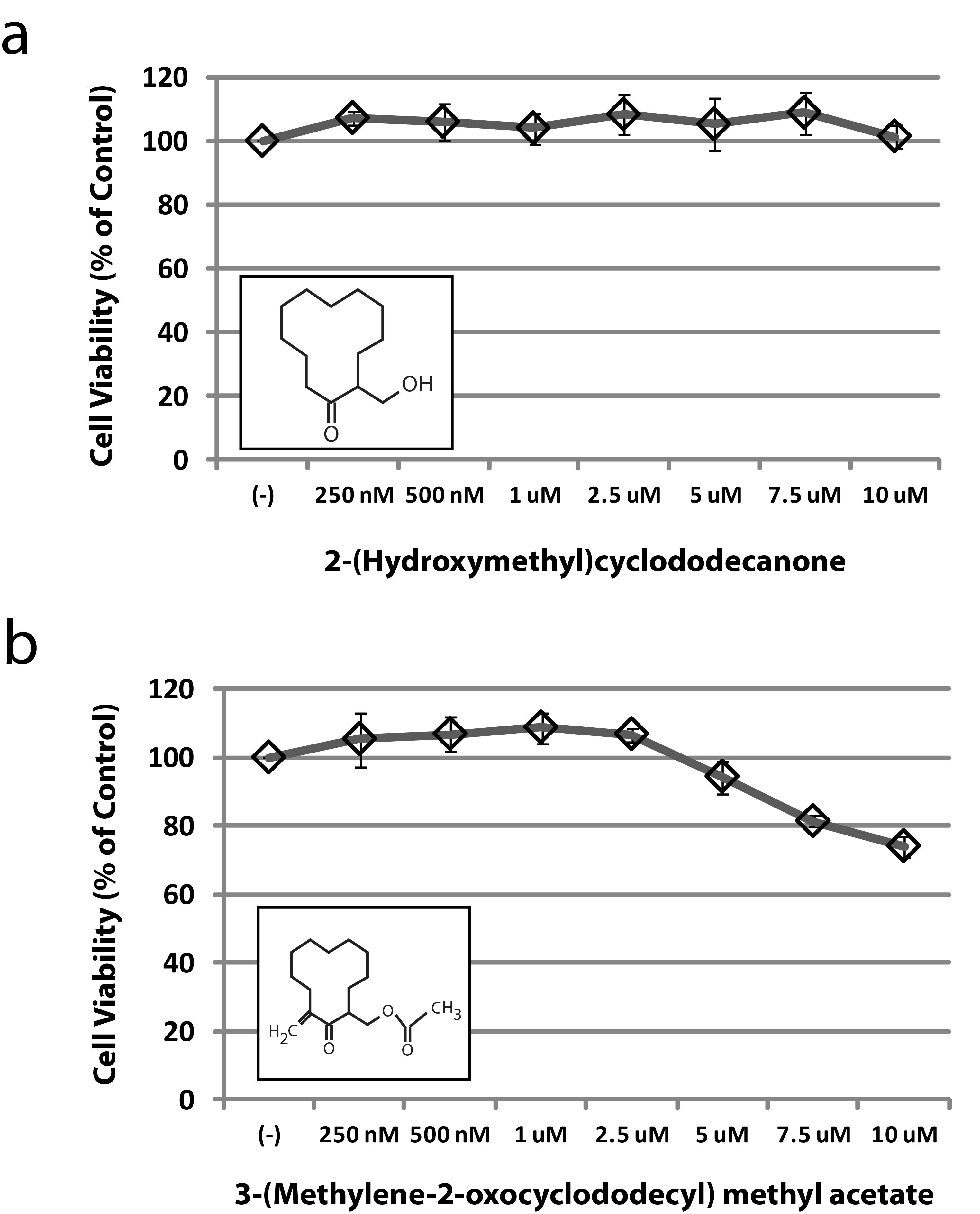
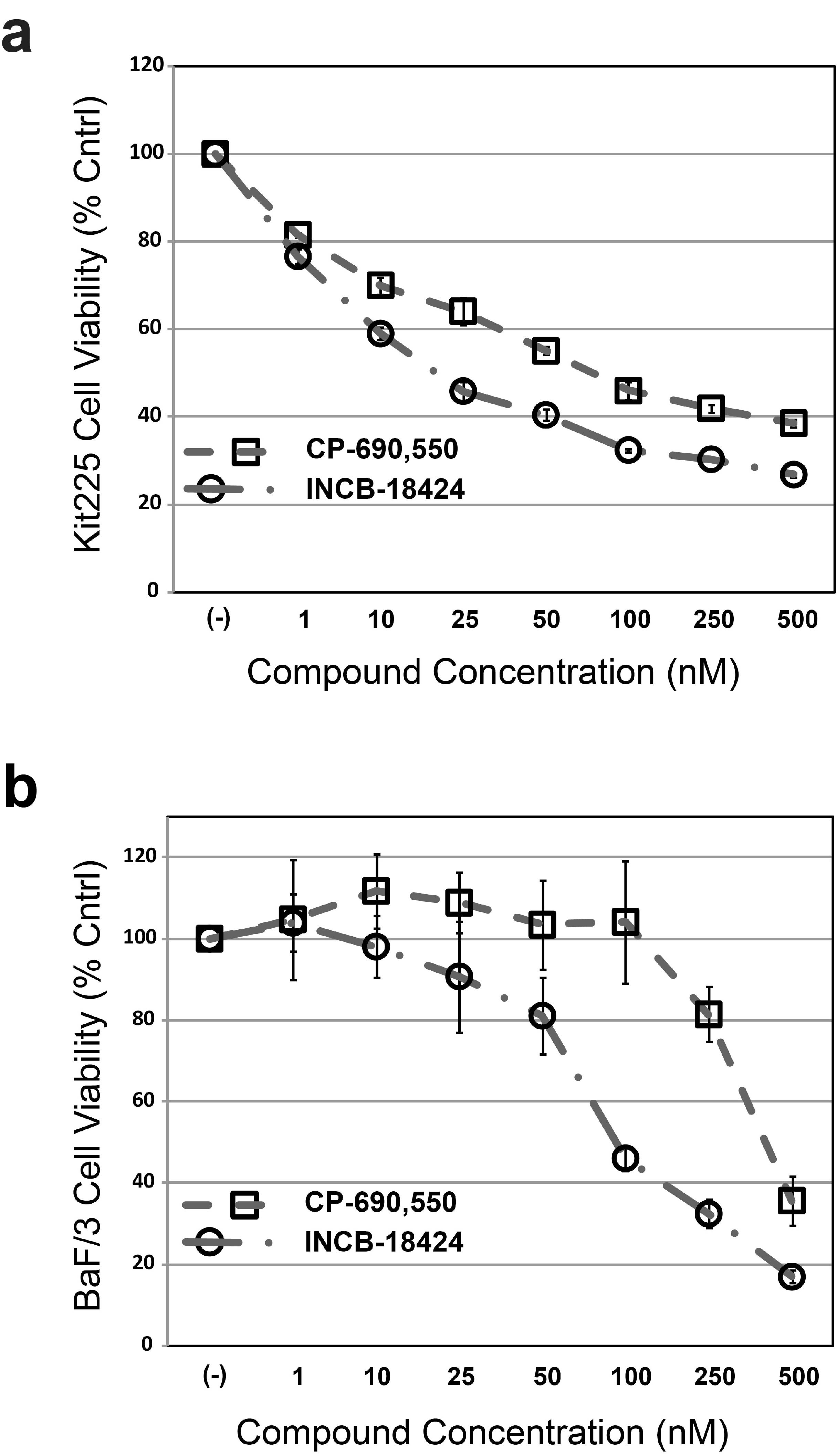
Through a series of structure-activity relationship (SAR) studies based upon the chemical scaffold of the established JAK3 inhibitor NC1153 12, we identified a novel, stable, selective, and efficacious inhibitor of JAK3 that was designated EP009 (Fig. 1a, Supplementary Fig. S1). In vitro kinase assays revealed EP009 to be a low micromolar inhibitor of JAK3 (Fig. 1b) with limited off-target effects against a panel of 92 human kinases (Supplementary Table S1). The IL-2-dependent T-cell line Kit225 and the IL-3-dependent pro-B-cell line BaF/3 were utilized to test EP009 specificity for JAK3 versus JAK2, respectively. In Kit225 cells, EP009 reduced IL-2-mediated JAK3 tyrosine phosphorylation with a cellular IC50 between 10 and 20 μM (Fig. 1c, upper panel). In contrast, EP009 had no detectable effect on IL-3-induced JAK2 tyrosine phosphorylation in BaF/3 cells up to 50 μM (Fig. 1c, lower panel). Additionally, EP009 treatment reduced Kit225 cell viability with a LD50 of 5.0 μM at 72 hours, while no effect on BaF/3 cell viability was detected (Fig. 1d). SAR studies revealed that deletion of the C12 methylene or substitution at the C2 hydroxymethyl groups of the cyclododecanone ring significantly reduced the efficacy of EP009 (Supplementary Fig. S2). Under the same experimental conditions, JAK inhibitors tofacitinib (CP-690,550) and ruxolitinib (INCB-18424) reduced both Kit225 and BaF/3 cell viability (Supplementary Fig. S3). Thus, although the IC50 values suggest that EP009 is not as potent as other JAK inhibitors, the degree of kinase selectivity differentiates it from what is typically obtained from an ATP-competitive inhibitor. Ongoing studies seek to delineate the underlying mechanism of JAK3 inhibition by EP009 through application of biochemical, biophysical, and structural studies.
Figure 1.

EP009 is a selective inhibitor of JAK3 with anti-cancer activity. (a) Chemical structure of EP009 (M.W. 224.34). (b) In vitro autokinase analysis of immunopurified JAK3 treated with vehicle (DMSO; lanes a and b) or ascending concentrations of EP009 (0–10 μM; lanes c–f). The reactions were incubated in the absence (lane a) or presence (lanes b–f) of 1 μM ATP prior to separation by 7.5% SDS-PAGE and subsequently Western blotted (WB) with anti-phosphotyrosine (α-pY). The blot was stripped and reprobed with α-JAK3 to confirm equivalent loading. (c) Kit225 (upper panel) or BaF/3 (lower panel) cells cultured in the presence of IL-2 or IL-3, respectively, were treated with vehicle (PBS; lane a) or increasing amounts of EP009 (0–50 μM; lanes b–h) for 12 hours. Cells were then lysed, clarified, and immunoprecipitated (IP) with anti-JAK3 (α-JAK3) or anti-JAK2 (α-JAK2), and subjected to WB analysis with α-pY. Blots were stripped and reprobed with corresponding antibody to verify equivalent protein loading. (d–h) The indicated cells were cultured with increasing amounts of EP009 (0–10 μM) for 72 hours and cell viability measured with the MTS tetrazolium salt assay. Values represent mean absorbance (OD490-OD650 nm) normalized to vehicle (PBS) treated control cells, while error bars represent the standard deviation (n = 3). (g, h) Additionally, total cell lysates were separated by 7.5% SDS-PAGE and subjected to WB analysis with α-JAK3. Blots were then stripped and reprobed with α-GAPDH to verify equivalent protein loading. Representative data from three independent experiments are shown.
To assess possible off-target effects of EP009 on renal, hepatic and peripheral lymphocytic systems, we examined its impact on human cells including the kidney cell line HEK293, liver cell line HEPG2, and primary naïve PBMCs, respectively. Cell treatments with 10 μM EP009 for 72 hours resulted in no detectable loss of cell viability (Fig. 1e), suggesting a favorable in vitro cytotoxicity profile for EP009. Thus, preclinical investigations into the efficacy of EP009 against T-cell malignancies that display constitutively activated JAK3 were initiated using representative T-NHL anaplastic large cell lymphoma (ALCL) cell lines (Supplementary Fig. S4)13. EP009 treatment led to a concentration-dependent reduction in viability of NPM-ALK-positive SU-DHL-1 and SUP-M2 ALCL cells with an LD50 of 5 μM at 72 hours (Fig. 1f). In contrast, EP009 had no effect on cell viability of the EML4-ALK-positive, but JAK3-negative, non-small cell lung cancer cell line H2228, further supporting the selectivity of EP009 toward JAK3 and its functional role in T-NHL ALCL. Similarly, we examined the effects of EP009 on the viability of human cutaneous T-cell lymphoma (CTCL) cell lines H9 and HH. EP009 reduced viability of HH cells with a LD50 between 2.5 and 5 μM at 72 hours, however identical concentrations had limited effects on H9 cells (Fig. 1g). Western blot analysis of total cell lysates revealed that while JAK3 protein expression was readily detectable in the EP009 sensitive HH cells, its presence in the EP009 refractory H9 cells was below the level of detection (Fig. 1g, upper panel). These data further support the hypothesis that EP009 selectively disrupts JAK3-dependent tumor cell viability.
The in vitro results using tumor T-cell lines prompted us to assess the ex vivo efficacy of EP009 against primary cultures of leukemia patient tumor cells. The cytotoxic effect of EP009 was evaluated in PBMCs obtained from two patients diagnosed with relapsed T-ALL (Supplementary Table S2). EP009 treatment led to a concentration-dependent reduction in viability of T-ALL-P1 cells with an LD50 between 2.5 and 5 μM at 72 hours (Fig. 1h). In contrast, EP009 had no effect on T-ALL-P2 cell viability at 72 hours. Western blot and densitometry analysis of total cell lysates revealed 80% less JAK3 protein expression in the TALL-P2 cells, suggesting a correlation between the level of JAK3 protein expression and EP009 sensitivity. Taken together, these findings indicate that EP009 represents a viable therapeutic strategy for the treatment of JAK3 dependent T-cell malignancies.
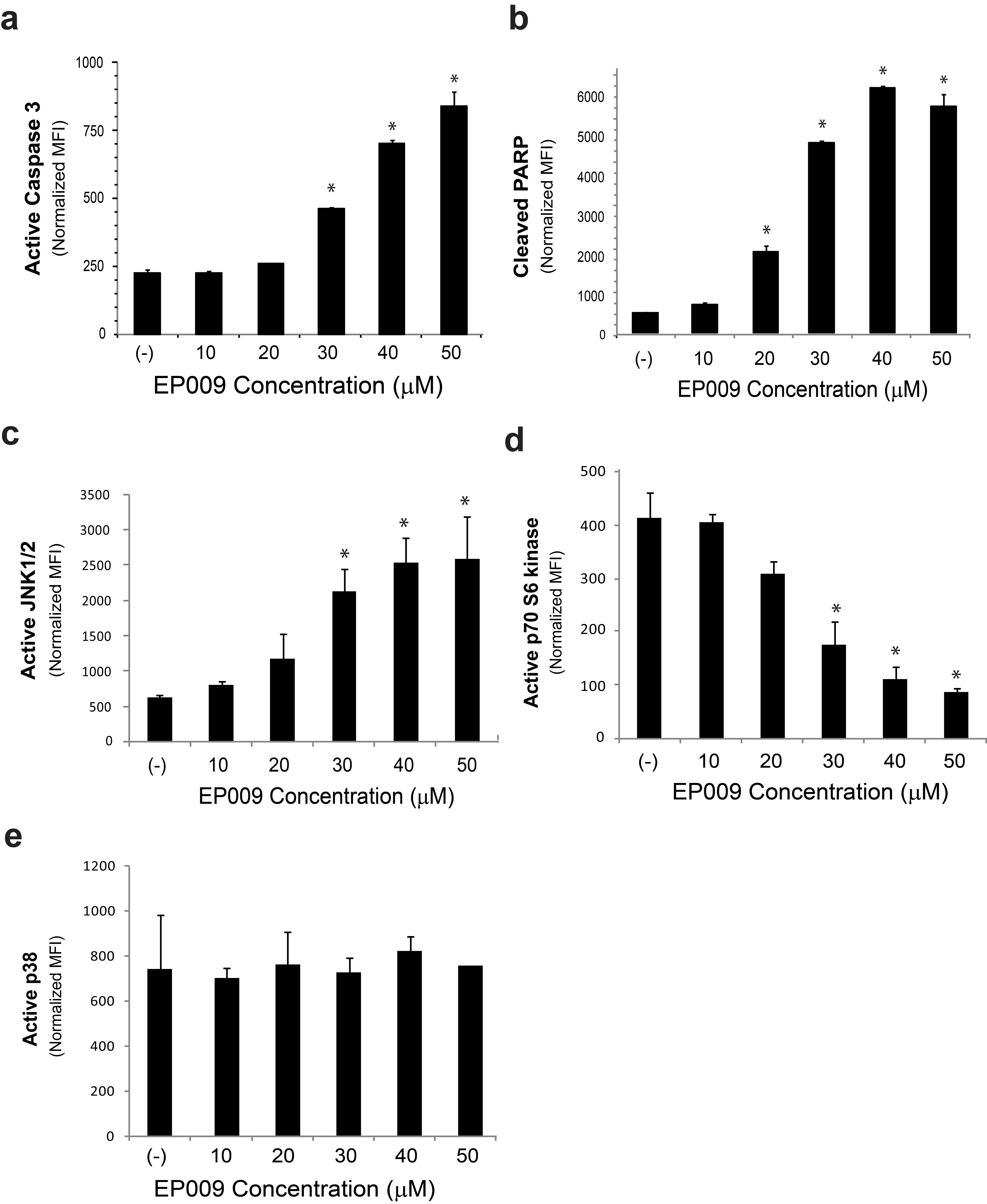
To further explore the functional consequences and in vivo activity of EP009 mediated JAK3 inhibition, we utilized the human T-NHL ALCL model system. Constitutive tyrosine phosphorylation and activation of JAK3/STAT3 in SU-DHL-1 cells was inhibited by EP009 in a dose-dependent manner (Fig. 2a and 2b). The decrease in JAK3/STAT3 activation was associated with loss of cell viability that was attributed to induction of apoptosis as confirmed by increased caspase 3 activation and PARP cleavage (Supplementary Fig. S5a and 5b). Additionally, EP009 cytotoxicity correlated with activation of JNK1/2, but not p38, mediated stress signaling and inactivation of p70 S6 kinase mediated growth and survival signaling in SU-DHL-1 (Supplementary Fig. S5c, 5d and 5e). These findings highlight the importance of stress generation through JAK3 inhibition that promotes apoptosis in T-NHL cells.
Figure 2.

EP009 inhibits JAK3/STAT3 signaling and reduces tumor growth in a murine xenograft model of human T-cell lymphoma. (a) Representative Western blot analysis of JAK3 tyrosine phosphorylation in SU-DHL-1 cells treated with vehicle (PBS; lane a) or increasing amounts of EP009 (0–40 μM; lanes b-e) for 12 hours. (b) Luminex multiplex analysis of tyrosine phosphorylated STATs from SU-DHL-1 cells treated with vehicle (PBS) or increasing amounts of EP009 (0–40 μM) for 12 hours. Values represent pSTAT mean fluorescence intensity (MFI) normalized to corresponding GAPDH MFI, while error bars represent the standard deviation (n = 2). Statistical significance was determined using Student’s t-test. (*, p < 0.05). Representative data from three independent experiments. (c) Sprague Dawley rats were administered EP009 by oral gavage (200 mg/kg) and pharmacokinetics measured by analysis of plasma concentrations at indicated time points. Values represent mean concentrations, while error bars represent the standard deviation (n=5). (d) Therapeutic study of EP009 in SU-DHL-1 model in SCID/NOD mice. Treatments with oral EP009 given at 100 mg/kg (n= 8, square) and 200 mg/kg (n=8, triangle) inhibited the SU-DHL-1 lymphoma growth significantly as seen by tumor size compared to the control group (n=8, circle) * p<0.05; ** p<0.01. (e) Haematoxylin and eosin (H&E) and immunohistochemistry staining for p-STAT3 in representative SU-DHL-1 tumors excised from vehicle- and EP009-treated (200 mg/kg) SCID/NOD mice. Images are shown at 400X magnification. (f) Histopathological evaluation of p-STAT3 positivity in SU-DHL-1 tumors excised from vehicle- and EP009-treated (200 mg/kg) SCID/NOD mice. Values represent mean percentage of p-STAT3 positive cells per field, while error bars represent the standard deviation (n=3) ** p<0.01.
To assess the clinical potential of EP009, its bioavailability was determined following oral administration (200 mg/kg dosing) to Sprague Dawley rats. Pharmacokinetic analysis showed rapid bioavailability with peak blood concentrations (~6 μM) at 30 min and was detectable up to eight hours (Fig. 2c). Thus, tumor xenografts should be exposed to inhibitory concentrations of EP009 previously observed in the cell-based studies. Whether repetitive dosing results in greater tissue accumulation and EP009 concentrations remains to be determined. The therapeutic efficacy of EP009 was further evaluated using a SCID/NOD murine xenograft model of human T-NHL ALCL. Treatment of mice bearing SU-DHL-1 tumors with EP009 given orally at 100 or 200 mg/kg resulted in significant tumor inhibition (>50% reduction; p<0.01) versus placebo control groups (Fig. 2d). Tumor responses were evident three weeks post EP009 treatment suggesting greater tissue accumulation is required for inhibiting tumor cell growth. However, once initiated, tumor responses were maintained for the duration of the treatment. Consistent with the in vitro results, EP009 treatment was associated with reduced levels of Tyr705-phosphorylated STAT3 (p<0.01) compared to vehicle treated control tumors (Fig. 2e and 2f). Considering the effects of EP009 on ALCL cells in vitro and in vivo, these results suggest that constitutive activation of JAK3 represents a viable therapeutic target to treat malignant T-cell lymphoma. Indeed, supporting evidence indicates that JAK3 is a secondary oncogenic driver in ALCL that is induced by autocrine cytokine signaling mechanisms via IL-9 14 and IL-21 15. Therefore, EP009 mediated inhibition of JAK3 would serve as a logical therapeutic strategy for intervention in hematopoietic malignancies in primary response to JAK3 activating alleles, and secondary responses where JAK3 acts as an oncogenic driver, such as ALK-positive T-NHL ALCL.
In conclusion, EP009 is a selective and orally active JAK3 inhibitor that displays a favorable cytotoxicity profile with therapeutic efficacy against JAK3-driven tumor T-cells in vitro, in vivo, and ex vivo. Current cancer drug development is focused on targeted therapies which selectively uncouple cell signaling pathways required for tumor cell growth and survival. The data generated and described in this study support the role of JAK3 as a viable and relevant molecular target in the treatment of T-cell malignancies where compounds such as EP009 can be effective anti-neoplastic agents.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. G. Inghirami, University of Torino, IT for kindly providing the Karpas299, SUP-M2, DEL and SU-DHL1 cell lines. This work was supported in part by grants from the National Institutes of Health (NIH) 1R43RR032354-01 (F.P.), Edward N. and Margaret G. Marsh Foundation (J.A.R, R.A.K), Lizanell and Colbert Coldwell Foundation (R.A.K), Associazione Italiana per la Ricerca sul Cancro (AIRC) IG 11675 (F.C.), and made possible by grants G12MD007592 to the Border Biomedical Research Center (BBRC) and P20MD002287-06 to the Hispanic Health Disparities Research Center (HHDRC) from the National Institutes on Minority Health and Health Disparities (NIMHD), a component of the NIH.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests.
The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of NIMHD or NIH.
Supplementary information is available at Leukemia’s website.
References
- 1.Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol. 2008;8:380–390. doi: 10.1038/nri2304. [DOI] [PubMed] [Google Scholar]
- 2.Schrappe M, Hunger SP, Pui CH, Saha V, Gaynon PS, Baruchel A, et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med. 2012;366:1371–1381. doi: 10.1056/NEJMoa1110169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bains T, Heinrich MC, Loriaux MM, Beadling C, Nelson D, Warrick A, et al. Newly described activating JAK3 mutations in T-cell acute lymphoblastic leukemia. Leukemia. 2012;26:2144–2146. doi: 10.1038/leu.2012.74. [DOI] [PubMed] [Google Scholar]
- 4.Cornejo MG, Kharas MG, Werneck MB, Le Bras S, Moore SA, Ball B, et al. Constitutive JAK3 activation induces lymphoproliferative syndromes in murine bone marrow transplantation models. Blood. 2009;113:2746–2754. doi: 10.1182/blood-2008-06-164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai R, Rassidakis GZ, Lin Q, Atwell C, Medeiros LJ, Amin HM. Jak3 activation is significantly associated with ALK expression in anaplastic large cell lymphoma. Hum Pathol. 2005;36:939–944. doi: 10.1016/j.humpath.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 6.Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270:794–797. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 8.Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O’Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–142. doi: 10.1111/j.1600-065X.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 10.Kremer JM, Cohen S, Wilkinson BE, Connell CA, French JL, Gomez-Reino J, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64:970–981. doi: 10.1002/art.33419. [DOI] [PubMed] [Google Scholar]
- 11.Borie DC, Changelian PS, Larson MJ, Si MS, Paniagua R, Higgins JP, et al. Immunosuppression by the JAK3 inhibitor CP-690,550 delays rejection and significantly prolongs kidney allograft survival in nonhuman primates. Transplantation. 2005;79:791–801. doi: 10.1097/01.tp.0000157117.30290.6f. [DOI] [PubMed] [Google Scholar]
- 12.Stepkowski SM, Kao J, Wang ME, Tejpal N, Podder H, Furian L, et al. The Mannich base NC1153 promotes long-term allograft survival and spares the recipient from multiple toxicities. J Immunol. 2005;175:4236–4246. doi: 10.4049/jimmunol.175.7.4236. [DOI] [PubMed] [Google Scholar]
- 13.Amin HM, Medeiros LJ, Ma Y, Feretzaki M, Das P, Leventaki V, et al. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene. 2003;22:5399–5407. doi: 10.1038/sj.onc.1206849. [DOI] [PubMed] [Google Scholar]
- 14.Qiu L, Lai R, Lin Q, Lau E, Thomazy DM, Calame D, et al. Autocrine release of interleukin-9 promotes Jak3-dependent survival of ALK+ anaplastic large-cell lymphoma cells. Blood. 2006;108:2407–2415. doi: 10.1182/blood-2006-04-020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dien Bard J, Gelebart P, Anand M, Zak Z, Hegazy SA, Amin HM, et al. IL-21 contributes to JAK3/STAT3 activation and promotes cell growth in ALK-positive anaplastic large cell lymphoma. Am J Pathol. 2009;175:825–834. doi: 10.2353/ajpath.2009.080982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.