

Dear Editor,
Created in 2001 and retained in 2008, the World Health Organization (WHO) now recognizes a distinct category of myelodysplastic/myeloproliferative neoplasms (MDS/MPN), for those patients at diagnosis with clinical, morphologic and laboratory features which overlap both those of MDS and MPN.(1) Four hematopathologic diagnoses exist within this category; chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia BCR-ABL1 negative (aCML), juvenile myelomonocytic leukemia (JMML), and myelodysplastic/myeloproliferative neoplasm, unclassifiable (MDS/MPN-U).
Of the four “overlap” MDS/MPN syndromes, MDS/MPN-U is the least well characterized.(2) It encompasses patients with features of both MDS and MPN at presentation that do not satisfy criteria for CMML, JMML, or aCML. MDS/MPN-U is formally defined as patients with no preceding history of MDS or MPN, no recent cytotoxic growth factor therapy, no Philadelphia chromosome, BCR-ABL1 fusion gene, PDGFRA, PDGFRB or isolated del(5q), t(3;3)(q21;q26) or inv(3)(q21q26), and with dysplastic features in ≥1 hematopoietic cell line, <20% blasts in the blood and bone marrow, prominent myeloproliferative features (i.e., platelet count ≥450×109/L or white blood cell count ≥13×109/L, with or without splenomegaly); or de novo disease with mixed myeloproliferative and myelodysplastic features which cannot be assigned to any other category of MDS, MPN or of MPS/MPN.(3) While there are no identifying cytogenetic or molecular features of MDS/MPN-U, recurrent mutations are found within this category and notably include JAK2-V617F in approximately 25% of patients. Additionally, JAK2-V617F mutations are present in up to 60% of MDS/MPN-U patients with refractory anemia with ringed sideroblasts and thrombocytosis (RARS-T), a pathologic diagnosis currently residing as a provisional entity within the MDS/MPN-U category.(4, 5)
MDS/MPN-U is a rare diagnosis, making up less than 5% of all myeloid disorders.(6) Accordingly, clinical characteristics and the natural history of patients with MDS/MPN-U are not well established, though poor prognosis among patients with MDS/MPN-U (without RARS-T) has been suggested in small series to date.(5, 6) No standard prognostic or treatment algorithms for MDS/MPN-U exist. Our aim was to evaluate patients with a confirmed diagnosis of MDS/MPN-U without RARS-T, in order to provide insights into the nature of this unique myeloid overlap syndrome with implications to appropriate treatment strategies.
All patients with MDS/MPNs from January 1987 to February 2013 at the University of Texas MD Anderson Cancer Center were reviewed. Patients seen prior to 2006 with a diagnosis of MDS/MPD or MPD-unclassifiable were reviewed by two independent hematopathologists and diagnoses were modified, when appropriate, according to the current WHO criteria.(3) Patients were excluded if the de novo presence of both myelodysplastic and myeloproliferative features at diagnostic presentation was unable to be confirmed. All patients with RARS-T were excluded per 2008 definition.(3) In total, 85 patients with a diagnosis of MDS/MPN-U, without RARS-T, were included. All patients were tested and were negative for the BCR/ABL translocation by fluorescence in-situ hybridization (FISH) and/or polymerase chain reaction (PCR), and JAK2-V617F was assessed by standard PCR technique. Peripheral and bone marrow monocyte counts were assessed in all patients, and all patients were classified according to the International Prognostic Scoring System (IPSS) for MDS(7), the International Working Group (IWG) IPSS for myelofibrosis(8), the IPSS-Revised (IPSS-R),(9) and the MDA Global MDS model.(10) This retrospective chart review protocol was approved by the University of Texas MD Anderson Institutional Review Board. Overall survival (OS) was analyzed using the Kaplan-Meier method and compared by log-rank test.
Characteristics of the 85 MDS/MPN-U patients are summarized in Table 1. MDS/MPN-U patients were predominantly over the age of sixty (92%) with a median age of 70 years, and 61 (72%) were male. Thirty patients (35%) had splenomegaly at presentation, 11 patients (13%) had thrombocytosis (>450 × 109/L), 15 patients (18%) had leukocytosis (>13 × 109/L) and median absolute monocyte count was 0.37 x109/L. Of 56 patients with known JAK2 status, 17 patients (30%) had a JAK2-V617 mutation. The majority of patients had either diploid cytogenetics (49%) or trisomy 8 as the sole abnormality (15%); 10 patients (12%) had a complex karyotype and 20 patients (23%) had other abnormalities including deletion(12p), trisomy 9, deletion (20q), or deletion (7q). One patient had isolated isochromosome (17q).
Table 1.
Baseline clinicopathologic features of MDS/MPN-U patients (n=85)
| Variable | Total # | % | Median OS (months) | P-value |
|---|---|---|---|---|
| Age | ||||
| Median | 70 (22-90) | |||
| <60 years | 7 | 8% | 50.2 | <0.001 |
| ≥60 years | 78 | 92% | 12.2 | |
| Sex | ||||
| Male | 61 | 72% | 15.4 | 0.556 |
| Female | 24 | 28% | 12.4 | |
| ECOG PS | ||||
| 0 | 51 | 60% | 12.5 | 0.884 |
| ≥1 | 34 | 40% | 12.4 | |
| Splenomegaly | ||||
| Yes | 30 | 35% | 11.6 | 0.777 |
| No | 55 | 65% | 15.5 | |
| Constitutional Symptoms | ||||
| Yes | 59 | 69% | 12.4 | 0.771 |
| No | 26 | 31% | 12.2 | |
| Prior Malignancy | ||||
| Yes | 13 | 15% | 11.9 | 0.242 |
| No | 72 | 85% | 12.5 | |
| WBC count (x109/L) | ||||
| Median | 17 (1-141) | |||
| <13 | 39 | 46% | 11.5 | 0.567 |
| ≥13 | 15 | 18% | 17.5 | |
| ≥25 | 31 | 36% | 12.4 | |
| HGB (g/dl) | ||||
| Median | 10 (5-15) | |||
| <10 | 39 | 46% | 12.4 | 0.504 |
| ≥10 | 46 | 54% | 12.4 | |
| PLT count (×109/L) | ||||
| Median | 85 (6-1168) | |||
| <450 | 74 | 87% | 11.9 | <0.001 |
| ≥450 | 11 | 13% | 52.5 | |
| PB Blast % | ||||
| Median | 1 (0-16) | |||
| ≥1% | 48 | 56% | 11.7 | 0.023 |
| None | 37 | 44% | 23.1 | |
| BM Blast % | ||||
| Median | 3 (0-17) | |||
| ≤5% | 60 | 71% | 15.7 | 0.017 |
| 6-10% | 17 | 20% | 11.7 | |
| >10% | 8 | 9% | 4.5 | |
| Cytogenetics | ||||
| Diploid | 42 | 49% | 15.7 | 0.224 |
| +8 | 13 | 15% | 11.9 | |
| Complex | 10 | 12% | 8.3 | |
| Other | 20 | 23% | 24.7 | |
| JAK2-V617F | ||||
| Median Allele Burden: | 48 (1-95) | |||
| Positive | 17 | 20% | 8.9 | 0.251 |
| Negative | 39 | 46% | 17.7 | |
| Unknown | 29 | 34% | ||
| IWG-MF Score | ||||
| Low | 2 | 2% | < 2 valid cases | 0.183 |
| Int-1 | 17 | 20% | 20.2 | |
| Int-2 | 18 | 21% | 15.4 | |
| High | 48 | 57% | 11.9 | |
| MDS IPSS Score | ||||
| Low | 23 | 27% | 15.4 | 0.039 |
| Int-1 | 35 | 41% | 17.7 | |
| Int-2 | 19 | 22% | 8.3 | |
| High | 8 | 9% | 6.9 | |
| IPSS-R Score | ||||
| Very Low | 7 | 9% | 15.4 | 0.061 |
| Low | 27 | 33% | 29.5 | |
| Int | 19 | 23% | 15.7 | |
| High | 23 | 28% | 11.5 | |
| Very High | 5 | 6% | 6.8 | |
| MDA-Risk Score | ||||
| Low | 16 | 20% | 50.2 | 0.004 |
| Int-1 | 17 | 21% | 12.5 | |
| Int-2 | 28 | 35% | 11.9 | |
| High | 20 | 25% | 11.4 |
ECOG indicates Eastern Cooperative Oncology Group, PS indicates performance status, PB indicates peripheral blood, BM indicates bone marrow, WBC indicates white blood cell, HGB indicates hemoglobin, PLT indicates platelet, IWG-MF indicates International Working Group - Myelofibrosis Score, IPSS indicates International Prognostic Scoring System, MDS indicates myelodysplastic syndrome, MDA indicates MD Anderson
Median OS was 12.4 months (0.3–138.7 months). The four clinical variables associated with favorable outcome included age <60 years (p<0.001), thrombocytosis (p<0.001), lack of circulating blasts (p=0.023) and ≤5% bone marrow blasts (p=0.017). In multivariate analysis, only thrombocytosis ≥450 × 109/L retained prognostic significance (p=0.001). Due to the defining presence of both MDS and MPN-like features, both MDS and MPN prognostic scoring systems were applied [Table 1]. While neither the MF-IPSS score nor IPSS-R score provided prognostic information (p=0.18 and p=0.06, respectively), the MDS-IPSS score imparted a statistically significant classification to our MDS/MPN-U cohort, with a median OS by MDS-IPSS score of 15.4, 17.7, 8.3 and 6.9 months, respectively (p=0.039) [Figure 1]. The MDA global MDS model also imparted a significant prognostic classification with a median OS of 50.2, 12.5, 11.9 and 11.4 months (p=0.004).
Figure 1.
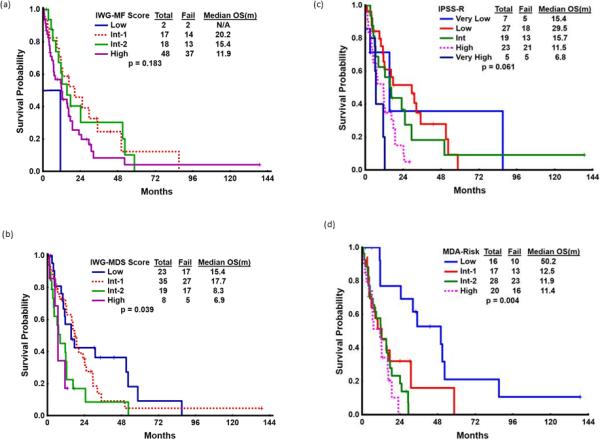
Overall Survival by (a) MF-IPSS, (b) MDS-IPSS, (c) IPSS-R and (d) MDA global model classification systems
Thirty-six patients (42%) received hypomethylating agent (HMA) therapy, and 13 patients (15%) an immunomodulatory approach (i.e. thalidomide, lenalidomide, interferon-α or antithymocyte globulin (ATG) with cyclosporine). Five patients (6%) received an allogeneic stem cell transplant (SCT), and five patients (6%) underwent splenectomy. HMA-treated patients had an OS of 16.4 versus 11.5 months with other approaches (p=0.57), while patients receiving immunotherapy had a non-significant trend towards a worse OS of 11.4 versus 15.4 months (p=0.07). Neither allogeneic SCT nor splenectomy impacted survival (OS 12.4 versus 12.5 months, p=0.16) and (51.1 versus 10.0 months, p=0.23), respectively.
For comparison purposes, the MDS/MPN-U population was compared to MDS (n=2241) and primary myelofibrosis (PMF) (n=600) MDA cohorts within the same time period [Supplementary Table 1]. While certain features of MDS/MPN-U aligned with either PMF or MDS, the MDS/MPN-U category was unique from MDS or PMF in almost every clinicopathologic characteristic. In general, MDS/MPN-U patients were older, more likely to be male, and more likely to have leukocytosis ≥25 ×109/L at diagnosis. Isolated trisomy 8 was seen more frequently in the MDS/MPN-U group (15%) than in either PMF (4%) or MDS (5%). Consistent with prior reports of poor prognosis, MDS/MPN-U patients had inferior survival of 12.4 months, compared to 16.0 months with MDS and 41.5 months with PMF (p<0.001) [Supplementary Figure 2].
MDS/MPN-U is an infrequent diagnosis with inadequate characterization. We describe the largest cohort of MDS/MPN-U analyzed to date, and several insights are worthy of discussion. Several limitations must nevertheless also be considered. There may be selection bias in the MDS/MPN-U diagnosis, as this is a rare entity which requires hematopathologist vigilance for accurate diagnosis. Whether MDS/MPN-U is differentially diagnosed in the community versus academic centers has not been systematically evaluated. Some patients who may fit the diagnostic criteria for this overlap syndrome may have instead been classified as MDS-U and not captured within our analysis; this may have occurred at increased frequency prior to the official MDS/MPN category designation in 2001. Conversely, patients diagnosed as MDS/MPN-U without verifiable diagnostic information were excluded to ensure all patients were accurately diagnosed. OS was measured from initial diagnostic presentation at our institution, as this was the time-point at which all clinicopathologic variables were reported and the MDS/MPN-U diagnosis was established and/or confirmed. Approximately half of the cohort had been diagnosed with a myeloid malignancy >3 months prior to MDACC presentation (48 patients, 56%), the majority (79%) of whom received observation, hydoxyurea, erythropoietin stimulating agents, or prior therapy was unknown. In patients who did initiate treatment prior to referral, prior treatment did not impact patient outcome.
Overall, we confirm the poor prognosis of MDS/MPN-U without RARS-T, with an OS of 12.4 months from presentation. Consistent with prior reports,(6) JAK2-V617F mutations were not prognostic. Age, peripheral blast percentage, bone marrow blast percentage, platelet count, MDS-IPSS and MDA global score provided statistical significance in univariate analysis; only thrombocytosis was prognostic in the multivariate model. The improved OS of 52.5 months in patients with thrombocytosis is perhaps related to the increased morbidity and mortality observed with thrombocytopenia in myeloid malignancies and particularly MDS.(11, 12) The 11 patients with thrombocytosis were otherwise a diverse group including 3 with circulating blasts, 2 with >5% bone marrow blasts, 2 with splenomegaly, 7 with diploid cytogenetics, and 1 with JAK2-V617F mutation. Whether these patients share a specific genetic phenotype is conceivable, and molecular characterization of this subgroup is ongoing.
Our analysis validates the diagnosis of MDS/MPN-U as a unique pathologic entity, with distinctive features such as an increased (15%) incidence of isolated trisomy(8). Despite the relative frequency of trisomy(8) in myeloid malignancies, remarkably little is known about the pathogenic basis of this abnormality.(13) Similar ongoing molecular analysis will investigate whether trisomy(8) associates with particular somatic mutations in this cohort.
Despite statistical significance, the MDS-IPSS model is not ideal, as the majority (68%) of MDS/MPN-U patients had lower risk scores according to the MDS-IPSS, and yet had poorer survival than their lower-risk MDS counterparts. Furthermore, the improved survival seen in the Int-1 category compared to the low-risk category is contrary to expectation. One prognostic model of clinicopathologic variables has recently been developed from a cohort (n=92) of patients with either MDS-U or MDS/MPN-U, interestingly this cohort did not find platelet count to be of prognostic importance.(14) The MDA global model also provided a significant tool for the MDS/MPN-U cohort (p=0.004). It is noteworthy that the MDA model was originally validated within 176 patients with CMML and leukocytosis, suggesting this may be an appropriate prognostic model to use in MDS/MPN patient populations.
No treatment regimen significantly improved response. Given their relative novelty, only 2 patients received JAK2-inhibitor therapy. In view of the JAK2-V617 mutations present among MDS/MPN-U patients signifying overactivity of JAK/STAT signaling, JAK2-inhibitor therapy may ultimately prove effective, and indeed a clinical trial incorporating the combination of ruxolitinib and azacitidine for patients with MDS/MPN-U is ongoing at our institution.
Supplementary Material
Footnotes
Conflict of Interest Disclosure: The authors declare no competing financial interest
Supplementary information is available at Leukemia's website.
References
- 1.Swerdlow SHCE, Harris LH, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th Edition ed. International Agency for Research on Cancer (IARC); Lyon: 2008. [Google Scholar]
- 2.Orazi A, Germing U. The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2008 Jul;22(7):1308–19. doi: 10.1038/leu.2008.119. PubMed PMID: 18480833. Epub 2008/05/16. eng. [DOI] [PubMed] [Google Scholar]
- 3.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009 Jul 30;114(5):937–51. doi: 10.1182/blood-2009-03-209262. PubMed PMID: 19357394. Epub 2009/04/10. eng. [DOI] [PubMed] [Google Scholar]
- 4.Wang SA, Hasserjian RP, Loew JM, Sechman EV, Jones D, Hao S, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis harbors JAK2 mutation and shows overlapping myeloproliferative and myelodysplastic features. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2006 Sep;20(9):1641–4. doi: 10.1038/sj.leu.2404316. PubMed PMID: 16871284. Epub 2006/07/28. eng. [DOI] [PubMed] [Google Scholar]
- 5.Atallah E, Nussenzveig R, Yin CC, Bueso-Ramos C, Tam C, Manshouri T, et al. Prognostic interaction between thrombocytosis and JAK2 V617F mutation in the WHO subcategories of myelodysplastic/myeloproliferative disease-unclassifiable and refractory anemia with ringed sideroblasts and marked thrombocytosis. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2008 Jun;22(6):1295–8. doi: 10.1038/sj.leu.2405054. PubMed PMID: 18059483. Epub 2007/12/07. eng. [DOI] [PubMed] [Google Scholar]
- 6.Cannella L, Breccia M, Latagliata R, Frustaci A, Alimena G. Clinical and prognostic features of patients with myelodysplastic/myeloproliferative syndrome categorized as unclassified (MDS/MPD-U) by WHO classification. Leukemia research. 2008 Mar;32(3):514–6. doi: 10.1016/j.leukres.2007.07.004. PubMed PMID: 17709138. Epub 2007/08/22. eng. [DOI] [PubMed] [Google Scholar]
- 7.Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997 Mar 15;89(6):2079–88. PubMed PMID: 9058730. Epub 1997/03/15. eng. [PubMed] [Google Scholar]
- 8.Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009 Mar 26;113(13):2895–901. doi: 10.1182/blood-2008-07-170449. PubMed PMID: 18988864. Epub 2008/11/08. eng. [DOI] [PubMed] [Google Scholar]
- 9.Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. Sep 20. 2012;120(12):2454–65. doi: 10.1182/blood-2012-03-420489. PubMed PMID: 22740453. Epub 2012/06/29. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kantarjian H, O'Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. Sep 15. 2008;113(6):1351–61. doi: 10.1002/cncr.23697. PubMed PMID: 18618511. Epub 2008/07/12. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kantarjian H, Giles F, List A, Lyons R, Sekeres MA, Pierce S, et al. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer. May 1. 2007;109(9):1705–14. doi: 10.1002/cncr.22602. PubMed PMID: 17366593. Epub 2007/03/17. eng. [DOI] [PubMed] [Google Scholar]
- 12.Germing U, Hildebrandt B, Pfeilstocker M, Nosslinger T, Valent P, Fonatsch C, et al. Refinement of the international prognostic scoring system (IPSS) by including LDH as an additional prognostic variable to improve risk assessment in patients with primary myelodysplastic syndromes (MDS). Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. Dec. 2005;19(12):2223–31. doi: 10.1038/sj.leu.2403963. PubMed PMID: 16193087. Epub 2005/09/30. eng. [DOI] [PubMed] [Google Scholar]
- 13.Mertens F, Johansson B, Heim S, Kristoffersson U, Mitelman F. Karyotypic patterns in chronic myeloproliferative disorders: report on 74 cases and review of the literature. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK. Mar. 1991;5(3):214–20. PubMed PMID: 2013980. [PubMed] [Google Scholar]
- 14.Liu Y, Tabarroki A, Visconte V, Hasrouni E, Bupathi M, Hamilton BK, et al. A Prognostic Scoring System for Unclassifiable MDS and MDS/MPN. ASH Annual Meeting Abstracts. 2012 2012 Nov 16;120(21):1701. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.