

Abstract
The initiation and progression of Alzheimer disease (AD) is a complex process not yet fully understood. While many hypotheses have been provided as to the cause of the disease, the exact mechanisms remain elusive and difficult to verify. Proteomic applications in disease models of AD have provided valuable insights into the molecular basis of this disorder, demonstrating that on a protein level, disease progression impacts numerous cellular processes such as energy production, cellular structure, signal transduction, synaptic function, mitochondrial function, cell cycle progression, and proteasome function. Each of these cellular functions contributes to the overall health of the cell, and the dysregulation of one or more could contribute to the pathology and clinical presentation in AD. In this review, foci reside primarily on the amyloid β-peptide (Aβ) induced oxidative stress hypothesis and the proteomic studies that have been conducted by our laboratory and others that contribute to the overall understanding of this devastating neurodegenerative disease.
Keywords: Alzheimer Disease, Amyloid-β, Redox proteomics, Oxidative stress, Methionine-35
1.0 Background
Alzheimer disease (AD), a severe, age-associated neurodegenerative disorder, affects many people aged from 65 years or older, with nearly half of those of age of 85 afflicted with this disorder. As the world population grows and life expectancies increase, the number of AD patients is growing at an ever-increasing rate since the most important contributing factor to AD is age. Current reports estimate that there about 5.1 million AD patients in the United States, and may increase up to 20 million by the year of 2050 [1].
The classic histopathological hallmarks of AD can be summarized as the accumulation of extracellular amyloid β-peptide (Aβ)-rich senile plaques (SPs), which are generated from the cleavage of amyloid precursor protein (APP), the accumulation of intracellular neurofibrillary tangles (NFTs), which are largely comprised of the aggregated form of hyperphosphorylated Tau, and synapse loss. Tau, a microtubule stabilizing protein, is hyperphosphorylated in AD neurons and causes microtubular abnormalities with consequent disruption of intra-neuronal trafficking. On a molecular basis, cell cycle changes and oxidative stress resulting from increases in ROS (Reactive Oxygen Species) and RNS (Reactive Nitrogen Species) have also been shown to play a detrimental role in AD [2, 3]. Not all AD cases are sporadic, with a small percentage being the result of hereditary on-set AD.
Classical AD progression can be categorized into four stages: preclinical AD (PCAD), mild cognitive impairment (MCI), early-onset AD (EAD), and late-stage AD (LAD). Many individuals with PCAD have a high amyloid plaque burden, yet function normally. PCAD, however, is difficult to study, as PCAD itself does not show evidence of being fatal and brain samples are only obtained after a death by another means. However, what research has been conducted has been helpful in understanding the earliest of AD progressions.
MCI has been categorized as the transition stage between normal cognition and EAD/AD and can be further sub-divided into amnestic MCI (aMCI) and non-amnestic MCI (naMCI). Pathologically, differences in amyloid load and NFTs vary from patient to patient, yet recent research has shown that a striking difference in aMCI progression to AD (38%) compared to naMCI (20%). naMCI patients present with higher concentrations of cerebral spinal fluid (CSF) Aβ(1-42) as well as less severe hippocampal atrophy that may play a role in these findings [4].
Patients with LAD present with the highest levels of Aβ peptide and NFTs. Applying imaging techniques such as MRI, various degrees of degeneration are observed for all stages of clinical AD. Positron emission tomography (PET) technology used to probe regional glucose utilization within the brain suggest severe energy deficiency for PCAD and MCI patients, which, considering that glucose is as the main energy resource for brain, demonstrates that the brain is under energy deprivation, consistent with the progression of AD.
2.0 ROS
The initiation and propagation of ROS and RNS generation has been shown to play a major role in the pathogenesis of AD [5]; thus, it is important to understand the origin of these oxidants, as well as their modes of action. ROS and RNS include, among many others, the superoxide radical anion (O2−·), hydrogen peroxide (H2O2), hydroxyl radical (·OH), nitric oxide (·NO), and peroxynitrite (ONOO−), most of which are free radicals. ROS/RNS play necessary and beneficial roles in many biological processes [6]. Oxidative stress is a condition in which there is an imbalance of these reactive species and antioxidant defenses [6–8]. In addition to free radicals produced from environmental exposure, excess production of free radicals or other reactive species and a failure of antioxidant defense systems to adequately handle the ROS/RNS load results in damage to biomolecules including proteins, lipids, and genetic material [3, 9–13].
Human mitochondria process oxygen at great potential self-risk due to production and leakage of damaging O2−· (Figure 1), which is elevated due to an age-related decrease in the efficiency of electron transport chain reactions, primarily from Complex I [6, 14–17]. Reactive O2−· has been shown to oxidize biomolecules directly through lipid peroxidation and protein oxidation [18–20] and indirectly inducing oxidative/nitrosative stress in the brain through production of other reactive species, as well as through cellular signaling routes such as that of inflammatory cytokine tumor necrosis factor alpha (TNF-α) [20–24]. The mitochondrial resident superoxide scavenger, manganese superoxide dismutase (MnSOD) in the matrix, and Cu/ZnSOD in the inter-membrane space, reacts with O2−· producing H2O2. H2O2, though only a weak oxidant, becomes much more damaging in the presence of free copper (I) or iron (II) ions through Fenton chemistry by which H2O2 is reduced to the extremely reactive ·OH radical [9, 20]. The ·OH radical is responsible for much of the downstream indirect damage from superoxide. Hard nucleophiles such as the hydroxyl radical may attack biomolecules at carbonyl moieties resulting in structural and functional changes.
Figure 1.

ROS formation involving mitochondria-derived superoxide free radicals and subsequent processing by MnSOD and Fenton Chemistry and RNS formation following NO production via i-NOS and reaction of NO with superoxide free radicals. See text for further details.
Free radicals in close proximity to allylic hydrogen atoms on biomolecules abstract such hydrogens leaving a carbon centered radical. When this process affects poly-unsaturated fatty acids (PUFAs) in the lipid bilayer, which are particularly rich in neurons, a subsequent reaction of this radical with molecular oxygen forms a peroxyl radical leading to further abstraction other allylic hydrogens from nearby biomolecules and consequent chain reaction propogation. Once initiated, lipid peroxidation can lead to increased production of reactive alkenals in the bilayer, loss of lipid asymmetry, and apoptosis [25–28]. A major reactive alkenal produced is 4-hydroxynonenal (HNE). HNE is primarily produced in the brain via lipid peroxidation of arachidonic acid, an abundant omega-6 PUFA found in the lipid bilayer in brain [27]. Arachidonic acid is released through cleavage of a phospholipid by phospholipase A2 (PLA2) and serves as a second messenger involved in cellular signaling and the regulation of signaling enzymes [29–31]. HNE binds proteins by Michael addition to certain amino acids resulting in protein dysfunction [2]. Aβ itself has been shown to be a source of ROS and an initiator of free radical damage to biomolecules in brain including lipid peroxidation [32–39].
A free radical in its natural form, ·NO is synthesized from L-arginine by endothelial, neuronal, or inducible nitric oxide synthases (NOS). ·NO has beneficial effects such as a biological mediator in several processes including neurotransmission, vascular smooth muscle relaxation as well as having antitumor, and antimicrobial activities [40, 41] but becomes toxic at high concentrations [42]. ·NO and O2−· exert more damaging effects when they react to form ONOO− [13]. Although the reaction of ·NO and molecular oxygen or O2−· may proceed relatively slow in the cytoplasm, the ability of these radical molecules to diffuse through lipid membranes, such as the mitochondrial membranes where large amounts of oxygen reside, has been shown to accelerate the rates ·NO autoxidation many times over [43, 44] This increase in autoxidation may be attributed to the ‘lens effect’ that focuses ·NO and O2·- within hydrophobic compartments, not only decreasing the distance they must travel to react with each other and their target proteins, but also increasing the rate of autoxidation [43].
Nitration of tyrosine occurs from the reaction ·NO with O2−· in the presence of CO2 by radical-radical recombination producing the reactive intermediate peroxynitrite, leading to tyrosine nitration by ·NO2 radical at the 3-position sterically blocking the phosphorylation site at the 4-position thereby altering regulation of protein activity by tyrosine kinases and leading to changes in protein function [45–49]. In addition to ·NO2, ·OH is another potential breakdown product of ONOO− [49].
MnSOD itself has been shown to be susceptible to tyrosine nitration in the active site of the enzyme [22, 46, 50], potentially altering the affinity of MnSOD for the O2−· substrate by changing the redox potential of the active site [45]. Tyrosine nitration leads to damaged MnSOD and subsequent mitochondrial dysfunction [22, 50].
Increases in 3-nitrotyrosine (3NT) and protein-bound HNE have been found even in brain of early AD subjects [51, 52]. The brain is rich in PUFA and has areas rich in iron. These facts coupled with high oxygen usage and a low antioxidant capacity, make the brain particularly susceptible to oxidative damage.
3.0 APP Processing
Central to the Aβ-induced oxidative stress hypothesis, is the generation of Aβ from its precursor protein, APP, a type I transmembrane protein that is conserved and expressed in many tissues. The isoform of APP preferentially expressed in the CNS is 695 amino acids in length and is heavily concentrated at the synaptic cleft [53]. The exact physiological role of APP remains undetermined, but this protein is thought to play a role in cell growth, neurite outgrowth, cell adhesion, cell signaling and cell survival [53–55]. The proteolytic processing of APP primarily occurs during its anterograde transport along neuronal axons by one of two pathways: non-amyloidogenic and amyloidogenic, the latter producing the neurotoxic Aβ fragment implicated in AD [56]. Amyloidogenic and non-amyloidogenic pathways are mediated by the actions of β- and γ- and α-secretases, respectively (Figure 2).
Figure 2.

Proteolytic processing of APP via the non-amyloidogenic and amyloidogenic pathways. The latter produces neurotoxic and oxidative stress-producing Aβ-peptide. See text for further details.
Proteolytic cleavage of APP at position 17 by α-secretase initiates the non-amyloidogenic pathway, producing a membrane-bound C-terminal fragment (CTFα) and the large, soluble fragment, APPsα, from the N-terminal domain of APP [57]. APPSα has been demonstrated to have neurotropic and neuroprotective effects [58, 59]. The primary α-secretase in the CNS is a disintegrin and metalloproteinase 10 (ADAM10), a membrane anchored and zinc dependent protease that has been revealed to be regulated by synaptic activity [60]. Further, synapse-associated protein-97 (SAP97) is a necessary protein mediating the trafficking of ADAM10, thereby promoting the non-amyloidogenic pathway by binding to α-secretase and directing it to the post-synaptic membrane where it cleaves APP inside of the Aβ domain of APP. This process negates the amyloidogenic pathway [61]. Moreover, SAP97 expression and activity have been shown to be altered in AD [62, 63]. Additionally, clathrin-mediated endocytosis (CME) of ADAM10 is a regulatory process that has been shown to diminish the non-amyloidogenic processing of APP by removing it from its cellular proximity – the plasma membrane [60].
The amyloidogenic pathway is initiated by the proteolytic action of β-secretase at position 671, thereby producing, the large, soluble APPsβ (β-secretase-cleaved soluble APP) and the C-terminal fragment (CTFβ), C99 [64]. The APPsβ fragment may have multiple detrimental effects, such as impairing anterograde axonal transport leading to axonal dystrophy and neuronal cell death, as well as binding to death receptor 6 to recruit caspase 6 to initiate the extrinsic pathway of apoptosis [65, 66]. The predominant neuronal β-secretase is β-site APP cleaving enzyme (BACE1), a type I transmembrane aspartyl protease – an enzyme that has been shown to be elevated in AD [67, 68]. Further, hydrogen peroxide stimulated oxidative stress increases the activity of the enzyme; and, the presence of 4-hydroxynonenal has been reported to increase the expression of BACE1 [64]. BACE1 has been associated with lipid rafts. Moreover, an increase of membrane cholesterol reportedly recruits APP to lipid rafts, which increases APP-BACE1 proximity, promoting the amyloidogenic processing of APP [69]. In contrast, non-amyloidogenic proteolysis occurs outside of lipid rafts.
The next step involved in both the non-amyloidogenic pathway and amyloidogenic pathway in the proteolytic processing of APP is the cleavage of the CTFα and CTFβ, fragments, respectively, by γ-secretase. Gamma secretase is an intra-membrane protease complex consisting of a quartet of proteins: presenilin, nicastrin, anterior pharynx-defective 1 (APH1) and presenilin enhancer 2 (PEN2) [70]. Further, γ-secretase is hypothesized to proteolytically cleave the remaining APP fragment at multiple sites in a step-wise manner within the transmembrane domain (TMD) of CTFα and CTFβ, thereby releasing p3 (non-amyloidogenic pathway), Aβ (amyloidogenic pathway) and APP intracellular domain (AICD) fragments [71]. In the amyloidogenic pathway in particular, this begins with the cleavage at the ε-site at either position 48 or 49 on C99, which releases the AICD fragment while the Aβ remains membrane-bound [70]. Next, it is thought that γ-secretase further cleaves the Aβ fragment every other 3 or 4 amino acids from the ζ-site to the γ-site until the Aβ fragment is freed from the membrane [70]. This leads to the production of Aβ fragments of varying sizes, from 37 to 46 amino acids in length (with heterogeneous C-termini), including neurotoxic Aβ(1-42), which can easily oligomerize and become more prone to aggregation [72]. Furthermore, mutations in APP and the PS1 protein in γ-secretase can lead to changes in this cleavage pattern [71].
4.0 Aβ-Induced Oxidative Stress Model
It has long been established that the role of oxidative stress in AD is a critical one that leads to the damage of vital cellular components such as proteins, lipids, and nucleic acids [20, 27, 73, 74]. This damage if left unchecked is a primary reason for the eventual degeneration of neurons, possibly through apoptotic means [75]. The Aβ-induced oxidative stress hypothesis proposed by our laboratory and others [76, 77], states that Aβ and the damage it initiates are the principal means underlying this injurious increase in oxidative stress observed in AD brain.
As discussed above, APP may be processed into two major isoforms of Aβ by way of β- and γ-secreteases; Aβ(1-40) and Aβ(1-42). Studies have demonstrated that as AD progresses, the levels of Aβ(1-40) in CSF remains relatively constant, while in contrast, levels of Aβ(1-42) in CSF decrease but levels of Aβ(1-42) increase in senile plaques [78]. This distribution has been attributed to the possibility of an efflux deficiency for Aβ(1-42) which may play a role in plaque formation [79]. Previously, plaques were perceived to be the primary pathogenic element of AD, yet more recently evidence provided insights into the notion that plaques may be an extra-cellular storage site for cells to deposit excess Aβ, suggesting the real damaging agent, may be a much smaller aggregate form of Aβ(1-42) oligomers [80, 81]. Research has shown that while plaques do not correlate with cognitive dysfunction in AD, soluble oligomers do [82]. More recently, researchers have discovered in AD that distinct assemblies of Aβ oligomers impair cholinergic neurotransmission [83].
The Aβ-induced oxidative stress hypothesis places the majority of the causative effect of increased cellular oxidative stress upon oligomeric Aβ(1-42), as it is believed that only the oligomers are viable to insert into the lipid bilayer wherein they may form alpha-helices to begin the proposed catalytic ROS production that may lead to the lipid peroxidation and protein oxidation found in AD [84].
4.1 Met-35 and Aβ-induced oxidative stress
While studies have found that oligomeric Aβ(1-42) correlates with increased oxidative stress, the exact method by which this occurs is still under debate [82]. Strong evidence has been put forth by our lab and others that implicates the metionine-35 (Met-35) residue of Aβ(1-42) in the process of ROS generation. Since Aβ(1-42) is cleaved from APP, a transmembrane protein, our lab proposes that Aβ(1-42) oligomers are capable of re-entering the lipid bilayer where they may adopt an alpha-helical structure. In doing so, Met-35 would interact with the carbonyl of Ile-31 according to the i+4 rule of alpha-helices [85]. It is hypothesized that because the oxygen of the carbonyl group is more electronegative than the sulfur of Met-35, the electron lone pairs on Met-35 are primed for oxidation by an extrinsic factor.
Once the Aβ oligomers insert into the bilayer, the hydrophobic environment lends itself towards the stabilization of a one-electron oxidation of an already primed Met to the sulfuranyl free radical [MetS+] [86]. It is this sulfuranyl free radical that is hypothesized as being the initiator in a series of free radical chain reactions that take place within the lipid bilayer that generate lipid peroxidation products and oxidatively modified membrane proteins [86, 87] (Figure 3). Substitution of amino acids in critical positions within the Aβ(1-42) peptide negate the injurious effects of Aβ(1-42). For example, the substitution of Gly with Asp at residue 37 of Aβ(1-42) imparts a negative charge to the Aβ(1-42) peptide that excludes the peptide from the lipid bilayer [88]. Another substitution that replaces Ile-31 with the known α-helix breaker, proline, and again the oxidative stress of the peptide is prevented, consistent with our hypothesis that the α-helical structure of Aβ(1-42) in the lipid bilayer is important for the production of oxidative stress [85].
Figure 3.
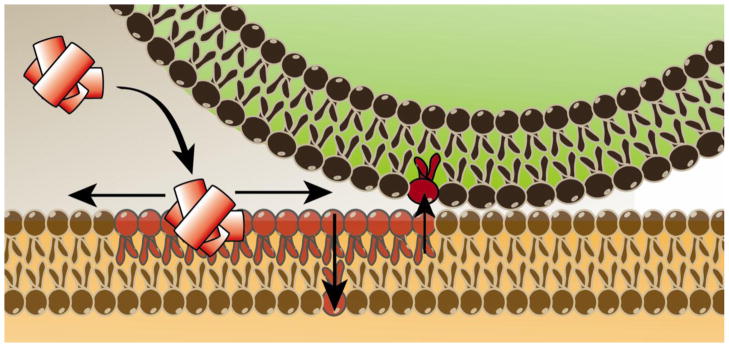
Insertion of oligomeric Aβ(1-42), a peptide rich in hydrophobic amino acids, into lipid bilayers leads to lipid peroxidation reactions with subsequent protein modification from lipid peroxidation products. See text for further details.
Transgenic Caenorhabditis elegans that express human Aβ(1-42) demonstrated increased oxidative stress that was nullified by the substitution of Met-35 with another sulfur containing amino acid, Cys, in an attempt to demonstrate the differences in chemistry of the two sulfur atoms and their associated residencies (thioether vs. thiol) [89]. In an in vitro study, Met-35 in Aβ(1-42) was substituted by norleucine, i.e., a methylene moiety for the S-atom of Met, to produce [Aβ(1-42)M35NLE]. This substitution produced a mutant peptide with an amino acid of similar length and hydrophobicity as the original Met-35. Aβ(1-42)M35NLE was unable to induce toxicity through oxidative stress by way of free radical generation [89–92].
The J20 mouse, which is a transgenic mouse with human APP containing Swedish (KM670/671NL) and Indiana (V717F) mutations, showed elevated Aβ(1-40/42) deposition and increased oxidative stress in brain [93]. Introduction of a third mutation to APP, Met631Leu, corresponding to the Met-35 residue of Aβ(1-42), resulted in no oxidative stress in brain of these mice at 9 months of age [94]. This results demonstrated in vivo in a mammalian model what had been seen earlier in a worm model: Met-35 of Aβ(1-42) is essential for oxidative stress in vivo in AD models, and presumably in AD brain as well.
Important to note are other findings that provide evidence contrary to the Met-35 centric hypothesis such as research conducted that used Aβ(25-35) instead of Aβ(1-42) with a substitution of Met-35 with norleucine at the c-terminal position that did not abrogate the oxidative induced by the peptide [95]. These data, however, should be read with the understanding that a C-terminal Met displays altered chemistry from a Met within the α-helix [96].
5.0 Proteomics Applications in AD and Models Thereof
Proteomics is the study of the proteome, meaning that proteomics studies view the entirety of all proteins present in a given system at any given point in time. Proteomics is far more complex than genomics as it includes all isoforms of a protein, their structure and post-translational modifications as well as protein-protein interactions [97]. In addition, the proteome is not static; it is subject to change during development and in response to various events such as oxidative stress, disease or drug administration. Therefore, proteomics can be applied to compare the proteome of control vs. treated samples or healthy controls vs. a disease state. Knowledge of the affected proteins can help in gathering insights into pathways and cellular mechanisms of a disease and also can help in developing interventions or therapeutic strategies. In addition to providing information on up- or down-regulated proteins (expression proteomics), proteomics techniques can be applied to look at changes in post-translational modifications (e.g., phosphoproteomics). Furthermore, our laboratory pioneered a proteomics technique, redox proteomics (Figure 4), that can specifically identify differentially oxidized proteins in a given sample [98–100].
Figure 4.

Schematic illustration of the principal steps involved in redox proteomics used to identify oxidatively modified proteins. See text for further details.
Gel-based proteomic studies generally consist of two main steps: In the first step, the sample is separated, e.g. by two-dimensional gel electrophoresis by which the proteins are separated based on their net charge, or isoelectric point, and subsequently by their migration rate in a polyacrylamide gel. The second step consists of identifying the proteins identified by mass spectrometry and data base inquiry. For redox proteomics, an additional step is used in which gel electrophoresis is followed by Western blot analysis with oxidation marker-specific antibodies (for comprehensive reviews see [98, 100].
Proteomics has been used extensively by our laboratory and others in the field to analyze the effects of Aβ-mediated oxidative stress in AD models as well as brains from subjects of different stages of AD. Some of these studies and their findings are summarized below.
5.1 Aβ in cell culture
Early studies have shown that Aβ(25-35) can produce free radicals in solution [101] or synaptic membranes [102] and that the addition of (pre-incubated/aggregated) Aβ(1-40) to neurons increases intracellular ROS formation and increases the levels of protein carbonyls [33, 37, 89]. Many proteomic studies have used this approach, the addition of Aβ-peptides to a system, to identify proteins that are affected by Aβ-induced oxidative stress. These studies have shown that Aβ-peptides change protein levels and lead to the oxidation of specific enzymes.
Treatment of neurons with oligomeric Aβ(1-42) decreased the levels metabolic enzymes involved in energy production (e.g., citrate synthase) and increased molecular chaperones such as Hsp70 [103]. In primary neurons Aβ(1-42) treatment also lead to the oxidation (measured as protein carbonyls) of proteins associated with energy metabolism (e.g., GAPDH) as well as regulatory and structural proteins [104–106]. Interestingly, increasing the cells’ antioxidant defense systems by pre-incubation of cells with a glutathione precursor or mimetic significantly reduced overall Aβ-induced protein oxidation (measured as protein carbonyls) as well as specifically inhibited the oxidation of four enzymes: GAPDH, 14-3-3 zeta, malate dehydrogenase and pyruvate kinase [104, 105]. In a different study, pretreatment with Vitamin E, a lipid-soluble free radical scavenger also inhibited protein oxidation and lipid peroxidation caused by Aβ(1-42) incubation [33, 107].
Since inflammation is observed in AD brain, a recent study analyzed the effects of Aβ(25-35) on microglia and found that Aβ(25-35) activated immortalized microglia and induced down-regulation of metabolic enzymes, redox proteins (e.g, Peroxiredoxin 3 and 4) and chaperones [108]. A caveat of these and other studies that used Aβ(25-35) is that the C-terminal Met-35 of Ab(25-35) has a different chemistry than intrachain Met-35 in AD-relevant Aβ(1-42) [96] It is our view that only the AD-relevant peptides should be used in studies of importance to AD, since (a) such peptides are known in AD brain, but (b) Aβ(25-35) has not been detected in AD and is not relevant to the disease per se but only of academic interest [96].
5.2 C. Elegans AD Model
Transgenic C. elegans expressing human Aβ(1-42) have been previously shown to present with increased oxidative stress prior to Aβ fibrillar deposition [109]. Using this information, another study of transgenic C. elegans with the application of redox proteomics was successful in identifying a number of specifically oxidized proteins [110]. Of the oxidized proteins found, many are important in energetic metabolism (i.e., malate dehydrogenase and Acyl-CoA dehydrogenase), antioxidant defense (i.e., glutathione S-transferase), and proteasome function (i.e., proteasome beta subunit).
5.3 AD Mouse Models
Since the discovery of mutations causing familial AD, different transgenic mouse models have been established to study cellular mechanisms relevant to AD. The human double-mutant APP/PS-1 knock-in mice carry a PS-1 mutation found in familial AD as well as a mutation in the APP gene to humanize the mouse Aβ sequence. Gene expression is controlled by the endogenous mouse promoters for APP and PS-1 [111]. These mice show increased levels of protein oxidation and lipid peroxidation [112] as well as changes in protein expression when compared to wild-type mice. In particular, proteins involved in energy metabolism such as GAPDH and enolase had decreased levels in APP/PS-1 mice [112–114]. Additional redox proteomics studies comparing APP/PS-1 to wild-type mice showed an age-related significant oxidation of enolase and other proteins (e.g, 14-3-3, actin and Pin-1) [115]. Oral administration of the glutathione precursor N-acetyl cysteine decreased brain protein oxidation and lead to increased levels of enolase indicating that antioxidants, as shown in cell culture studies, could ameliorate the deleterious effects of Aβ-induced oxidation in brain in vivo [113–115].
The SAMP8 mouse, or senescence-accelerated prone mouse, is a mouse model of AD used that show age-dependent learning and memory deficits and Aβ accumulation [116], as well as increased oxidative stress [117]. Redox proteomics of 12 month old SAMP8 mice has been used to identify many proteins that are specifically carbonylated in this mouse model for oxidation which include: α-enolase, collapsin response mediator protein-2 (CRMP-2/DRP-2), lactate dehydrogenase (LDH-2), α-spectrin, and creatine kinase (CK). Expression proteomics was also used to determine a decreased expression of critical proteins such as LDH-2, triosphosphate isomerase (TPI), α-spectrin, and neurofilament (NF-L) [118]. Many of these proteins affected are vital to both the production of energy as well as the structural organization of the cell, both of which are vital to the neuron, a cell with high energy demand that must maintain the ability to transport vesicles and mitochondrion the length of the cell and to reorganize itself when constructing and deconstructing synapses.
5.4 Aβ injection in Animal Models
Intracerebral injection of Aβ(1-40) lead to behavioral deficits in rats and decreased levels of different proteins, among others proteins associated with ATP production and cytoskeletal structure [119]. Another study used Aβ(1-42) injections into the nucleus basalis magnocellularis (NBM) of rats followed by a proteomic analysis. The cortex, NBM, and hippocampus were all shown to exhibit significant protein oxidation, affecting proteins such as glutamine synthetase, 14-3-3 zeta, beta-synuclein, pyruvate dehydrogenase, phosphoglycerate mutase 1 and glyceraldehyde-3-phosphate dehydrogenase [38]. While other groups have reported results from similar experiments, such as intrahippocampal injections of Aβ [120], the results of Boyd-Kimball et al. are significant on their own as they highlight that injection of Aβ(1-42) into one brain region has consequences for other regions of the brain [38].
5.5 Human studies
While animal models are adequate and convenient substitutions for human disease, the results have to be interpreted with respect to the facts that the cell biology of humans and the majority of laboratory animal specimens have differences that can make a direct comparison difficult. This inherent difficulty is what makes human proteomic studies so valuable. Proteomic studies in brains of subjects with various stages of AD have identified proteins involved in a variety of cellular pathways to be expressed at differential levels in AD brain when compared to controls. At the University of Kentucky, we are fortunate to have autopsy-derived specimens obtained at a short post-mortem interval (PMI), typically 2–4 h, making comparison to living brain as feasible as possible. In contrast, long PMIs have sometime been reported in the literature, and one wonders what can be realistically and meaningfully determined with such samples?
Proteomics studies from our laboratory have shown that some proteins, like Pin-1 and β-tubulin, were significantly decreased while others, like enolase and heat shock protein70, were significantly increased in AD [121]. A proteomics study focusing only the detergent-insoluble proteins also found significant changes in samples from AD subjects when compared to controls. Amongst others, protein levels of 14-3-3 and GAPDH were altered in the detergent-insoluble fractions [122]. Supporting the notion that oxidation of proteins may render them less soluble and thus more prone to aggregation, GAPDH (among others) was shown to be oxidized in AD [123]. A number of redox proteomics studies from our laboratory and collaborators have identified a variety of proteins that are extensively oxidized in AD brain, including, among others, carbonylation of ubiquitin carboxy-terminal hydrolase L-1 (UCHL-1) [124], nitration of enolase [123] and carbonylation and loss of CRMP-2 [125], a protein found to be intricately involved in axonal growth and plasticity, a process heavily impaired in AD, while also being associated with neurofibrillary tangles [126]. In a similar fashion to protein Tau in AD brain, CRMP-2 has been shown to not only be phosphorylated by GSK3β and CDK5 [127], but is also localized to NFTs [126, 128]. A systematic depletion of CRMP-2 via insoluble tangle formation would likely have detrimental effects on the ability of an AD affected neuron to maintain or repair the synaptic connections lost during the disease progression.
5.5 Summary of Proteomic Data
In summary, proteomic results from cell culture to animal models to human studies identified a number of proteins to be affected by the presence of Aβ, supporting the notion that Aβ-mediated oxidative stress contributes to the perturbations seen in a variety of cellular mechanisms in AD. Since some proteomics studies identified a number of afflicted proteins, rather than discussing an exhaustive list of proteins, only a few commonly recurring ones have been mentioned in this review. Throughout the studies of the various animal models and AD brain, a common theme emerges in the form of affected cellular functions. The proteins that show either altered levels or oxidation or both can be summarized into following functional groups:
Energy metabolism (i.e., GAPDH, enolase, ATP synthase)
Signal transduction (i.e., 14-3-3)
Synaptic function (i.e., SNAPs)
Structural proteins (i.e., actin, tubulin, DRP2/CRMP2)
Proteasome function and protein clearance (i.e., UCHL-1)
Stress response/chaperones (i.e., HSPs and Peroxiredoxins)
Cell cycle, Ab production and tau hyperphosphorylation (i.e., Pin-1)
Mitochondrial function (i.e., ATP synthase, VDAC)
This overview provides a glimpse into the AD-affected proteome, information that if not for the use of proteomics would have been difficult or nearly impossible to collect. Many of the proteins affected by Aβ presented herein contribute to vital cellular functions, and the dysfunction or loss of these proteins are consistent with the pathology and clinical presentation in AD. For instance, the fact that GAPDH and enolase, both glycolytic enzymes with multiple additional functions such as cell signaling [129, 130], are affected by Aβ, supports a role of Aβ in the observed energy impairment in AD [131] while other proteins such as CRMP2 play important roles in cellular organization and structure.
6.0 Dietary antioxidants and AD biomarkers
Proteomic changes due to Aβ-mediated oxidative stress can be significantly modulated by pretreatment of cells with different antioxidants. In agreement with cell culture studies, treatment with antioxidants ameliorates negative effects associated with Aβ in different animal models of AD. Unfortunately, clinical trials and epidemiological studies in AD have produced inconsistent results. While in the Rotterdam study high dietary intake of Vitamins C and E was associated with lower risk for AD [132], a study in elderly subjects found no association between carotenes, vitamins C or E and a decreased risk of AD [133]. Vitamin E supplementation in MCI patients proved to have no benefit [134]. While it has been shown that antioxidant based treatment in synaptosomes and rodents appear to have ameliorating effects in regards to Aβ induced oxidative stress production [33, 135, 136], the lack of efficacy in human studies indicates a lack of a complete understanding in either the mechanisms of Aβ induced oxidative stress or of the inability of current antioxidant therapies to be effective where they are needed. Regardless, this avenue of research must be thoroughly investigated.
As seen in animal studies (APP/PS-1 mice) [113] the starting point of the intervention influences the outcome, and since Aβ-mediated oxidative stress is present long before any clinical and pathological hallmarks of AD are detectable, it may prove to be necessary to start therapies years if not decades before symptoms are evident. The availability of reliable AD biomarkers would improve AD diagnostics dramatically; not only could AD be diagnosed unequivocally ante mortem, but it may enable earlier diagnosis and therefore allow for earlier treatment of people at risk of developing AD. Additionally, the effects of interventional strategies and drug efficacy could be monitored. Unfortunately, reliable biomarkers for AD diagnostics are yet unavailable. Due to its proximity to the brain, much research has focused on cerebrospinal fluid for biomarker identification (e.g. [137]) but the caveat with this strategy is the necessity of a lumbar puncture to obtain the sample. An easily accessible source for potential biomarkers is blood. Given that 80–85 percent of the plasma proteome consists of albumin and IgGs, AD biomarkers in blood likely would be far less concentrated than in cerebrospinal fluid. To overcome this challenge, a recent study depleted plasma samples of highly abundant proteins and applied a series of fractionation steps before proteomic identification of potential plasma biomarkers [138].
One other approach may be to look at other blood elements. For example, our laboratory recently demonstrated elevated oxidative stress in mitochondria isolated from peripheral lymphocytes from patients with AD [139] and aMCI [140]. The latter study also demonstrated, using proteomics, differential levels of mitochondrial proteins in both AD and aMCI that are consistent with the known mitochondrial alterations in these disorders. Consonant with these findings, and noting that oxidation plays an important role AD coupled with the need for reliable biomarkers for AD, including differentially oxidized proteins, redox proteomics may be a promising approach (for further comprehensive reviews see [141, 142]).
7.0 Conclusion
Redox proteomics has become an invaluable tool in not only the study of AD, but in many other areas of study in which oxidative stress is believed to be central to disease onset [98]. While the ability to treat a disease is of grave importance, the understanding of how a disease is initiated and progresses may have implications into many different diseases with similar pathology. Data gathered from proteomics studies play a role in this understanding of disease pathology, providing valuable information into the molecular basis of the cell that might otherwise go unnoticed. In its application to AD, proteomics has shown that energy metabolism and structural proteins, among others, are primarily affected by the increase in oxidative stress observed in AD. These cellular processes are important in their maintenance of a healthy cellular environment, while their absence has been demonstrated to be deleterious. Further proteomics studies into the efficacy of antioxidant treatment prior to disease onset should be conducted in order to fully test the oxidative stress hypothesis of AD. Moreover, while biomarkers and antioxidant treatment may possibly contribute to a better disease outcome, general awareness of preventative lifestyle changes by the general populace may also play its role in disease prevention, as many lifestyle factors have been implicated in cognitive performance, which may delay or possibly prevent disease onset [143–145].
Highlights.
Alzheimer disease (AD), Amyloid-β processing, and ROS implicated in AD are examined
The Aβ peptide induced oxidative stress hypothesis of AD is discussed
Met-35 of Amyloid-β peptide and its role in oxidative stress in AD is discussed
An overview of redox proteomics and applications to AD and AD models is presented
Antioxidant treatment and biomarker research relevant to AD is presented
Acknowledgments
This work was supported in part by a NIH grant to D.A.B. [AG-05119].
Abbreviations
- 3NT
3-nitrotyrosine
- HNE
4-hydroxynonenal
- AD
Alzheimer disease
- aMCI
Amnestic mild cognitive impairment
- APP
Amyloid precursor protein
- AICD
Amyloid precursor protein intracellular domain
- Aβ
Amyloid-β
- BACE1
Beta-secretase 1
- CTF
C-terminal fragment
- EAD
Early onset Alzheimer disease
- LAD
Late-stage Alzheimer disease
- MCI
Mild cognitive impairment
- NFTs
Neurofibrillary tangles
- naMCI
Non-amnestic mild cognitive impairment
- ONOO−
Peroxynitrite
- PUFA
Polyunsaturated fatty acid
- PET
Positron emission tomography
- PCAD
Pre-clinical Alzheimer disease
- PS-1
Presenilin-1
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SAMP8
Senescence-accelerated prone mouse 8
- sAPPα
Soluble amyloid precursor protein α
- sAPPβ
Soluble amyloid precursor protein β
- O2−·
Superoxide
- SOD
Superoxide dismutase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Archives of neurology. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free radical biology & medicine. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 3.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Current neuropharmacology. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vos SJ, van Rossum IA, Verhey F, Knol DL, Soininen H, Wahlund LO, Hampel H, Tsolaki M, Minthon L, Frisoni GB, Froelich L, Nobili F, van der Flier W, Blennow K, Wolz R, Scheltens P, Visser PJ. Prediction of Alzheimer disease in subjects with amnestic and nonamnestic MCI. Neurology. 2013;80:1124–1132. doi: 10.1212/WNL.0b013e318288690c. [DOI] [PubMed] [Google Scholar]
- 5.Sultana R, Butterfield DA. Role of oxidative stress in the progression of Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2010;19:341–353. doi: 10.3233/JAD-2010-1222. [DOI] [PubMed] [Google Scholar]
- 6.Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free radical biology & medicine. 2012;62:170–185. doi: 10.1016/j.freeradbiomed.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Sies H. Oxidative stress: oxidants and antioxidants. Experimental physiology. 1997;82:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 8.Schulz E, Wenzel P, Munzel T, Daiber A. Mitochondrial Redox Signaling: Interaction of Mitochondrial Reactive Oxygen Species with Other Sources of Oxidative Stress. Antioxidants & redox signaling. 2012 doi: 10.1089/ars.2012.4609. (ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halliwell B. Free radicals and antioxidants: updating a personal view. Nutrition reviews. 2012;70:257–265. doi: 10.1111/j.1753-4887.2012.00476.x. [DOI] [PubMed] [Google Scholar]
- 10.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. The international journal of biochemistry & cell biology. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Butterfield DA, Dalle-Donne I. Redox proteomics. Antioxidants & redox signaling. 2012;17:1487–1489. doi: 10.1089/ars.2012.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hensley K, Williamson KS, Floyd RA. Measurement of 3-nitrotyrosine and 5-nitro-gamma-tocopherol by high-performance liquid chromatography with electrochemical detection. Free radical biology & medicine. 2000;28:520–528. doi: 10.1016/s0891-5849(00)00155-6. [DOI] [PubMed] [Google Scholar]
- 13.Williamson KS, Gabbita SP, Mou S, West M, Pye QN, Markesbery WR, Cooney RV, Grammas P, Reimann-Philipp U, Floyd RA, Hensley K. The nitration product 5-nitro-gamma-tocopherol is increased in the Alzheimer brain. Nitric oxide: biology and chemistry/official journal of the Nitric Oxide Society. 2002;6:221–227. doi: 10.1006/niox.2001.0399. [DOI] [PubMed] [Google Scholar]
- 14.Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxidants & redox signaling. 2005;7:1140–1149. doi: 10.1089/ars.2005.7.1140. [DOI] [PubMed] [Google Scholar]
- 15.Calabrese V, Lodi R, Tonon C, D’Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, Stella AM, Butterfield DA. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. Journal of the neurological sciences. 2005;233:145–162. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 16.De Giusti VC, Caldiz CI, Ennis IE, Perez NG, Cingolani HE, Aiello EA. Mitochondrial reactive oxygen species (ROS) as signaling molecules of intracellular pathways triggered by the cardiac renin-angiotensin II-aldosterone system (RAAS) Frontiers in physiology. 2013;4:126. doi: 10.3389/fphys.2013.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scialo F, Mallikarjun V, Stefanatos R, Sanz A. Regulation of Lifespan by the Mitochondrial Electron Transport Chain: Reactive Oxygen Species-Dependent and Reactive Oxygen Species-Independent Mechanisms. Antioxidants & redox signaling. 2012 doi: 10.1089/ars.2012.4900. (ahead of print) [DOI] [PubMed] [Google Scholar]
- 18.Fridovich I. Biological effects of the superoxide radical. Archives of biochemistry and biophysics. 1986;247:1–11. doi: 10.1016/0003-9861(86)90526-6. [DOI] [PubMed] [Google Scholar]
- 19.Deby C, Goutier R. New perspectives on the biochemistry of superoxide anion and the efficiency of superoxide dismutases. Biochemical pharmacology. 1990;39:399–405. doi: 10.1016/0006-2952(90)90043-k. [DOI] [PubMed] [Google Scholar]
- 20.Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. The Biochemical journal. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klamt F, Gottfried C, Tramontina F, Dal-Pizzol F, Da Frota ML, Jr, Moreira JC, Dias RD, Moriguchi E, Wofchuk S, Souza DO. Time-related increase in mitochondrial superoxide production, biomolecule damage and antioxidant enzyme activities in cortical astrocyte cultures. Neuroreport. 2002;13:1515–1518. doi: 10.1097/00001756-200208270-00005. [DOI] [PubMed] [Google Scholar]
- 22.Tangpong J, Sompol P, Vore M, St Clair W, Butterfield DA, St Clair DK. Tumor necrosis factor alpha-mediated nitric oxide production enhances manganese superoxide dismutase nitration and mitochondrial dysfunction in primary neurons: an insight into the role of glial cells. Neuroscience. 2008;151:622–629. doi: 10.1016/j.neuroscience.2007.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aluise CD, Miriyala S, Noel T, Sultana R, Jungsuwadee P, Taylor TJ, Cai J, Pierce WM, Vore M, Moscow JA, St Clair DK, Butterfield DA. 2-Mercaptoethane sulfonate prevents doxorubicin-induced plasma protein oxidation and TNF-alpha release: implications for the reactive oxygen species-mediated mechanisms of chemobrain. Free radical biology & medicine. 2011;50:1630–1638. doi: 10.1016/j.freeradbiomed.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Dean RT, Fu S, Stocker R, Davies MJ. Biochemistry and pathology of radical-mediated protein oxidation. The Biochemical journal. 1997;324(Pt 1):1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bader Lange ML, St Clair D, Markesbery WR, Studzinski CM, Murphy MP, Butterfield DA. Age-related loss of phospholipid asymmetry in APP(NLh)/APP(NLh) x PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice: relevance to Alzheimer disease. Neurobiology of disease. 2010;38:104–115. doi: 10.1016/j.nbd.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castegna A, Lauderback CM, Mohmmad-Abdul H, Butterfield DA. Modulation of phospholipid asymmetry in synaptosomal membranes by the lipid peroxidation products, 4-hydroxynonenal and acrolein: implications for Alzheimer’s disease. Brain research. 2004;1004:193–197. doi: 10.1016/j.brainres.2004.01.036. [DOI] [PubMed] [Google Scholar]
- 27.Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochimica et biophysica acta. 2010;1801:924–929. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perluigi M, Coccia R, Butterfield DA. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies. Antioxidants & redox signaling. 2012;17:1590–1609. doi: 10.1089/ars.2011.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frisardi V, Panza F, Seripa D, Farooqui T, Farooqui AA. Glycerophospholipids and glycerophospholipid-derived lipid mediators: a complex meshwork in Alzheimer’s disease pathology. Progress in lipid research. 2011;50:313–330. doi: 10.1016/j.plipres.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Jones PM, Persaud SJ. Arachidonic acid as a second messenger in glucose-induced insulin secretion from pancreatic beta-cells. The Journal of endocrinology. 1993;137:7–14. doi: 10.1677/joe.0.1370007. [DOI] [PubMed] [Google Scholar]
- 31.Prentki M, Matschinsky FM. Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiological reviews. 1987;67:1185–1248. doi: 10.1152/physrev.1987.67.4.1185. [DOI] [PubMed] [Google Scholar]
- 32.Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. Journal of structural biology. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 33.Yatin SM, Varadarajan S, Butterfield DA. Vitamin E Prevents Alzheimer’s Amyloid beta-Peptide (1-42)-Induced Neuronal Protein Oxidation and Reactive Oxygen Species Production. Journal of Alzheimer’s disease: JAD. 2000;2:123–131. doi: 10.3233/jad-2000-2212. [DOI] [PubMed] [Google Scholar]
- 34.Butterfield DA. beta-Amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer’s disease. Chemical research in toxicology. 1997;10:495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 35.Butterfield DAS, ER Protein Oxidation Processes in Aging Brain. Advances in Cell Aging and Gerontology. 1997;2:161–191. [Google Scholar]
- 36.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1-42. Journal of neurochemistry. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 37.Harris ME, Hensley K, Butterfield DA, Leedle RA, Carney JM. Direct evidence of oxidative injury produced by the Alzheimer’s beta-amyloid peptide (1-40) in cultured hippocampal neurons. Experimental neurology. 1995;131:193–202. doi: 10.1016/0014-4886(95)90041-1. [DOI] [PubMed] [Google Scholar]
- 38.Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1-42) into rat brain: implications for Alzheimer’s disease. Neuroscience. 2005;132:313–324. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 39.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiology of aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 40.Shinde UA, Mehta AA, Goyal RK. Nitric oxide: a molecule of the millennium. Indian journal of experimental biology. 2000;38:201–210. [PubMed] [Google Scholar]
- 41.Xu W, Charles IG, Moncada S, Gorman P, Sheer D, Liu L, Emson P. Mapping of the genes encoding human inducible and endothelial nitric oxide synthase (NOS2 and NOS3) to the pericentric region of chromosome 17 and to chromosome 7, respectively. Genomics. 1994;21:419–422. doi: 10.1006/geno.1994.1286. [DOI] [PubMed] [Google Scholar]
- 42.McCarty MF. Down-regulation of microglial activation may represent a practical strategy for combating neurodegenerative disorders. Medical hypotheses. 2006;67:251–269. doi: 10.1016/j.mehy.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 43.Moller MN, Li Q, Lancaster JR, Jr, Denicola A. Acceleration of nitric oxide autoxidation and nitrosation by membranes. IUBMB life. 2007;59:243–248. doi: 10.1080/15216540701311147. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, Miller MJ, Joshi MS, Thomas DD, Lancaster JR., Jr Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feeney MB, Schoneich C. Tyrosine modifications in aging. Antioxidants & redox signaling. 2012;17:1571–1579. doi: 10.1089/ars.2012.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- 47.Surmeli NB, Litterman NK, Miller AF, Groves JT. Peroxynitrite mediates active site tyrosine nitration in manganese superoxide dismutase. Evidence of a role for the carbonate radical anion. Journal of the American Chemical Society. 2010;132:17174–17185. doi: 10.1021/ja105684w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ischiropoulos H. Protein tyrosine nitration--an update. Archives of biochemistry and biophysics. 2009;484:117–121. doi: 10.1016/j.abb.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 49.Amirmansour C, Vallance P, Bogle RG. Tyrosine nitration in blood vessels occurs with increasing nitric oxide concentration. British journal of pharmacology. 1999;127:788–794. doi: 10.1038/sj.bjp.0702590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Surmeli NB, Litterman NK, Miller AF, Groves JT. Peroxynitrite Mediates Active Site Tyrosine Nitration in Manganese Superoxide Dismutase. Evidence of a Role for the Carbonate Radical Anion. J Am Chem Soc. 2010;132:17174–17185. doi: 10.1021/ja105684w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reed TT, Pierce WM, Jr, Turner DM, Markesbery WR, Butterfield DA. Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. Journal of cellular and molecular medicine. 2009;13:2019–2029. doi: 10.1111/j.1582-4934.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reed TT, Pierce WM, Markesbery WR, Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain research. 2009;1274:66–76. doi: 10.1016/j.brainres.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 53.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annual review of neuroscience. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng H, Koo EH. The amyloid precursor protein: beyond amyloid. Molecular neurodegeneration. 2006;1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Billnitzer AJ, Barskaya I, Yin C, Perez RG. APP independent and dependent effects on neurite outgrowth are modulated by the receptor associated protein (RAP) Journal of neurochemistry. 2013;124:123–132. doi: 10.1111/jnc.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grimm MO, Haupenthal VJ, Rothhaar TL, Zimmer VC, Grosgen S, Hundsdorfer B, Lehmann J, Grimm HS, Hartmann T. Effect of Different Phospholipids on alpha-Secretase Activity in the Non-Amyloidogenic Pathway of Alzheimer’s Disease. International journal of molecular sciences. 2013;14:5879–5898. doi: 10.3390/ijms14035879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 58.Hartlage-Rubsamen M, Waniek A, Rossner S. Munc13 genotype regulates secretory amyloid precursor protein processing via postsynaptic glutamate receptors. International journal of developmental neuroscience: the official journal of the International Society for Developmental Neuroscience. 2013;31:36–45. doi: 10.1016/j.ijdevneu.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 59.Gustafsen C, Glerup S, Pallesen LT, Olsen D, Andersen OM, Nykjaer A, Madsen P, Petersen CM. Sortilin and SorLA display distinct roles in processing and trafficking of amyloid precursor protein. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:64–71. doi: 10.1523/JNEUROSCI.2371-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lichtenthaler SF. Alpha-secretase cleavage of the amyloid precursor protein: proteolysis regulated by signaling pathways and protein trafficking. Current Alzheimer research. 2012;9:165–177. doi: 10.2174/156720512799361655. [DOI] [PubMed] [Google Scholar]
- 61.Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A, Epis R, Borroni B, Cattabeni F, Sala C, Padovani A, Di Luca M. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marcello E, Epis R, Saraceno C, Gardoni F, Borroni B, Cattabeni F, Padovani A, Di Luca M. SAP97-mediated local trafficking is altered in Alzheimer disease patients’ hippocampus. Neurobiology of aging. 2012;33:422, e421–410. doi: 10.1016/j.neurobiolaging.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 63.Vingtdeux V, Marambaud P. Identification and biology of alpha-secretase. Journal of neurochemistry. 2012;120(Suppl 1):34–45. doi: 10.1111/j.1471-4159.2011.07477.x. [DOI] [PubMed] [Google Scholar]
- 64.Ortega F, Stott J, Visser SA, Bendtsen C. Interplay between alpha-, beta-, and gamma-secretases determines biphasic amyloid-beta protein level in the presence of a gamma-secretase inhibitor. The Journal of biological chemistry. 2013;288:785–792. doi: 10.1074/jbc.M112.419135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodrigues EM, Weissmiller AM, Goldstein LS. Enhanced beta-secretase processing alters APP axonal transport and leads to axonal defects. Human molecular genetics. 2012;21:4587–4601. doi: 10.1093/hmg/dds297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Sutinen EM, Pirttila T, Anderson G, Salminen A, Ojala JO. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-beta production in human neuron-like cells. Journal of neuroinflammation. 2012;9:199. doi: 10.1186/1742-2094-9-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venugopal C, Demos CM, Rao KS, Pappolla MA, Sambamurti K. Beta-secretase: structure, function, and evolution. CNS & neurological disorders drug targets. 2008;7:278–294. doi: 10.2174/187152708784936626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marquer C, Devauges V, Cossec JC, Liot G, Lecart S, Saudou F, Duyckaerts C, Leveque-Fort S, Potier MC. Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25:1295–1305. doi: 10.1096/fj.10-168633. [DOI] [PubMed] [Google Scholar]
- 70.Steiner H, Fluhrer R, Haass C. Intramembrane proteolysis by gamma-secretase. The Journal of biological chemistry. 2008;283:29627–29631. doi: 10.1074/jbc.R800010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crump CJ, Johnson DS, Li YM. Development and Mechanism of gamma-Secretase Modulators for Alzheimer’s Disease. Biochemistry. 2013 doi: 10.1021/bi400377p. (ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tate B, McKee TD, Loureiro RM, Dumin JA, Xia W, Pojasek K, Austin WF, Fuller NO, Hubbs JL, Shen R, Jonker J, Ives J, Bronk BS. Modulation of gamma-secretase for the treatment of Alzheimer’s disease. International journal of Alzheimer’s disease. 2012;2012:210756. doi: 10.1155/2012/210756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weidner AM, Bradley MA, Beckett TL, Niedowicz DM, Dowling AL, Matveev SV, LeVine H, 3rd, Lovell MA, Murphy MP. RNA oxidation adducts 8-OHG and 8-OHA change with Abeta42 levels in late-stage Alzheimer’s disease. PloS one. 2011;6:e24930. doi: 10.1371/journal.pone.0024930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Markesbery WR, Lovell MA. DNA oxidation in Alzheimer’s disease. Antioxidants & redox signaling. 2006;8:2039–2045. doi: 10.1089/ars.2006.8.2039. [DOI] [PubMed] [Google Scholar]
- 75.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 76.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends in molecular medicine. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 77.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free radical biology & medicine. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 78.Tamaoka A. Characterization of amyloid beta protein species in the plasma, cerebrospinal fluid and brains of patients with Alzheimer’s disease. Nihon Ronen Igakkai zasshi Japanese journal of geriatrics. 1998;35:273–277. doi: 10.3143/geriatrics.35.273. [DOI] [PubMed] [Google Scholar]
- 79.Gustafson DR, Skoog I, Rosengren L, Zetterberg H, Blennow K. Cerebrospinal fluid beta-amyloid 1-42 concentration may predict cognitive decline in older women. Journal of neurology, neurosurgery, and psychiatry. 2007;78:461–464. doi: 10.1136/jnnp.2006.100529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Glabe CC. Amyloid accumulation and pathogensis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Sub-cellular biochemistry. 2005;38:167–177. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- 81.Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein and peptide letters. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 82.Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiology of disease. 2009;35:352–358. doi: 10.1016/j.nbd.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bao F, Wicklund L, Lacor PN, Klein WL, Nordberg A, Marutle A. Different beta-amyloid oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity. Neurobiology of aging. 2012;33:825, e821–813. doi: 10.1016/j.neurobiolaging.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 84.Butterfield DA, Swomley AM, Sultana R. Amyloid beta-Peptide (1-42)-Induced Oxidative Stress in Alzheimer Disease: Importance in Disease Pathogenesis and Progression. Antioxidants & redox signaling. 2013 doi: 10.1089/ars.2012.5027. (ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanski J, Aksenova M, Schoneich C, Butterfield DA. Substitution of isoleucine-31 by helical-breaking proline abolishes oxidative stress and neurotoxic properties of Alzheimer’s amyloid beta-peptide. Free radical biology & medicine. 2002;32:1205–1211. doi: 10.1016/s0891-5849(02)00821-3. [DOI] [PubMed] [Google Scholar]
- 86.Butterfield DA, Kanski J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer’s amyloid beta-peptide 1-42. Peptides. 2002;23:1299–1309. doi: 10.1016/s0196-9781(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 87.Butterfield DA, Sultana R. Methionine-35 of abeta(1-42): importance for oxidative stress in Alzheimer disease. Journal of amino acids. 2011;2011:198430. doi: 10.4061/2011/198430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kanski J, Aksenova M, Butterfield DA. The hydrophobic environment of Met35 of Alzheimer’s Abeta(1-42) is important for the neurotoxic and oxidative properties of the peptide. Neurotox Res. 2002;4:219–223. doi: 10.1080/10298420290023945. [DOI] [PubMed] [Google Scholar]
- 89.Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1-42) Neurobiol Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. discussion 339–342. [DOI] [PubMed] [Google Scholar]
- 90.Murray MM, Bernstein SL, Nyugen V, Condron MM, Teplow DB, Bowers MT. Amyloid beta protein: Abeta40 inhibits Abeta42 oligomerization. Journal of the American Chemical Society. 2009;131:6316–6317. doi: 10.1021/ja8092604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Clementi ME, Pezzotti M, Orsini F, Sampaolese B, Mezzogori D, Grassi C, Giardina B, Misiti F. Alzheimer’s amyloid beta-peptide (1-42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochemical and biophysical research communications. 2006;342:206–213. doi: 10.1016/j.bbrc.2006.01.137. [DOI] [PubMed] [Google Scholar]
- 92.Dai XL, Sun YX, Jiang ZF. Attenuated cytotoxicity but enhanced betafibril of a mutant amyloid beta-peptide with a methionine to cysteine substitution. FEBS letters. 2007;581:1269–1274. doi: 10.1016/j.febslet.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 93.Robinson RA, Lange MB, Sultana R, Galvan V, Fombonne J, Gorostiza O, Zhang J, Warrier G, Cai J, Pierce WM, Bredesen DE, Butterfield DA. Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: effects of the amyloid-beta peptide of amyloid precursor protein. Neuroscience. 2011;177:207–222. doi: 10.1016/j.neuroscience.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang J, Sultana R, Bredesen DE. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free radical biology & medicine. 2010;48:136–144. doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. Journal of neurochemistry. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 96.Varadarajan S, Kanski J, Aksenova M, Lauderback C, Butterfield DA. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer’s A beta(1--42) and A beta(25--35) Journal of the American Chemical Society. 2001;123:5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- 97.Tyers M, Mann M. From genomics to proteomics. Nature. 2003;422:193–197. doi: 10.1038/nature01510. [DOI] [PubMed] [Google Scholar]
- 98.Butterfield DA, Perluigi M, Reed T, Muharib T, Hughes CP, Robinson RA, Sultana R. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxidants & redox signaling. 2012;17:1610–1655. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sultana R, Butterfield DA. Oxidative Modification of Brain Proteins in Alzheimer’s Disease: Perspective on Future Studies Based on Results of Redox Proteomics Studies. Journal of Alzheimers Disease. 2013;33:S243–S251. doi: 10.3233/JAD-2012-129018. [DOI] [PubMed] [Google Scholar]
- 100.Di Domenico F, Butterfield DA, Robinson RAS. Mass Spectrometry and Redox Proteomics: Applications in Disease. Mass Spectrometry Reviews. 2013 doi: 10.1002/mas.21374. in press. [DOI] [PubMed] [Google Scholar]
- 101.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Butterfield DA, Hensley K, Harris M, Mattson M, Carney J. beta-Amyloid peptide free radical fragments initiate synaptosomal lipoperoxidation in a sequence-specific fashion: implications to Alzheimer’s disease. Biochemical and biophysical research communications. 1994;200:710–715. doi: 10.1006/bbrc.1994.1508. [DOI] [PubMed] [Google Scholar]
- 103.Foldi I, Datki ZL, Szabo Z, Bozso Z, Penke B, Janaky T. Proteomic study of the toxic effect of oligomeric Abeta1-42 in situ prepared from ‘iso-Abeta1-42’. Journal of neurochemistry. 2011;117:691–702. doi: 10.1111/j.1471-4159.2011.07238.x. [DOI] [PubMed] [Google Scholar]
- 104.Boyd-Kimball D, Sultana R, Poon HF, Mohmmad-Abdul H, Lynn BC, Klein JB, Butterfield DA. Gamma-glutamylcysteine ethyl ester protection of proteins from Abeta(1-42)-mediated oxidative stress in neuronal cell culture: a proteomics approach. Journal of neuroscience research. 2005;79:707–713. doi: 10.1002/jnr.20393. [DOI] [PubMed] [Google Scholar]
- 105.Sultana R, Newman SF, Abdul HM, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Protective effect of D609 against amyloid-beta1-42-induced oxidative modification of neuronal proteins: redox proteomics study. Journal of neuroscience research. 2006;84:409–417. doi: 10.1002/jnr.20876. [DOI] [PubMed] [Google Scholar]
- 106.Thomas SN, Soreghan BA, Nistor M, Sarsoza F, Head E, Yang AJ. Reduced neuronal expression of synaptic transmission modulator HNK-1/neural cell adhesion molecule as a potential consequence of amyloid beta-mediated oxidative stress: a proteomic approach. J Neurochem. 2005;92:705–717. doi: 10.1111/j.1471-4159.2004.02892.x. [DOI] [PubMed] [Google Scholar]
- 107.Boyd-Kimball D, Sultana R, Mohmmad-Abdul H, Butterfield DA. Rodent Abeta(1-42) exhibits oxidative stress properties similar to those of human Abeta(1-42): Implications for proposed mechanisms of toxicity. Journal of Alzheimer’s disease: JAD. 2004;6:515–525. doi: 10.3233/jad-2004-6509. [DOI] [PubMed] [Google Scholar]
- 108.Di Francesco L, Correani V, Fabrizi C, Fumagalli L, Mazzanti M, Maras B, Schinina ME. 14-3-3 epsilon marks the amyloid-stimulated microglia long-term activation. Proteomics. 2012;12:124–134. doi: 10.1002/pmic.201100113. [DOI] [PubMed] [Google Scholar]
- 109.Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiology of aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 110.Boyd-Kimball D, Poon HF, Lynn BC, Cai J, Pierce WM, Jr, Klein JB, Ferguson J, Link CD, Butterfield DA. Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Abeta(1-42): implications for Alzheimer’s disease. Neurobiology of aging. 2006;27:1239–1249. doi: 10.1016/j.neurobiolaging.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 111.Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK. Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. The American journal of pathology. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free radical biology & medicine. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huang Q, Aluise CD, Joshi G, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: toward therapeutic modulation of mild cognitive impairment. Journal of neuroscience research. 2010;88:2618–2629. doi: 10.1002/jnr.22422. [DOI] [PubMed] [Google Scholar]
- 114.Robinson RA, Joshi G, Huang Q, Sultana R, Baker AS, Cai J, Pierce W, St Clair DK, Markesbery WR, Butterfield DA. Proteomic analysis of brain proteins in APP/PS-1 human double mutant knock-in mice with increasing amyloid beta-peptide deposition: insights into the effects of in vivo treatment with N-acetylcysteine as a potential therapeutic intervention in mild cognitive impairment and Alzheimer’s disease. Proteomics. 2011;11:4243–4256. doi: 10.1002/pmic.201000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sultana R, Robinson RA, Di Domenico F, Abdul HM, St Clair DK, Markesbery WR, Cai J, Pierce WM, Butterfield DA. Proteomic identification of specifically carbonylated brain proteins in APP(NLh)/APP(NLh) x PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice model of Alzheimer disease as a function of age. Journal of proteomics. 2011;74:2430–2440. doi: 10.1016/j.jprot.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yagi H, Katoh S, Akiguchi I, Takeda T. Age-related deterioration of ability of acquisition in memory and learning in senescence accelerated mouse: SAM-P/8 as an animal model of disturbances in recent memory. Brain research. 1988;474:86–93. doi: 10.1016/0006-8993(88)90671-3. [DOI] [PubMed] [Google Scholar]
- 117.Butterfield DA, Poon HF. The senescence-accelerated prone mouse (SAMP8): a model of age-related cognitive decline with relevance to alterations of the gene expression and protein abnormalities in Alzheimer’s disease. Experimental gerontology. 2005;40:774–783. doi: 10.1016/j.exger.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 118.Poon HF, Castegna A, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, Morley JE, Klein JB, Butterfield DA. Quantitative proteomics analysis of specific protein expression and oxidative modification in aged senescence-accelerated-prone 8 mice brain. Neuroscience. 2004;126:915–926. doi: 10.1016/j.neuroscience.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 119.Shi X, Lu X, Zhan L, Liu L, Sun M, Gong X, Sui H, Niu X, Liu S, Zheng L, Chen J, Zhou Y. Rat hippocampal proteomic alterations following intrahippocampal injection of amyloid beta peptide (1-40) Neuroscience letters. 2011;500:87–91. doi: 10.1016/j.neulet.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 120.Yang GF, Wang LN, Zhao X, Nie YH. Two-dimensional electrophoregram for proteomic analysis of rat brain with intrahippocampal amyloid beta injection and normal rat brain. Di 1 jun yi da xue xue bao = Academic journal of the first medical college of PLA. 2004;24:553–555. [PubMed] [Google Scholar]
- 121.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. Journal of Alzheimer’s disease: JAD. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 122.Woltjer RL, Cimino PJ, Boutte AM, Schantz AM, Montine KS, Larson EB, Bird T, Quinn JF, Zhang J, Montine TJ. Proteomic determination of widespread detergent-insolubility including Abeta but not tau early in the pathogenesis of Alzheimer’s disease. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1923–1925. doi: 10.1096/fj.05-4263fje. [DOI] [PubMed] [Google Scholar]
- 123.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiology of disease. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 124.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free radical biology & medicine. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 125.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 126.Yoshida H, Watanabe A, Ihara Y. Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer’s disease. The Journal of biological chemistry. 1998;273:9761–9768. doi: 10.1074/jbc.273.16.9761. [DOI] [PubMed] [Google Scholar]
- 127.Li T, Hawkes C, Qureshi HY, Kar S, Paudel HK. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry-Us. 2006;45:3134–3145. doi: 10.1021/bi051635j. [DOI] [PubMed] [Google Scholar]
- 128.Cole AR, Noble W, van Aalten L, Plattner F, Meimaridou R, Hogan D, Taylor M, LaFrancois J, Gunn-Moore F, Verkhratsky A, Oddo S, LaFerla F, Giese KP, Dineley KT, Duff K, Richardson JC, Yan SD, Hanger DP, Allan SM, Sutherland C. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J Neurochem. 2007;103:1132–1144. doi: 10.1111/j.1471-4159.2007.04829.x. [DOI] [PubMed] [Google Scholar]
- 129.Butterfield DA, Lange ML. Multifunctional roles of enolase in Alzheimer’s disease brain: beyond altered glucose metabolism. Journal of neurochemistry. 2009;111:915–933. doi: 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Butterfield DA, Hardas SS, Lange ML. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: many pathways to neurodegeneration. Journal of Alzheimer’s disease: JAD. 2010;20:369–393. doi: 10.3233/JAD-2010-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hoyer S. Brain glucose and energy metabolism abnormalities in sporadic Alzheimer disease. Causes and consequences: an update. Experimental gerontology. 2000;35:1363–1372. doi: 10.1016/s0531-5565(00)00156-x. [DOI] [PubMed] [Google Scholar]
- 132.Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA: the journal of the American Medical Association. 2002;287:3223–3229. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 133.Luchsinger JA, Tang MX, Shea S, Mayeux R. Antioxidant vitamin intake and risk of Alzheimer disease. Archives of neurology. 2003;60:203–208. doi: 10.1001/archneur.60.2.203. [DOI] [PubMed] [Google Scholar]
- 134.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ. Vitamin E and donepezil for the treatment of mild cognitive impairment. The New England journal of medicine. 2005;352:2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 135.Joshi G, Perluigi M, Sultana R, Agrippino R, Calabrese V, Butterfield DA. In vivo protection of synaptosomes by ferulic acid ethyl ester (FAEE) from oxidative stress mediated by 2,2-azobis(2-amidino-propane)dihydrochloride (AAPH) or Fe(2+)/H(2)O(2): insight into mechanisms of neuroprotection and relevance to oxidative stress-related neurodegenerative disorders. Neurochemistry international. 2006;48:318–327. doi: 10.1016/j.neuint.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 136.Grossi C, Rigacci S, Ambrosini S, Ed Dami T, Luccarini I, Traini C, Failli P, Berti A, Casamenti F, Stefani M. The Polyphenol Oleuropein Aglycone Protects TgCRND8 Mice against Ass Plaque Pathology. PloS one. 2013;8:e71702. doi: 10.1371/journal.pone.0071702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Finehout EJ, Franck Z, Choe LH, Relkin N, Lee KH. Cerebrospinal fluid proteomic biomarkers for Alzheimer’s disease. Annals of neurology. 2007;61:120–129. doi: 10.1002/ana.21038. [DOI] [PubMed] [Google Scholar]
- 138.Henkel AW, Muller K, Lewczuk P, Muller T, Marcus K, Kornhuber J, Wiltfang J. Multidimensional plasma protein separation technique for identification of potential Alzheimer’s disease plasma biomarkers: a pilot study. J Neural Transm. 2012;119:779–788. doi: 10.1007/s00702-012-0781-3. [DOI] [PubMed] [Google Scholar]
- 139.Sultana R, Mecocci P, Mangialasche F, Cecchetti R, Baglioni M, Butterfield DA. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. Journal of Alzheimer’s disease: JAD. 2011;24:77–84. doi: 10.3233/JAD-2011-101425. [DOI] [PubMed] [Google Scholar]
- 140.Sultana R, Baglioni M, Cecchetti R, Cai J, Klein JB, Bastiani P, Ruggiero C, Mecocci P, DAB Oxidative Stress in the Proteomics Analysis of Mitochondria Isolated from Peripheral Lymphocytes in Mild Cognitive Impairment: Towards Biomarker Development and Better Understanding of the Progression and Pathogenesis of Alzheimer Disease. Free radical biology & medicine. 2013 doi: 10.1016/j.freeradbiomed.2013.08.001. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Di Domenico F, Coccia R, Butterfield DA, Perluigi M. Circulating biomarkers of protein oxidation for Alzheimer disease: Expectations within limits. Biochimica et biophysica acta. 2011;1814:1785–1795. doi: 10.1016/j.bbapap.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 142.Bachi A, Dalle-Donne I, Scaloni A. Redox proteomics: chemical principles, methodological approaches and biological/biomedical promises. Chemical reviews. 2013;113:596–698. doi: 10.1021/cr300073p. [DOI] [PubMed] [Google Scholar]
- 143.Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Roberts RO, Lowe VJ, Kantarci K, Senjem ML, Gunter JL, Boeve BF, Petersen RC, Jack CR., Jr Effect of lifestyle activities on Alzheimer disease biomarkers and cognition. Annals of neurology. 2012;72:730–738. doi: 10.1002/ana.23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dao AT, Zagaar MA, Levine AT, Salim S, Eriksen JL, Alkadhi KA. Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Current Alzheimer research. 2013;10:507–515. doi: 10.2174/1567205011310050006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shah R. The role of nutrition and diet in Alzheimer disease: a systematic review. Journal of the American Medical Directors Association. 2013;14:398–402. doi: 10.1016/j.jamda.2013.01.014. [DOI] [PubMed] [Google Scholar]