

Abstract
The heterogeneous delivery of drugs in tumors is an established process contributing to variability in treatment outcome. Despite the general acceptance of variable delivery, the study of the underlying causes is challenging given the complex tumor microenvironment including intra- and inter-tumor heterogeneity. The difficulty in studying this distribution is even more significant for small molecule drugs where radiolabeled compounds or mass spectrometry detection lack the spatial and temporal resolution required to quantify the kinetics of drug distribution in vivo. In this work, we take advantage of the synthesis of fluorescent drug conjugates that retain their target binding but are designed with different physiochemical and thus pharmacokinetic properties. Using these probes, we followed the drug distribution in cell culture and tumor xenografts with temporal resolution of seconds and subcellular spatial resolution. These measurements, including in vivo permeability of small molecule drugs, can be used directly in predictive pharmacokinetic models for the design of therapeutics and companion imaging agents as demonstrated by a finite element model.
Keywords: Drug distribution, tumor heterogeneity, finite element modeling, drug permeability, intracellular imaging agent
Introduction
Heterogeneous drug distribution is a well-known phenomenon in the delivery of therapeutics to tumors (1–6), and the manipulation of delivery has a direct impact on clinical treatment of tumors (7). Variable delivery also has a pivotal but often less appreciated role in the development of targeted imaging agents (8–11). Despite the importance and impact of this heterogeneity on therapeutic outcome and imaging agent design, the study of the underlying causes is complicated by the complex and variable tumor microenvironment (4). At the whole tumor level, heterogeneous vascularization and blood flow (12), gradients in oxygen (13), nutrients (14), and waste products (15), multiple cell types (16), genetic (17), and epigenetic (18) variability in protein expression (19) all contribute to complex patterns in distribution. At the cellular level, heterogeneity can be due to differences in MDR expression levels among different clones (20), mosaic tumors (17), different cell states (18), nutrient supply (21), and acquired drug resistances, among others. Ultimately, the final distribution is determined by both these tumor characteristics and the drug specific properties, such as molecular weight, charge, lipophilicity, target and non-target binding.
The study of small molecule distribution has been complicated by a lack of high spatial and temporal resolution methods for tracking molecules in tumors. The two most common ex vivo methods are autoradiography of tritiated or other radiolabeled compounds and organ based detection using LCMS. Typically these experiments provide data over several hours with tissue level distribution (6, 22–25) and an upper limit of sampling rate of minutes with cellular resolution. The relatively fast distribution of some drugs has required that in vitro methods, such as multi-layered cells or tumor spheroids (26–30), be developed to measure transport parameters. In vivo methods lack the high spatial and temporal resolution necessary for many of these drugs. Mathematical models have also played a large role due to their ability to easily decouple the drug and tumor specific effects and aid in experimental design (31–35). However, for small molecule drugs, the tissue, cellular, and subcellular experimental data for mathematical model validation is often lacking, providing a significant obstacle to the use of these models.
Given the challenges associated with studying small molecule distribution in tumors, in concert with the recent technological advances in intravital and preclinical tumor biology research (36–38), we have been developing new imaging techniques to follow drugs at higher spatial and temporal resolution in the tumor microenvironment. For example, we have derived therapeutic drug precursors with BODIPY FL to develop companion imaging drugs (39–41). This has led to new insight into the pharmacokinetics of some drugs at the single cell level in vivo. However, the attachment of the different fluorochromes also offers the unique opportunity to study variable PK given differences in the physiochemical and fluorescent properties (Figure 1). In particular, using a relatively small footprint BODIPY FL on one molecule and a larger BODIPY 650 fluorophore on another, the two drugs can be used to study molecular effects in the same tissue without complicating local environment factors.
Figure 1. Molecular Properties and Cellular Imaging.
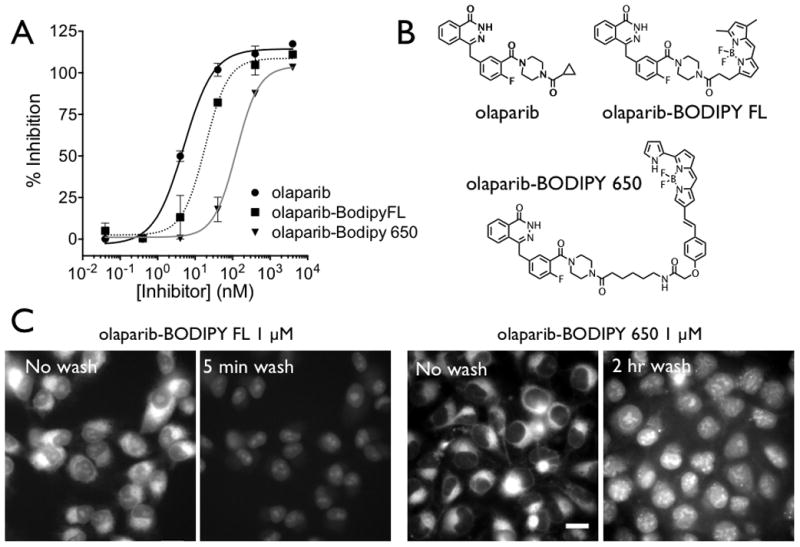
(A) The measured inhibitory concentrations of the three PARP inhibitors with the structure shown in (B). (C) In vitro images of olaparib-BODIPY FL and olaparib-BODIPY 650 show similar patterns of uptake. Before washing, the majority of the probe is located in the perinuclear endoplasmic reticulum, and after washing, the nuclear signal (with higher PARP concentrations in the nucleolus) is more prominent. The kinetics of distribution are significantly different between the two probes, with the olaparib-BODIPY 650 rates around 10 times slower. The ratio of uptake in the ER to the nucleus before washing is also much higher with the olaparib-BODIPY 650 probe. The contrast is identical for olaparib-BODIPY FL before and after the wash, but the contrast was increased after 2 hrs of washing to show the nuclear specific staining which is much lower than the perinuclear signal before washing (Figure S2) Scale bar = 20 μm
In this paper, we use two different PARP inhibitor companion imaging drugs to investigate the specific effects of molecular weight and lipophilicity on target versus non-target localization and tumor vessel permeability. The physiochemical properties have a significant effect on cellular uptake and wash-out kinetics, tumor vessel permeability, and target versus off-target ratio. A mathematical model is developed using a finite element mesh to predict the distribution in the tissue with in vivo imaging data used to validate the results.
Materials and Methods
Synthesis of Probes
Unless otherwise noted, all reagents were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification. BODIPY FL and BODIPY 650/665 succinimidyl ester were purchased from Invitrogen (Carlsbad, CA). Olaparib (AZD2281) was purchased from Selleck Chemicals (Houston, TX). 4-[[4-Fluoro-3-(piperazine-1-carbonyl)phenyl]methyl]-2H-phthalazin-1-one and olaparib-BODIPY FL were synthesized as described elsewhere (40–42). HPLC-ESI-MS analyses and HPLC purifications were performed on a Waters (Milford, MA) LC-MS system. For LC-ESI-MS analyses, a Waters XTerra® C18 5 μm column was used. For preparative runs, an Atlantis® Prep T3 OBDTM 5 μM column was used. High-resolution ESI mass spectra were obtained on a Bruker Daltonics APEXIV 4.7 Tesla Fourier Transform mass spectrometer (FT-ICR-MS) in the Department of Chemistry Instrumentation Facility at the Massachusetts Institute of Technology. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Varian AS-400 (400 MHz) spectrometer. Chemical shifts for protons are reported in parts per million (ppm) and are referenced against the dimethylsulfoxide lock signal (1H, 2.50 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constants (Hz) and integration.
Synthesis of Olaparib-BODIPY 650
Triethylamine (11 μL, 78 μmol) was added to a solution of BODIPY 650/665 succinimidyl ester (5.0 mg, 7.8 μmol) and 4-[[4-fluoro-3-(piperazine-1-carbonyl)phenyl]methyl]-2H-phthalazin-1-one (5.7 mg, 15.6 μmol) in dimethyl sulfoxide (500 μL). The reaction mixture was stirred for 2 hours at room temperature and purified via HPLC to yield the title compound as a blue solid (5.5 mg, 6.1 μmol, 78%). 1H NMR (400 MHz, DMSO-d6) δ = 12.59 (s, 1H), 11.45 (s, 1H), 8.30-8.25 (m, 1H), 8.11 (t, 3JHH = 5.2, 1H), 7.95 (d, 3JHH = 7.7, 1H), 7.88 (t, 3JHH = 7.3, 1H), 7.82 (t, 3JHH = 7.5, 1H), 7.58-7.04 (m, 16H), 6.37 (m, 1H), 4.52 (s, 2H), 4.32 (s, 2H), 3.62-3.12 (m, 10H), 2.34-2.23 (m, 2H), 1.51-1.43 (m, 4H), 1.29-1.22 (m, 2H); 19F NMR (376 MHz, DMSO-d6) δ = −120.2 (s, 1F), −142.3 (q, JBF = 34 Hz, 2F); LC-ESI-MS(+) m/z (%) = 875.5 [M−F]+ (100), 894.5 [M+H]+ (35), 917.5 [M+Na]+ (15); LC-ESI-MS(−) m/z (%) = 893.4 [M−H]− (100); HRMS-ESI [M+H]+ m/z calcd. for [C49H47BF3N8O5]+ 895.3709, found 895.3732 [M+H]+.
Plasma protein binding
Plasma protein binding was measured using rapid equilibrium dialysis plates from Pierce (Rockford, IL) according to the manufacturer’s instructions. Briefly, each probe was diluted to a 10 μM concentration in 300 μL of mouse serum and allowed to equilibrate at 37°C on a shaker for 4 hours with 100 μL of phosphate buffered saline (PBS). After equilibration, samples from each compartment were mixed with additional plasma or PBS so the final buffer was 50% PBS and 50% plasma (to minimize protein binding effects on fluorescence). The fluorescence was measured using a Tecan Safire plate reader (Morrisville, NC).
PARP enzyme assay
PARP enzymatic activity was measured following the manufacturer’s instructions for the universal PARP colorimetric assay kit from Trevigen (Gaithersburg, MD). Briefly, increasing concentrations of olaparib, olaparib-BODIPY FL, or olaparib-BODIPY 650 (0–4 μM) were added to histone-coated strip wells in a 96-well plate. Inhibitors were then pre-incubated for 10 min at room temperature with 0.5U/well recombinant human PARP. Wells were then incubated for 1 hr with PARP cocktail and then washed twice with PBS containing 0.1% Triton X-100 and twice with PBS. Streptavidin-HRP was then added to each well and incubated for 1 hr. Wells were again washed twice with PBS-0.1% Triton X-100 and twice with PBS. The colorimetric reaction was developed upon addition of TACS-Sapphire for 15 min, followed by reaction termination with 0.2 M HC. Absorbance was measured on a TECAN Sapphire2 plate reader and enzyme inhibition curves were fit using non-linear regression dose-response inhibition equations from GraphPad (Prism).
Cell lines and imaging setup
For cell and xenograft experiments, HT-1080 cells were purchased from ATCC (Manassas, VA) in February of 2006. Cells were resuscitated in February of 2012 for experiments; no authentication was done by the authors. The cells were grown in DMEM supplemented with non-essential amino acids, 10% FBS, and 1% penicillin-streptomycin. These cells contain large nuclei and highly vascularized xenografts ideal for measuring subcellular distribution in vivo. Live cell imaging was done using an inverted Deltavision deconvolution microscope with a 40x objective (Applied Precision Instruments) and equipped with a 37°C heated stage and 5% CO2 live cell imaging setup. Media and solutions with probes were prewarmed to 37°C prior to addition through an access window. The appropriate dichroic mirrors and filters were used for imaging BODIPY FL and BODIPY 650 fluorescence.
Mouse xenografts
Xenografts were imaged as previously described (40), and all animal experiments were carried out in accordance with guidelines from the Institutional Subcommittee on Research Animal Care. Briefly, dorsal skin window chambers were surgically implanted in 6–8 week old nude mice. Approximately 3–4 million cells were suspended in a 50% Matrigel and 50% PBS solution and injected under the fascia and allowed to grow for 1–2 weeks. When tumors were vascularized and reached 1–2 mm in size, mice were anesthetized using 1–2% isoflurane in 2 L/min oxygen and a tail vein catheter was inserted. The mouse was moved to a heated microscope stage where the vital signs were continuously monitored. Angiosense-750 (Perkin Elmer) or 250 μg of 500 kDa amino-dextran labeled with Pacific Blue NHS ester (Life Technologies) was injected via tail vein and used to visualize the vasculature. Once the region of interest was selected, a time-lapse imaging sequence was initiated, followed by tail vein injection of the probe. 75 nmol of probe was formulated in 30 μL of 1:1 dimethylacetamide:solutol solution and 112.5 μL of phosphate buffered saline and sonicated.
Permeability measurements
Permeability measurements were taken using a single vessel method (43). Briefly, a time lapse imaging sequence (sequential scans every 10 seconds) was started prior to injection. After intravenous delivery of 75 nmol of both probes, regions of interest inside the vessel and outside the vessel were drawn to measure the plasma concentration and extravascular concentration over time. Taking into account the plasma fraction and protein binding (since the permeability is measured relative to the free drug concentration in the plasma), the permeability was calculated for each region. These measurements were repeated at several locations within a field of view for a minimum of three mice.
Mathematical modeling
Details of the mathematical model can be found in the supporting material, but briefly, the plasma concentration was set to a biexponential decay as measured in vivo. Due to high plasma protein binding, this concentration was held constant throughout the length of the vessels in agreement with theoretical (3) and experimental results (40). The product of the permeability and free drug concentration gradient across the vessel wall was set equal to the diffusive flux in the tissue for the boundary conditions. In the tissue, the rapid immobilization by non-specific protein binding and cellular uptake was assumed to be linear, so the effective diffusion coefficient was adjusted by the bound to free ratio (44). Free drug entered the nucleus and bound the target at a rate equal to that measured in cell culture. The wash-out kinetics were set to those in cell culture. These rates are a combination of transport to the target protein and binding/dissociation, so they are not necessarily equal to the intrinsic association/dissociation kinetics of the drug.
Results
Structure function studies during drug development show tolerance for a large number of substitutions at the cyclopropyl ring in olaparib (42). The charge neutral BODIPY dyes were used to ensure membrane permeability required to access the nuclear target protein. The structure and properties of the drugs are listed in Figure 1 and Table 1. Enzyme inhibition assays of olaparib-BODIPY FL and olaparib-BODIPY 650 showed nM IC50 values and expected decreasing affinities as cyclopropane substitutions increase in bulk size (40).
Table 1. Molecular, Cellular, and In vivo Properties.
Top: molecular weight, calculated logP, enzyme affinity, and fluorescence properties of the three PARP inhibitors. Middle: Cellular compartment uptake and clearance rates measured in vitro. Bottom: Plasma clearance of fluorescent probes. Clearance is similar, although the 1 min concentration of olaparib-BODIPY FL is significantly less even after injecting the same dose. Plasma protein binding for both probes was high but similar.
| Molecule | olaparib | olaparib-BODIPY FL | olaparib-BODIPY 650 |
|---|---|---|---|
| MW (g/mol) | 434 | 640 | 895 |
| clogP | 1.2 | 4.0 | 6.0 |
| PARP EC50 | 4.4 +/− 1.0 nM | 14 +/− 1.2 nM | 92 +/− 45 nM |
| Ex/Em (nm) | na | 502 / 510 | 646 / 660 |
| Perinuclear uptake half-life (at 1 μM) | 1.18 min | 21.2 min | |
| Nuclear uptake half-life (at 1 μM) | 1.14 min | 8.27 min | |
| Perinuclear clearance half-life | ~1.66 min | ~12.8 min | |
| Nuclear clearance half-life | 1.36 hr | 1.50 hr | |
| Perinuclear/nuclear ratio (equilibrium at 1 μM) | 6.0 | 130 | |
| Mouse plasma clearance | C0 = 7.0 μM A = 0.77 t1/2,alpha = 5.16 min t1/2,beta = 85.7 min |
C0 = 22.8 μM A = 0.91 t1/2,alpha = 7.68 min t1/2,beta = 92.5 min |
|
| Mouse plasma protein binding | 99.23% (+/− 0.11%) | 99.22% (+/− 0.66%) | |
After validating target binding of the two agents, the ability of the drugs to localize to the nucleus in vitro was investigated. HT1080 cells were incubated with 1 μM of each drug in real time using an inverted microscope and glass coverslip bottom dishes. Using an epifluorescence microscope, images were acquired every second to capture the rapid uptake (half-life 71 seconds for olaparib-BODIPY FL) and wash-out of the small molecule companion imaging drug. After the drug accumulated within cells, they were washed twice and incubated with fresh media (without drug). PARP1 is located in the nucleus with a higher concentration in the nucleolus, enabling rapid identification of target bound drug by drawing subcellular regions of interest. Figure 1C shows the cells immediately after the drug was removed (no wash). Both drugs accumulated significantly in the perinuclear region, most likely the endoplasmic reticulum (ER). Within five minutes of washing, the non-specific staining of the ER with the BODIPY FL drug disappeared, while the target specific nuclear staining remained. However, the larger BODIPY 650 version required over 2 hours of washing to reduce the ER staining, although the target specific labeling remained. The uptake and wash-out kinetics for the cells were measured and fit to uptake and clearance curves (Supplementary Materials and Methods).
Based on the target affinity and assuming passive transport across the plasma membrane, a simple model of nuclear versus ER uptake was developed to capture the relative cellular uptake. Specific nuclear uptake was saturable based on the affinity and estimated nuclear concentration of PARP of 3 μM (Supplementary Table S1). Using the relative concentration of the nucleus and free drug concentration, a linear (non-saturable) uptake in the ER was assumed, which is consistent with equilibrium models of small molecules partitioning into lipid membranes(45, 46). From these estimates, low concentrations of the BODIPY FL drug would specifically stain the nucleus, while the BODIPY 650 version would not obtain a higher concentration in the nucleus at equilibrium based on the higher uptake in the ER relative to the lower affinity and specific binding. Because the BODIPY FL version starts to wash out in seconds, cells were imaged in the presence of drug in the media (no washing). Since the ER concentration is higher than free drug in solution, it is still possible to obtain a cellular image using widefield microscopy under these conditions (Figure 2). As predicted by the cell kinetic study, the 10 and 100 nM concentrations gave a higher nuclear signal for the BODIPY FL drug, but the 1 μM concentration resulted in a higher ER signal. The higher lipophilicity and ER uptake and the lower affinity of the BODIPY 650 labeled drug resulted in higher ER signal at all concentrations, even though nuclear specific staining could be obtained by washing the cells. Note that the overall intensity of the images varied dramatically, so the exposure times are different, but the relative intensity behaved as expected.
Figure 2. Specific Cell Uptake.
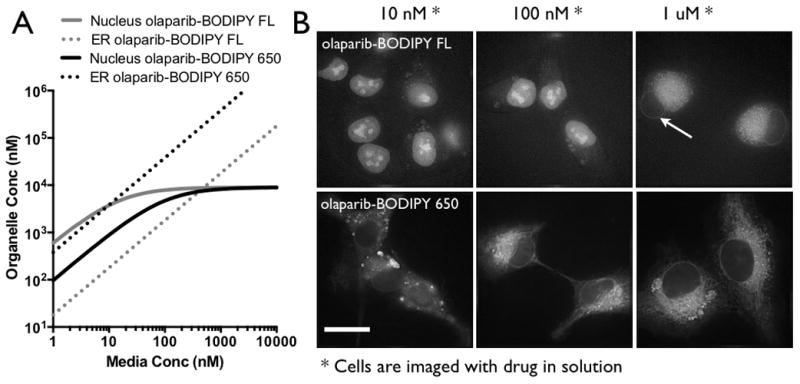
(A) A simple equilibrium model derived from the kinetic data predicts that the olaparib-BODIPY FL yields a higher nuclear signal at low concentrations and higher ER signal at high concentrations. The greater uptake in the ER for olaparib-BODIPY 650 indicates that at equilibrium, the ER signal will always be higher than the nucleus. (B) Widefield fluorescence images of the cells in equilibrium with probe in the media are consistent with the equilibrium model. Note that the image contrast was changed between the different concentrations due to the order of magnitude difference in signal intensity. Arrow points to lower uptake in nucleus. Scale bar = 20 μm
Recent advances in intravital microscopy techniques allow us to measure subcellular drug concentrations at many frames per minute. This improvement in time resolution prompted us to measure the distribution of the BODIPY FL and BODIPY 650 conjugated olaparib after intravenous injection. Vascular permeability is one of the most important parameters determining the relationship between tumor physiology and drug uptake, and this value can vary by over 5 orders of magnitude between large nanoparticles and small molecules such as oxygen (3). Using intravital microscopy in dorsal window chambers of HT-1080 xenografts, the permeability was measured simultaneously for both drugs in tumor blood vessels. In all cases, the permeability of the BODIPY FL drug was higher than the BODIPY 650 drug despite similar plasma protein binding for both (Figure 3A). Plotting this data alongside a compilation of permeability for proteins and nanoparticles (32), the olaparib-BODIPY 650 permeability (0.46 +/− 0.27 μm/s) follows the same trend as these hydrophilic molecules that primarily extravasate along a paracellular path (Figure 3B). However, the BODIPY FL version has a much higher permeability (4.7 +/− 2.5 μm/s) than would be expected for this trend. The molecular weight of these drugs only differs by 255 Da and plasma protein binding is not statistically significantly different, but the permeability difference is significant (p = 0.001). Given the rapid cellular uptake and wash-out kinetics of the BODIPY FL drug, we hypothesize that the transcellular route for extravasation is significant for a molecule of this size. For reference, oxygen is not shown on the graph but has been estimated at 5×10−2 cm/s with a radius of ~0.3 nm. Plasma clearance was qualitatively similar between the two agents, although the peak concentration was higher for the larger drug. The dose for both drugs was 75 nmol, so it is likely that the rapid cellular uptake of BODIPY FL drug (1.2 min half-life in vitro) lowered the concentration prior to the initial 1 min time point (Table 1).
Figure 3. Tumor Vessel Permeability.
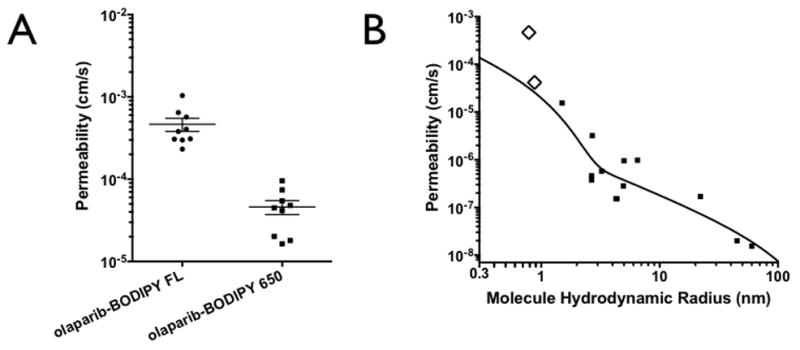
(A) Permeability of olaparib-BODIPY FL and olaparib-BODIPY 650 in HT-1080 xenografts. The olaparib-BODIPY FL consistently resulted in an approximately 10-fold higher permeability than olaparib-BODIPY 650. (B) The two olaparib derivatives, shown with open diamonds, compared to a variety of proteins and polymers with varying size (hydrodynamic radius). The black line is a two-pore model fit for paracellular transport(32).
Given the challenges of measuring drug distribution within tumors, mathematical models play an important role in predicting the impact of variability in drug properties and the tumor microenvironment (32, 47, 48). To take advantage of the insight provided by this modeling, we used a finite element model (FEM) of drug distribution to compare uptake of both drugs. For comparison with experimental results, a vascular probe was injected to define the vessels within the microscopic field of view (Figure S1), and a Delaunay triangulation algorithm was used to create a mesh in the extravascular space. Due to the high plasma protein binding of these drugs, the blood flow rate through each vessel has a negligible impact (3), so the plasma concentration was set equal to the systemic concentration. The experimentally measured permeability, non-specific uptake ratio, and target binding were used in the simulations to predict the distribution at various times after injection. The lower non-specific uptake and faster washout of the olaparib-BODIPY FL is predicted to give nuclear specific labeling as seen in the in vivo microscope image after intravenous injection of the companion imaging drug (Figure S1). The higher non-specific uptake and slower cellular wash-out of the olaparib-BODIPY 650 is predicted to result in higher non-specific uptake at 218 min than nuclear bound drug (Figure S1), which was validated with the intravital image (Figure 4). The experimental results validate the model predictions, where the nuclear target bound drug is less than the non-specific cellular uptake, even though wash-out in vitro was shown to result in nuclear specific labeling (Figure 1).
Figure 4. In Vivo Subcellular Distribution.
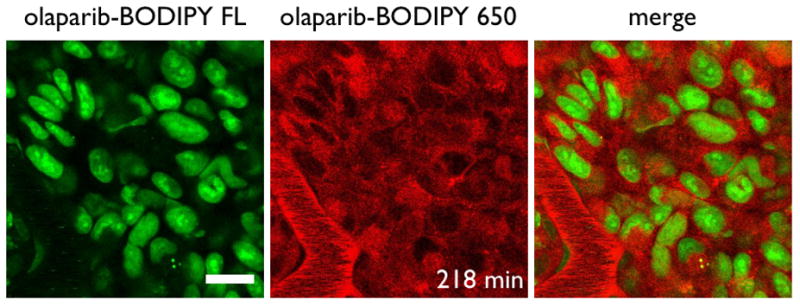
Olaparib-BODIPY FL shows predominantly nuclear and nucleolus uptake after 218 min while olaparib-BODIPY 650 is located primarily in the perinuclear region. Scale bar = 20 μm
The finite element simulation was able to capture the differences in subcellular uptake seen with the in vivo experiments at long time points after the diffusion gradients had dissipated. To further validate the model, the simulation results were compared at earlier times after injection (Figure 5). Minutes after injection, the drug diffuses away from the vessels, and the olaparib-BODIPY FL shows approximately equal non-specific and nuclear targeting at 8 min in both the simulation (top) and in vivo microscope image (bottom). By 25 min, the non-specific uptake has cleared enough to identify individual nuclei in the image. In contrast, the higher non-specific uptake of the olaparaib-BODIPY 650 results in poorer distribution and much higher non-specific uptake early after injection. Due to the higher plasma signal (Table 1), the confocal microscope settings must be reduced to not saturate the blood vessel signal, resulting in less apparent signal in the tissue. However, the drug does not penetrate as far as the olaparib-BODIPY FL in the experimental image (bottom). By 25 min, the olaparib-BODIPY 650 simulation shows increased penetration but still a higher non-specific signal than target signal (top). By this time, the blood signal has diminished enough to distinguish the high perinuclear signal for olaparib-BODIPY 650.
Figure 5. Tumoral heterogeneity.
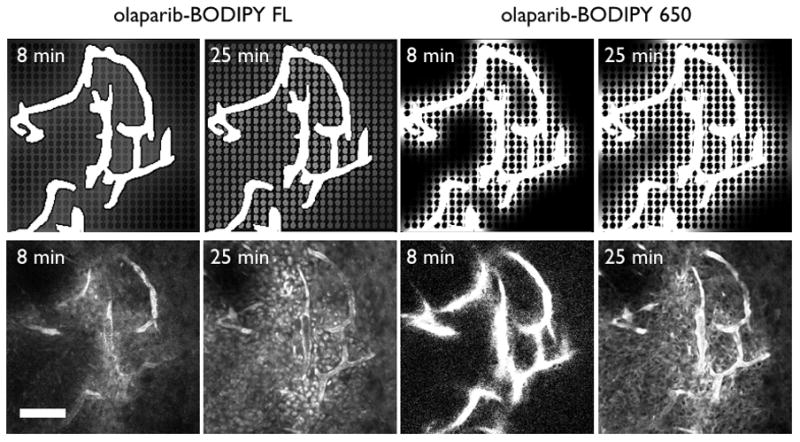
Finite element models of the probe distribution (top) during the first half hour show faster distribution for the olaparib-BODIPY FL with nuclear specific staining apparent after only 25 min. The higher non-specific immobilization of olaparib-BODIPY 650 results in more early time heterogeneity. In vivo images for the olaparib-BODIPY FL confirm the lack of nuclear signal at 8 min but presence of the signal by 25 min (bottom). Olaparib-BODIPY 650 has low penetration into the tissue at 8 min, and perinuclear staining readily apparent by 25 min. Scale bar = 100 μm
Discussion
Heterogeneous delivery of imaging probes and therapeutics to tumors is a complex problem that limits the design, development, and efficacy of these agents. While significant progress has been made in understanding the distribution of macromolecules and nanoparticles (4, 32, 49, 50), the techniques for studying small molecule distribution lack the subcellular spatial resolution and high sampling rate for directly observing the pertinent rates in vivo. In this work, we take advantage of the development of fluorescent drug conjugates that retain their cellular specificity and enzyme inhibition (Figure 1) while allowing the simultaneous imaging of multiple drugs with different physiochemical properties in vivo. The different fluorophores enable the imaging of multiple probes in the same tumor microenvironment, isolating the effect of drug properties from highly variable tumor physiology. This technique has allowed us to measure the kinetics of two small molecule drugs that bind their target with unprecedented temporal and spatial resolution.
As one specific example, this methodology allows the direct imaging of small molecule drug permeability across the tumor vasculature. Historically, this property was measured in isolated perfused tissues to determine the physiological permeability surface area product. However, without a simultaneous estimate of surface area, which can vary over an order of magnitude in tumors, the results cannot be used for tumor predictions where the permeability of tumor blood vessels is often higher than in healthy tissue (43). Fluorescence imaging can overcome this limitation by direct visualization of extravasation in the relevant tumor microenvironment, and previous results shed light on important trends, such as the increase in permeability with decreasing size (51, 52). Taking advantage of the fast sampling rate afforded by intravital fluorescence microscopy, we were able to measure significant differences in tumor vessel permeability given small changes in the structure of companion imaging drugs. These values are critical for predictive modeling of therapeutics and imaging agent design, but there is a paucity of data in the literature for in vivo small molecule drug permeability. While it is generally accepted that macromolecules have limited uptake due to low permeability and oxygen and some small molecule drugs readily cross the endothelium, the transition between these two regimes (permeability limited versus diffusion or blood flow limited uptake) is not well defined. Our data here indicate that the transition between the two regimes occurs for lipophilic small molecule drugs between 500 and 1000 Da, which has important implications for combination therapies with angiogenesis inhibitors that normalize the tumor vasculature (7). Our results here are consistent with the hypothesis that drugs with slow cellular uptake and washout have lower vascular permeability. The permeability of olaparib BODIPY650 matches the predicted value based on a two-pore model that only accounts for paracellular extravasation of macromolecules, while olaparib BODIPY FL has a higher permeability and much faster cellular uptake and washout. Additionally, previous observations for olaparib BODIPY FL(40) show that the vascular permeability of this molecule is the same in tumor vessels and mouse ear vessels. The paracellular extravasation of macromolecules is increased in tumors due to the action of VEGF and inflammatory molecules that increase permeability, and the lack of difference between these locations indicates that paracellular transport is not significant for olaparib BODIPY FL.
The olaparib scaffold is an ideal inhibitor for these studies given the high target expression of PARP (typically 106 proteins per cell (53)) and distinct subcellular distribution pattern. Target expression is localized to the nucleus while non-specific uptake occurred in the perinuclear region including the endoplasmic reticulum (Figure 2). It is therefore easy to identify non-specific versus target specific uptake. This pattern of non-specific uptake is not surprising for a lipophilic neutral molecule, which accumulates in the membrane, but the method allowed us to measure the in vivo kinetics of uptake into the different subcellular compartments. The ER distribution is in contrast to weak bases that are trapped in lysosomes upon protonation and positively charged small molecules that accumulate in the mitochondria (45). Given the lipophilic nature of these drugs, it was assumed that the non-specific uptake was non-saturable and linear with concentration (Figure 2), and the resulting prediction of uptake agreed with the cellular results. The observed subcellular distribution is important for cellular transport and metabolism, with p450 enzymes and transporters, such as p-glycoprotein, located within certain cellular compartments. The complex interplay with charge, pKa, molecular weight, and tissue physiology (e.g. anti-VEGF therapy) will be explored in future work.
Understanding the impact of physiochemical properties on tumoral distribution is important for both drug and imaging agent development. Empirical methods, such as Lipinski’s rule of 5 (54) or limits on physiochemical properties (e.g. using neural networks(55)) can provide guidance, but outliers and counterexamples exist(56). For example, canertinib, an irreversible EGFR tyrosine kinase inhibitor (TKI), obeys all the ‘rule of 5′ criteria but failed in clinical trials (57). The next generation variant, dacomitinib, violates one of the rules but has shown better phase II results (58, 59). A more precise method for predicting pharmacokinetic and pharmacodynamic requirements is needed.
An understanding of the transient changes in concentration after dosing is even more important for the development of imaging agents. While many therapeutics benefit from continuous dosing, allowing a steady-state or pseudo-steady state to develop, imaging agents are typically delivered as a single bolus dose. In several studies of radiolabelled EGFR tyrosine kinase inhibitors, 18F-gefitinib provided insight into the distribution of this clinically approved drug(9). However, it failed to localize based on EGFR target expression. A more hydrophilic radiolabelled EGFR TKI (60) did distribute differentially to higher expressing tumors. Interestingly, the predicted logP of these compounds (using the same software in all cases) is 5.6 for gefitinib and 4.4 for the radiolabelled PEGylated EGFR tyrosine kinase inhibitor. These are similar to the 6.0 value for olaparib-BODIPY 650 and 4.0 for olaparib-BODIPY FL reported here, which are also both able to hit their target but distribute differently. Although these results cannot be extrapolated to all agents, it may provide an ‘upper limit’ around a logP of ~5 for target specific retention. The lower limit is not explored in this work, although successful hypoxia imaging agents have logP values around 0 to 1 (61) while negative logP values, such as Oregon Green tetrazine (39) cannot penetrate cells to reach their target. Clearly, the molecular weight and hydrophilicity must be low enough to allow access to intracellular targets. It is also important to note in these examples that while some goals of therapeutic and imaging agent development are aligned (e.g. high affinity and specificity), others are at odds (rapid systemic clearance of imaging agents to reduce background versus sustained systemic levels of therapeutics to reduce dosing).
A deeper understanding of drug distribution for both small molecule agents and biologics is required as the lines between these two discrete fields become blurred. For example, Navitoclax, a ‘small molecule’ inhibitor of Bcl-2 family proteins is almost 975 Da in size, a molecular weight likely required for its disruption of protein-protein interactions. Smaller biologics include imetelstat (MW = 4610), a telomerase inhibitor, and stapled peptides (~2 kDa) such as BH3 domains (62), among many others. These new agents may present complex pharmacokinetic behavior that do not follow small molecule or biologic drug development patterns. A better mechanistic understanding of distribution, including the influence of blood flow, permeability, diffusion, cellular uptake, and target binding (3) will enable more rapid development of these types of drugs. Unfortunately, simple extrapolation of some experiments, such as the permeability of molecules less than 1 nm in Figure 3B, can provide misleading results since charge and protein binding can have a large effect on the paracellular versus transcellular routes of extravasation. Future work is required to make better predictions of drug uptake based on molecular properties.
Mathematical modeling will continue to play an increasing role in predicting the distribution of novel drug and imaging agents. With multiple simultaneous rates occurring during experiments, including plasma clearance, extravasation, diffusion, target binding, and metabolism, it becomes difficult to experimentally isolate the effect of any one parameter; however, this can easily be done with a validated model. This joint theoretical and experimental approach helped elucidate the effect of dose on antibody uptake (8), and the required timing after injection of hypoxic imaging agents (63). Even the clinically widespread FDG agent shows blood-flow dependent uptake at early times followed by glucose metabolism dependent localization at later times (64), an effect captured by mathematical models (3). The development of agents based on iterative synthesis and animal experiments is labor and cost intensive. Mathematical models provide a rapid and cost-effective way of focusing synthesis on desirable molecular properties to obtain sufficient uptake and distribution for therapeutics or efficient target binding and clearance for imaging agents. They can also be combined with pharmacodynamic models for prediction of therapeutic outcome. The mechanistic framework can be scaled to humans by changing the plasma clearance rates and concentration, thus providing insight into the distribution in the clinic, which is currently unobtainable by other methods. Finally, mathematical modeling and simulations also help in interpretation of experimental and clinical results.
In conclusion, a combination of novel fluorescent drug conjugates and in vivo imaging experiments have provided insight into the cellular and tumoral heterogeneity of small molecule drugs. The lipophilic PARP inhibitors studied here show a sharp transition in both cellular uptake and permeability between a 640 Da probe with a clogP of 4 and a 895 Da probe with clogP of 6. These measurements can be used to develop predictive mathematical simulations that help in understanding drug distribution in vivo at the preclinical and clinical stage, in designing better imaging agents and therapeutics, and in interpreting experimental results.
Supplementary Material
Acknowledgments
Financial Support: NIH Grants P50 CA086355 (R. Weissleder), R01 EB010011 (R. Weissleder) and K01 DK093766 (G.M. Thurber).
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nature Reviews Cancer. 2006;6:583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 2.Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev. 2008;60:1421–34. doi: 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thurber GM, Weissleder R. A Systems Approach for Tumor Pharmacokinetics. PLoS ONE. 2011;6:e24696. doi: 10.1371/journal.pone.0024696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jain RK. Transport of Molecules, Particles, and Cells in Solid Tumors. Annual Reviews in Biomedical Engineering. 1999;01:241–63. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 5.Patel KJ, Tannock IF. The influence of P-glycoprotein expression and its inhibitors on the distribution of doxorubicin in breast tumors. Bmc Cancer. 2009;9 doi: 10.1186/1471-2407-9-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyle AH, Huxham LA, Yeoman DM, Minchinton AI. Limited tissue penetration of taxanes: A mechanism for resistance in solid tumors. Clinical Cancer Research. 2007;13:2804–10. doi: 10.1158/1078-0432.CCR-06-1941. [DOI] [PubMed] [Google Scholar]
- 7.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiological Reviews. 2011;91:1071–121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thurber GM, Weissleder R. Quantitating antibody uptake in vivo: conditional dependence on antigen expression levels. Molecular imaging and biology. 2011;13:623–32. doi: 10.1007/s11307-010-0397-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su H, Seimbille Y, Ferl GZ, Bodenstein C, Fueger B, Kim KJ, et al. Evaluation of [F-18]gefitinib as a molecular imaging probe for the assessment of the epidermal growth factor receptor status in malignant tumors. Eur J Nucl Med Mol Imaging. 2008;35:1089–99. doi: 10.1007/s00259-007-0636-6. [DOI] [PubMed] [Google Scholar]
- 10.Tolmachev V, Stone-Elander S, Orlova A. Radiolabelled receptor-tyrosine-kinase targeting drugs for patient stratification and monitoring of therapy response: prospects and pitfalls. Lancet Oncology. 2010;11:992–1000. doi: 10.1016/S1470-2045(10)70088-7. [DOI] [PubMed] [Google Scholar]
- 11.Doubrovin M, Kochetkova T, Santos E, Veach DR, Smith-Jones P, Pillarsetty N, et al. I-124-Iodopyridopyrimidinone for PET of Abl Kinase-Expressing Tumors In Vivo. Journal of Nuclear Medicine. 2010;51:121–9. doi: 10.2967/jnumed.109.066126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pries AR, Cornelissen AJM, Sloot AA, Hinkeldey M, Dreher MR, Hopfner M, et al. Structural Adaptation and Heterogeneity of Normal and Tumor Microvascular Networks. PLoS Comput Biol. 2009;5:11. doi: 10.1371/journal.pcbi.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dewhirst MW, Klitzman B, Braun RD, Brizel DM, Haroon ZA, Secomb TW. Review of methods used to study oxygen transport at the microcirculatory level. Int J Cancer. 2000;90:237–55. [PubMed] [Google Scholar]
- 14.Schroeder T, Yuan H, Viglianti BL, Peltz C, Asopa S, Vujaskovic Z, et al. Spatial heterogeneity and oxygen dependence of glucose consumption in R3230Ac and fibrosarcomas of the Fischer 344 rat. Cancer Research. 2005;65:5163–71. doi: 10.1158/0008-5472.CAN-04-3900. [DOI] [PubMed] [Google Scholar]
- 15.Helmlinger G, Sckell A, Dellian M, Forbes N, Jain RK. Acid Production in Glycolysis-impaired Tumors Provides New Insights into Tumor Metabolism. Clinical Cancer Research. 2002;8:1284–91. [PubMed] [Google Scholar]
- 16.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–U4. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 17.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. New England Journal of Medicine. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–U144. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Souza R, Zahedi P, Badame RM, Allen C, Piquette-Miller M. Chemotherapy Dosing Schedule Influences Drug Resistance Development in Ovarian Cancer. Molecular Cancer Therapeutics. 2011;10:1289–99. doi: 10.1158/1535-7163.MCT-11-0058. [DOI] [PubMed] [Google Scholar]
- 20.Ramachandran C, Sauerteig A, Sridhar KS, Thurer RJ, Krishan A. MDR-1 Gene-Expression, Anthracycline Retention and Cytotoxicity in Human Lung-tumor Cells from Refractory Patients. Cancer Chemotherapy and Pharmacology. 1993;31:431–41. doi: 10.1007/BF00685031. [DOI] [PubMed] [Google Scholar]
- 21.Kim BJ, Forbes NS. Single-Cell Analysis Demonstrates How Nutrient Deprivation Creates Apoptotic and Quiescent Cell Populations in Tumor Cylindroids. Biotechnology and Bioengineering. 2008;101:797–810. doi: 10.1002/bit.21985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKillop D, Partridge EA, Kemp JV, Spence MP, Kendrew J, Barnett S, et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Molecular Cancer Therapeutics. 2005;4:641–9. doi: 10.1158/1535-7163.MCT-04-0329. [DOI] [PubMed] [Google Scholar]
- 23.Kuh HJ, Jang SH, Wientjes MG, Weaver JR, Au JLS. Determinants of paclitaxel penetration and accumulation in human solid tumor. Journal of Pharmacology and Experimental Therapeutics. 1999;290:871–80. [PubMed] [Google Scholar]
- 24.Lesser GJ, Grossman SA, Eller S, Rowinsky EK. The Distribution of Systemically Administered [H-3] Paclitaxel in Rats - A Quantitative Autoradiographic Study. Cancer Chemotherapy and Pharmacology. 1995;37:173–8. doi: 10.1007/BF00685646. [DOI] [PubMed] [Google Scholar]
- 25.Gangloff A, Hsueh WA, Kesner AL, Kiesewetter DO, Pio BS, Pegram MD, et al. Estimation of paclitaxel biodistribution and uptake in human-derived xenografts in vivo with F-18-fluoropaclitaxel. Journal of Nuclear Medicine. 2005;46:1866–71. [PubMed] [Google Scholar]
- 26.Tannock I, Lee C, Tunggal J, Cowan D, Egorin M. Limited Penetration of Anticancer Drugs through Tumor Tissue: A Potential Cause of Resistance of Solid Tumors to Chemotherapy. Clinical Cancer Research. 2002;8:878–84. [PubMed] [Google Scholar]
- 27.Minchinton AI, Wendt KR, Clow KA, Fryer KH. Multilayers of cells growing on a permeable support. An in vitro tumour model. Acta Oncol. 1997;36:13–6. doi: 10.3109/02841869709100724. [DOI] [PubMed] [Google Scholar]
- 28.Pruijn FB, Sturman JR, Liyanage HDS, Hicks KO, Hay MP, Wilson WR. Extravascular transport of drugs in tumor tissue: Effect of lipophilicity on diffusion of tirapazamine analogues in multicellular layer cultures. Journal of Medicinal Chemistry. 2005;48:1079–87. doi: 10.1021/jm049549p. [DOI] [PubMed] [Google Scholar]
- 29.Sutherland R. Cell and Environment Interactions in Tumor Microregions: The Multicell Spheroid Model. Science. 1988;240:177–84. doi: 10.1126/science.2451290. [DOI] [PubMed] [Google Scholar]
- 30.Toley BJ, Tropeano Lovatt ZG, Harrington JL, Forbes NS. Microfluidic technique to measure intratumoral transport and calculate drug efficacy shows that binding is essential for doxorubicin and release hampers Doxil. Integrative biology : quantitative biosciences from nano to macro. 2013;5 doi: 10.1039/c3ib40021b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sung C, Youle RJ, Dedrick RL. Pharmacokinetic Analysis of Immunotoxin Uptake in Solid Tumors - Role of Plasma Kinetics, Capillary-Permeability, and Binding. Cancer Research. 1990;50:7382–92. [PubMed] [Google Scholar]
- 32.Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Molecular Cancer Therapeutics. 2009;8:2861. doi: 10.1158/1535-7163.MCT-09-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El-Kareh AW, Secomb TW. Theoretical models for drug delivery to solid tumors. Critical Reviews in Biomedical Engineering. 1997;25:503–71. doi: 10.1615/critrevbiomedeng.v25.i6.20. [DOI] [PubMed] [Google Scholar]
- 34.Baxter L, Zhu H, Mackensen D, Jain RK. Physiologically Based Pharmacokinetic Model for Specific and Nonspecific Monoclonal Antibodies and Fragments in Normal Tissues and Human Tumor Xenografts in Nude Mice. Cancer Research. 1994;54:1517–28. [PubMed] [Google Scholar]
- 35.Venkatasubramanian R, Arenas RB, Henson MA, Forbes NS. Mechanistic modelling of dynamic MRI data predicts that tumour heterogeneity decreases therapeutic response. British Journal of Cancer. 2010;103:486–97. doi: 10.1038/sj.bjc.6605773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beerling E, Ritsma L, Vrisekoop N, Derksen PWB, van Rheenen J. Intravital microscopy: new insights into metastasis of tumors. Journal of Cell Science. 2011;124:299–310. doi: 10.1242/jcs.072728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bravo-Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Current Opinion in Cell Biology. 2012;24:277–83. doi: 10.1016/j.ceb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wyckoff J, Gligorijevic B, Entenberg D, Segall J, Condeelis J. High-resolution multiphoton imaging of tumors in vivo. Cold Spring Harbor protocols. 2011;2011:1167–84. doi: 10.1101/pdb.top065904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang KS, Budin G, Reiner T, Vinegoni C, Weissleder R. Bioorthogonal Imaging of Aurora Kinase A in Live Cells. Angewandte Chemie-International Edition. 2012;51:6598–603. doi: 10.1002/anie.201200994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T, Weissleder R. Single-cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo. Nature Communications. 2013;4:1504. doi: 10.1038/ncomms2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reiner T, Lacy J, Keliher EJ, Yang KS, Ullal A, Kohler RH, et al. Imaging Therapeutic PARP Inhibition In Vivo through Bioorthogonally Developed Companion Imaging Agents. Neoplasia. 2012;14:169. doi: 10.1593/neo.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, et al. 4- 3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl -2H-ph thalazin-1-one: A Novel Bioavailable Inhibitor of Poly(ADP-ribose) Polymerase-1. Journal of Medicinal Chemistry. 2008;51:6581–91. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 43.Gerlowski L, Jain RK. Microvascular Permeability of Normal and Neoplastic Tissues. Microvasc Res. 1986;31:288–305. doi: 10.1016/0026-2862(86)90018-x. [DOI] [PubMed] [Google Scholar]
- 44.Crank J. The Mathematics of Diffusion. 2. Oxford: Clarendon Press; 1975. [Google Scholar]
- 45.Rosania GR, Lee JW, Ding L, Yoon HS, Chang YT. Combinatorial approach to organelle-targeted fluorescent library based on the styryl scaffold. Journal of the American Chemical Society. 2003;125:1130–1. doi: 10.1021/ja027587x. [DOI] [PubMed] [Google Scholar]
- 46.Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. Journal of Pharmaceutical Sciences. 2002;91:129–56. doi: 10.1002/jps.10005. [DOI] [PubMed] [Google Scholar]
- 47.Rowland M, Peck C, Tucker G. Physiologically-Based Pharmacokinetics in Drug Development and Regulatory Science. In: Cho AK, editor. Annual Review of Pharmacology and Toxicology. Vol. 51. Palo Alto: Annual Reviews; 2011. pp. 45–73. [DOI] [PubMed] [Google Scholar]
- 48.Praxmarer M, Sung C, Bungay PM, van Osdol WW. Computational models of antibody-based tumor imaging and treatment protocols. Annals of Biomedical Engineering. 2001;29:340–58. doi: 10.1114/1.1359453. [DOI] [PubMed] [Google Scholar]
- 49.Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, et al. Design considerations for tumour-targeted nanoparticles. Nat Nanotechnol. 2010;5:42–7. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee H, Hoang B, Fonge H, Reilly RM, Allen C. In Vivo Distribution of Polymeric Nanoparticles at the Whole-Body, Tumor, and Cellular Levels. Pharmaceutical Research. 2010;27:2343–55. doi: 10.1007/s11095-010-0068-z. [DOI] [PubMed] [Google Scholar]
- 51.Dreher MR, Liu WG, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. Journal of the National Cancer Institute. 2006;98:335–44. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 52.Wu NZ, Klitzman B, Rosner G, Needham D, Dewhirst MW. Measurement of Material Extravasation in Microvascular Networks Using Fluorescence Video-Microscopy. Microvasc Res. 1993;46:231–53. doi: 10.1006/mvre.1993.1049. [DOI] [PubMed] [Google Scholar]
- 53.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochemical Journal. 1999;342:249–68. [PMC free article] [PubMed] [Google Scholar]
- 54.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 55.Taskinen J, Yliruusi J. Prediction of physicochemical properties based on neural network modelling. Adv Drug Deliv Rev. 2003;55:1163–83. doi: 10.1016/s0169-409x(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 56.Mager DE. Quantitative structure-pharmacokinetic/pharmacodynamic relationships. Adv Drug Deliv Rev. 2006;58:1326–56. doi: 10.1016/j.addr.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Rixe O, Franco SX, Yardley DA, Johnston SR, Martin M, Arun BK, et al. A randomized, phase II, dose-finding study of the pan-ErbB receptor tyrosine-kinase inhibitor CI-1033 in patients with pretreated metastatic breast cancer. Cancer Chemotherapy and Pharmacology. 2009;64:1139–48. doi: 10.1007/s00280-009-0975-z. [DOI] [PubMed] [Google Scholar]
- 58.Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, et al. Randomized Phase II Study of Dacomitinib (PF-00299804), an Irreversible Pan-Human Epidermal Growth Factor Receptor Inhibitor, Versus Erlotinib in Patients With Advanced Non-Small-Cell Lung Cancer. Journal of Clinical Oncology. 2012;30:3337–44. doi: 10.1200/JCO.2011.40.9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janne PA, Boss DS, Camidge DR, Britten CD, Engelman JA, Garon EB, et al. Phase I Dose-Escalation Study of the Pan-HER Inhibitor, PF299804, in Patients with Advanced Malignant Solid Tumors. Clinical Cancer Research. 2011;17:1131–9. doi: 10.1158/1078-0432.CCR-10-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeh HH, Ogawa K, Balatoni J, Mukhapadhyay U, Pal A, Gonzalez-Lepera C, et al. Molecular imaging of active mutant L858R EGF receptor (EGFR) kinase-expressing nonsmall cell lung carcinomas using PET/CT. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1603–8. doi: 10.1073/pnas.1010744108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krohn KA, Link JM, Mason RP. Molecular imaging of hypoxia. Journal of Nuclear Medicine. 2008;49:129S–48S. doi: 10.2967/jnumed.107.045914. [DOI] [PubMed] [Google Scholar]
- 62.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–70. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monnich D, Troost EGC, Kaanders J, Oyen WJG, Alber M, Thorwarth D. Modelling and simulation of (18)F fluoromisonidazole dynamics based on histology-derived microvessel maps. Physics in medicine and biology. 2011;56:2045–57. doi: 10.1088/0031-9155/56/7/009. [DOI] [PubMed] [Google Scholar]
- 64.Mullani NA, Herbst RS, O’Neil RG, Gould KL, Barron BJ, Abbruzzese JL. Tumor blood flow measured by PET dynamic imaging of first-pass F-18-FDG uptake: A comparison with O-15-Labeled water-measured blood flow. Journal of Nuclear Medicine. 2008;49:517–23. doi: 10.2967/jnumed.107.048504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.