

Abstract
In search for a novel chemotype to develop Topoisomerase I (Top1) inhibitors, the pyrazolo[1,5–a]quinazoline nucleus, structurally related to the indenoisoquinoline system precursor of well-known Top1 poisons, was variously decorated (i.e. a substituted phenyl ring at 2– or 3–position, a protonable side chain at 4– or 5–position) affording a number of Top1 inhibitors with cleavage patterns common to CPT and MJ–III–65. SARs data were rationalized by means of an advanced docking protocol.
Keywords: Topoisomerase I, pyrazoloquinazoline derivatives, structure–activity relationships, medicinal chemistry
Introduction
DNA topoisomerases (Top) are essential enzymes inducing DNA modification required during cellular processes such as replication, transcription, repair, etc.1 There are two major families of Top: Type I (Top1) and Type II (Top2) depending on whether they cleave only one or two DNA strands.2 Top1 relaxes supercoiled DNA by forming DNA single-strand breaks, and religates the broken strand, to rapidly restore intact duplex DNA.2 At this stage, the enzyme is particularly vulnerable to a group of anticancer agents, the Top1 poisons, that reversibly trap the Top1-mediated cleavage complex, leading to irreversible DNA strand breaks, activation of apoptosis and cell cycle arrest.2
Top1 inhibitors are a relatively new group of anticancer agents with a wide range of activity in hematological and solid tumors. Camptothecin (CPT I, Chart I),3 was the first small molecule identified as a Top1 inhibitor. Efforts to improve its toxicity profile and pharmacokinetics led to the development of two clinical water-soluble CPT derivatives, topotecan II (Chart I) and irinotecan,4 as well as novel compounds currently under clinical evaluation.5 However, CPTs are not ideal drugs as they display a number of limitations, including chemical instability,6 and potential induction of cellular resistance.7 To overcome the main drawbacks of CPTs, several chemical classes of non–CPT Top1 poisons were developed as promising antitumor drugs, including the phenanthridines III, and the indenoisoquinolines IV (Chart I).8,9
Chart I.

Structures of known Top1 inhibitors; design of pyrazoloquinazolines V.
As part of our program in search for new antiproliferative agents, in the last decades we extensively studied several polyheterocyclic systems.10–12 In the present study, we have directed our attention to the pyrazolo[1,5-a]quinazoline system V (Chart I),13,14 as a novel scaffold to develop non-CPT agents acting against Top1. Actually, the core of V would mimic the A, B and C rings of IV, while the phenyl hanging from the pyrazole portion of V could mimic the substituted D ring of IV (Chart I). Compounds bearing a phenyl alternatively at 2– or 3–position of pyrazolo[1,5–a]quinazoline system were designed (V, Chart I). Further, as a protonable chain linked at the 5– or 6–position of the indenoisoquinoline ring or at the 5– position of the benzophenanthridine system is a common feature of the most active derivatives,9 we decorated the pyrazoloquinazoline ring at the 4– or 5–position with aminoalkyl chains. In particular, we studied the influence on the Top1 inhibitory activity of: (i) the length of the chain, (ii) the nature of the linker between the ring and the chain; (iii) the nature of the terminal basic site. Interestingly, these compounds featuring a basic nitrogen in the side chain could be converted in the corresponding salts, thus increasing aqueous solubility that might facilitate their formulation.
In this study, derivatives 1–34 were prepared and evaluated for their ability to inhibit Top1 (Table 1), and an advanced docking protocol was employed to rationalize the biological results.
Table 1.
Topoisomerase I Inhibitory Activities of 1–34.
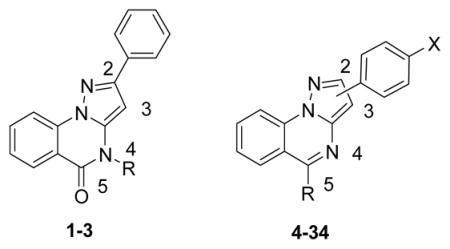
| ||||
|---|---|---|---|---|
| N | Phenyl position | Ra | X | Top1 Inhibition |
| 1 | - | A | - | 0/+ |
| 2 | - | B | - | 0 |
| 3 | - | C | - | 0 |
| 4 | 2 | D | H | 0/+ |
| 5 | 2 | E | H | 0/+ |
| 6 | 2 | F | H | 0 |
| 7 | 2 | H | H | 0 |
| 8 | 2 | J | H | 0 |
| 9 | 3 | D | OCH3 | + |
| 10 | 3 | E | OCH3 | 0/+ |
| 11 | 3 | F | OCH3 | 0/+ |
| 12 | 3 | G | OCH3 | + |
| 13 | 3 | H | OCH3 | 0/+ |
| 14 | 3 | I | OCH3 | 0/+ |
| 15 | 3 | J | OCH3 | 0 |
| 16 | 3 | D | CF3 | 0/+ |
| 17 | 3 | E | CF3 | 0 |
| 18 | 3 | F | CF3 | 0 |
| 19 | 3 | G | CF3 | + |
| 20 | 3 | H | CF3 | 0/+ |
| 21 | 3 | I | CF3 | 0/+ |
| 22 | 3 | J | CF3 | 0 |
| 23 | 3 | D | Cl | + |
| 24 | 3 | E | Cl | 0/+ |
| 25 | 3 | F | Cl | + |
| 26 | 3 | G | Cl | ++ |
| 27 | 3 | H | Cl | ++ |
| 28 | 3 | I | Cl | ++ |
| 29 | 3 | J | Cl | 0 |
| 30 | 3 | D | H | ++ |
| 31 | 3 | E | H | + |
| 32 | 3 | F | H | + |
| 33 | 3 | H | H | + |
| 34 | 3 | J | H | 0/+ |
A: (CH2)2N(CH3)2; B: (CH2)3N(CH3)2; C: (CH2)3–1–-imidazolyl; D: O(CH2)2N(CH3)2; E: O(CH2)2N(C2H5)2; F: O(CH2)3N(CH3)2; G: NH(CH2)2N(CH3)2; H: NH(CH2)2N(C2H5)2; I: NH(CH2)3N(CH3)2; J: NH(CH2)3–1–imidazolyl. The activity of the compounds to produce Top1-mediated DNA cleavage was expressed semi-quantitatively as follows: 0, not active; 0/+, trace of activity; +, weak activity; ++, moderate activity; +++, strong activity; ++++, activity equivalent to 1 μM I or 55.
Chemistry
The 2–phenyl–5H–pyrazolo[1,5-a][3,1]benzoxazin–5–one 36 was obtained by an improved microwaves (MW)- assisted reported procedure15,16 [experimental details, Scheme 1S, Supporting Information (SI)] and then reacted with the appropriate dialkylaminoalkylamine to obtain compounds 1–3 through the use of MW (Scheme 1).
Scheme 1.

Synthesis of pyrazoloquinazolines 1–3.
The phenylpyrazolo[1,5–a]quinazolin–5(4H)–ones 37–41 (Scheme 2) were obtained by improving known synthetic procedures (experimental details, Schemes 2S and 3S, SI).13,14 Treatment of 37, 41 with sodium hydride and addition of the appropriate dialkylaminoalkyl chloride gave derivatives 4–6, 30–32 (Scheme 2). The best yields in the preparation of 9–11, 16–18, 23–25, were obtained by a Mitsunobu reaction between 38–40 and the appropriate aminoalcohol (Scheme 2). The synthesis of compounds 7, 8, 12–15, 19–22, 26–29, 33, 34 includes the transformation of 37–41 in the corresponding 5–chloro derivatives 50–54 (Scheme 2, see SI for details), that were then reacted with the proper dialkylaminoalkylamines through the use of MW (Scheme 2). All the products (4–34) were purified by conversion into the corresponding hydrochlorides by treatment with ethanol hydrochloride in absolute ethanol.
Scheme 2.

Synthesis of pyrazoloquinazolines 4–34.
Biological Results and Discussion
Table 1 lists the relative potencies of the pyrazoloquinazolines 1–34 toward the production of Top1– mediated DNA cleavage, ranked by a systematic visual analysis of the number of cleavage sites and their respective intensity in each lane as compared to the positive control lanes containing I or 55 (MJ–III–65, Chart I)17,18 at 1 RM. A semi-quantitative ranking system is then used to rank the compounds from 0, not active; 0/+, trace of activity; +, weak activity; ++, moderate activity; +++, strong activity; to ++++, activity equivalent to 1 μM I or 55.
Compounds 1–3, that were synthesized due to their structural similarities with the indenoisoquinolines IV, showed null Top1 inhibitory activity (Table 1). So we turned out our attention to 2–phenylpyrazoloquinazolines bearing different protonable chains at the 5–position (4–8). Also these substitution patterns did not produce favorable effects on biological activity. Only derivatives 4 and 5 showed a scarce activity as Top1 inhibitors.
Compounds 9–34 were then designed by shifting the pendant phenyl from the 2– to the 3–position, and slightly expanding the variability at the 5–chain with respect to 1–8. Initially, an electron–donating OCH3 was inserted at p–position of the 3–phenyl (9–15), due to the well–known beneficial effect of this substituent on the potency of Top1 inhibitors.9 Then, variously (CF3, Cl, H) 4′– substituted compounds 16–34 were developed to expand the SAR. The presence of the 3–phenyl produced a general improvement in the biological activity (9–34, Table 1), suggesting a specific arrangement of the molecule that is favorable for Top1 inhibition. The presence of a 4′–OCH3 does not particularly favor the activity, yielding poorly active derivatives (9–15), whatever the nature of the chain at 5–position. An analogous effect is produced by a highly electron–withdrawing CF3 substituent (16–22). Conversely, the presence of the lipophilic and electron–withdrawing 4′–Cl (23–29) determined an enhancement in the biological activity. In this subclass, the nature of the 5–chain in terms of type (–O– or –NH–) and length of the linker, and terminal nitrogen containing group moderately influence Top1 inhibitory activity.
The imidazole–containing chain (29) scored the worst results, while the best activity was obtained with the dimethylaminoethylamino, dimethylaminopropylamino, and diethylaminoethylamino chains (26–28).
An analogous trend was observed when the 4′–position is unsubstituted (30–34), with the presence of a terminal imidazole nucleus that again results to be detrimental for the activity, whereas a 5–dimethylaminoethoxy moiety confers to 30 an increase in Top1 inhibition.
Figure 1 displays the Top1–mediated DNA cleavage patterns for compounds 2, 18, 24, 26–29, selected as representative of the whole series, along with those resulting from I and the indenoisoquinoline 55.17,18 It should be observed that compounds 26–28, differently from the poorly active 2, 18, 24, and 29 showed cleavage sites that are common to I and 55.
Figure 1.

Top1-mediated DNA cleavage induced by compounds 24, 26, 28, 27, 29, 2 and 18. Lane 1: DNA alone; lane 2: Top1 alone; lane 3: I, 1 RM:; lane 4: Indenoisoquinoline 55, 1 RM; lane 5–25: 24, 26, 28, 27, 29, 2 and 18 at 1, 10 and 100 RM respectively from left to right. Numbers and arrows on the left indicate arbitrary cleavage site positions.
Docking studies were performed to rationalize the Top1 inhibitory activity and the relative potencies of our pyrazoloquinazolines.
We selected the high resolution (2.10 Å) crystal structure of the human Top1 in complex with the poison Topotecan (II, Chart I) and covalent complex with a 22 bp DNA duplex (PDB code: 1K4T).19 In this structure II establishes direct H–bonds with E356, R364, K532 and D533, and additional water–mediated interactions with N722 and the phosphotyrosine 723 (P-Y723) (Figure S1a, SI). Thus, we decided to explicitly consider the waters molecules in docking calculations using the software Autodock4.2 (AD4).20 In this respect, Forli et al. have recently developed a new AD4 force field and hydration docking method that allows for the automated prediction of waters mediating ligand binding.21 To evaluate the predictive power of this approach in our system, we first ran self-docking calculations on the Top1–II–DNA complex. AD4 reproduced the experimental binding conformation as the lowest energy solution, with a ligand RMSD equal to 1.08 Å, also recapitulating the water–mediated interactions between II and the enzyme (Figure S1b, SI).
These results encouraged us to apply this protocol on our pyrazoloquinazolines. Among these, we selected compound 26 within the subclass displaying the most interesting pharmacological profile (26–28). According to docking results, 26 intercalates at the DNA cleavage site (Figure 2) stacking with its polyaromatic system between the downstream (−1) T–A and upstream (+1) G–C base pairs like other CPT–Top1 inhibitors, including II (Figure S2, SI).19
Figure 2.
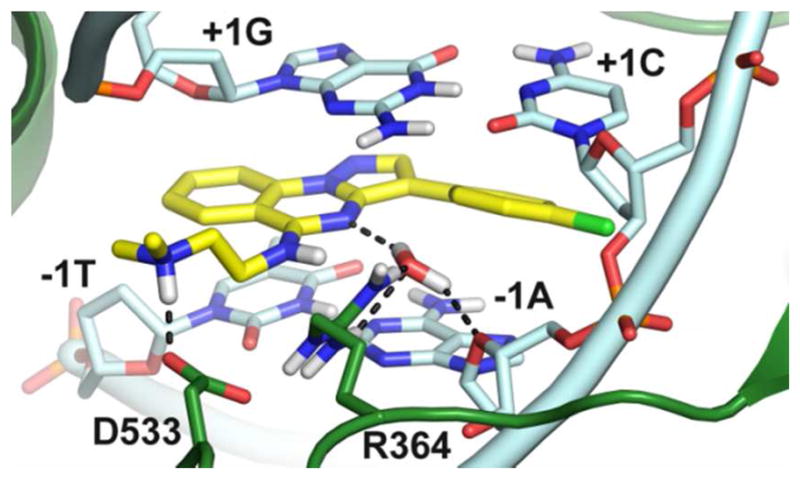
Binding mode of 26 (yellow sticks) at the Top1 (green cartoons) DNA (cyan cartoons) cleavage site. Residues important for ligand binding are highlighted as sticks. H–bonds are dashed black lines.
In particular, the pyrazoloquinazoline scaffold stacks between the −1T and the +1G of the scissile strand, with the N4 atom establishing water–mediated H–bonds with R364 and the ribose endocyclic oxygen (O5) of the −1 adenosine on the non–scissile strand. The 3–phenyl establishes well–oriented parallel–displaced interactions with the −1A and the +1C which should be lost when the phenyl is moved to position 2, thus explaining the lower activity of compounds 1–8.
As expected, in the 3–phenyl compounds enhancement of ligand–target charge–transfer interactions, through the introduction of an electron–withdrawing 4′–Cl (23–29), resulted in higher inhibitory potencies if compared to the unsubstitued 30–34. Also, the Cl seems to perfectly fit in the crevice formed by the −1A and +1C residues of the non–scissile strand (Figure S3, SI); this is further confirmed by the lower potency displayed by analogues featuring bulkier substituents in the same position such as the p–OCH3 and p–CF3 substituted 9–22.
In the docking pose predicted for 26, the 5– dimethylaminoethylamino branch extends outside the double–helix DNA towards a rather shallow protein pocket where the exocyclic NH group can donate a H– bond to the adjacent water molecule. Thus, it can be predicted that the lower Top1 inhibition rate displayed by compounds bearing exocyclic ether oxygen (23–25) might in part be ascribed to the loss of this water– mediated interaction. In this position, the terminal dimethylamino moiety of 26 establishes a tight salt bridge with the carboxylate group of D533. In this regard, the length of the aminoalkyl chain (12 vs 14, 19 vs 21, and 26 vs 28) or the alkyl on the terminal amine group (12 vs 13, 19 vs 20, and 26 vs 27) poorly influence the Top1 inhibitory potency. In this respect, longer but still flexible chains [the dimethylaminopropylamino (i.e. 14, 21, 28), the diethylaminoethylamino (i.e. 13, 20, 27) branches] can rearrange without steric restrictions to preserve the salt bridge with D533. However, the introduction of a more rigid and less basic substituent such as the imidazole (8, 15, 22, 29 and 34) should result in low or null Top1 inhibitory activity due to the loss of the ionic interaction described above.
In conclusion, we designed and synthesized a series of novel non–CPT Top1 inhibitors based on the phenylpyrazolo[ 1,5–a]quinazolin–5(4H)–one scaffold, structurally related to the indenoisoquinoline nucleus. SARs emerging from this series, together with the theoretical model for the 26/Top1/DNA ternary complex provided by hydrated docking calculations, allowed to identify the following structural requirements to gain Top1 inhibitory activity: (i) a properly–substituted 3–phenyl ring; (ii) a protonable dialkylaminoalkylamino chain at 5–position. Compounds 26–28 are among the most active Top1 inhibitors developed in this study showing cleavage patterns that are common to I and 55.
Taken together, all these findings highlight the pyrazoloquinazoline nucleus as a suitable scaffold to further expand the chemical diversity in Top1 inhibitors, and provided SAR data for the optimized design of new derivatives with improved biological activity.
Experimental Section
Chemistry
General directions are in the SI. Purity of tested compounds is ≥95% (combustion analysis).
General procedure for the synthesis of 2–phenylpyrazolo[1,5–a]quinazolin–5(4H)–ones 1–3
A mixture of 2–phenyl–5H–pyrazolo[1,5–a][3,1]benzoxazin– 5–one 36 (0.262 g, 1 mmol) and the proper alkylamine (0.9 mmol) in DMF (2 ml) was irradiated at a T=135 °C, P=100 PSI, power=150 W for 2 min. Products 1–3 (SI) crystallized from the reaction mixture by dilution with ice/water in the desired purity degree (≥95%).
General procedure for the synthesis of 5–(N,N–dialkylaminoalkoxy)–2(3)–phenylpyrazolo[1,5–a]quinazolines 4–6, and 30–32
Sodium hydride (1.1 mmol, 0.044 g, 60% dispersion in mineral oil) was added portion–wise to an ice–cooled solution of 37, 41 (1 mmol) in 10 ml of DMF, and the mixture was stirred at rt for 1 h. The appropriate dialkylaminoalkyl chloride (1.1 mmol) was added dropwise, and stirring was continued for 24 h at rt (TLC analysis). The reaction mixture was evaporated to dryness, and the residue triturated with ice/water and extracted with CHCl3. Evaporation of the organic phase yielded products 4–6, 30–32 in the desired purity degree (≥95%). Samples of 4–6, 30–32 were characterized as hydrochloride salts, obtained by treatment with ethanol hydrochloride in absolute ethanol (SI).
General procedure for the synthesis of 5– (substitutedalkylamino)–2(3)–phenylpyrazolo[1,5–a]– quinazolines 7, 8, 12–15, 19–22, 26–29, 33, and 34
A mixture of the appropriate 5–chloro derivatives 50–54 (1.2 mmol) and the proper alkylamine (2.4 mmol) were irradiated at a T=80 °C, P=100 PSI, power=100 W for 2 min, using aluminium oxide basis as solid support. Then, the mixture was dissolved in ethanol, the aluminium oxide basis was filtered off and the organic solvent was evaporated yielding compounds 7, 8, 12–15, 19–22, 26–29, 33, and 34 in the desired purity degree (≥95%). Samples of 7, 8, 12–15, 19–22, 26–29, 33, and 34 were characterized as hydrochloride salts, obtained by treatment with ethanol hydrochloride in absolute ethanol (SI).
General procedure for the synthesis of 5–(N,N– dialkylaminoalkoxy)–3–(4– substitutedphenyl)pyrazolo[1,5–a]quinazolines 9–11, 16–18, 23–25
The opportune pyrazolo[1,5–a]quinazolin– 5(4H)–ones 38–40 (0.62 mmol) was stirred with PPh3(0.340g, 1.3 mmol) in dry THF under nitrogen atmosphere for 5 min. DEAD (0.21 ml, 1.2 mmol) was then added dropwise. After 15 min, the appropriate aminoalcohol (0.63 mmol) was added and the mixture was stirred for 16 h at rt (TLC analysis). The reaction mixture was evaporated to dryness, and the residue was treated with water and extracted with CHCl3. Evaporation of the organic phase yielded products 9–11, 16–18 and 23–25 in the desired purity degree (≥ 95%). Samples of 9–11, 16–18 and 23–25 were characterized as hydrochloride salts, obtained by treatment with ethanol hydrochloride in absolute ethanol (SI).
Supplementary Material
Acknowledgments
Funding Sources
This work was financially supported by University of Pisa, “Sapienza” University of Rome, and the Center for Cancer Research, Intramural Program of the NCI, NIH.
ABBREVIATIONS
- CPT
camptothecin
- MW
microwaves
- SAR
structure-activity relationship
- Top1
Topoisomerase I
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. General chemistry directions; synthesis of compounds 35–41, and 46–54; yields, physical, and spectral data of compounds 1–34; biological tests; computational studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 2.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant Antitumor Agents. I. The isolation and structure of Campthotecin a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 4.Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8:641–661. [PubMed] [Google Scholar]
- 5.Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109:2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burke TG, Mi Z. The structural basis of camptothecin interactions with human serum albumin: impact on drug stability. J Med Chem. 1994;37:40–46. doi: 10.1021/jm00027a005. [DOI] [PubMed] [Google Scholar]
- 7.Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, Agama K, Nitiss JL, Redon C. Repair of topoisomerase I–mediated DNA damage. Prog Nucleic Acid Res Mol Biol. 2006;81:179–229. doi: 10.1016/S0079-6603(06)81005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 9.Beretta GL, Zuco V, Perego P, Zaffaroni N. Targeting DNA topoisomerase I with non–camptothecin poisons. Curr Med Chem. 2012;19:1238–1257. doi: 10.2174/092986712799320529. [DOI] [PubMed] [Google Scholar]
- 10.Da Settimo A, Da Settimo F, Marini AM, Primofiore G, Salerno S, Viola G, Dalla Via L, Marciani Magno S. Synthesis, DNA binding and in vitro antiproliferative activity of purinoquinazoline, pyridopyrimidopurine and pyridopyrimidobenzimidazole derivatives as potential antitumor agents. Eur J Med Chem. 1998;33:685–696. [Google Scholar]
- 11.Dalla Via L, Gia O, Marciani Magno S, Da Settimo A, Primofiore G, Da Settimo F, Simorini F, Marini AM. Dialkylaminoalkylindolonaphthyridines as potential antitumour agents: synthesis, cytotoxicity and DNA binding properties. Eur J Med Chem. 2002;37:475–486. doi: 10.1016/s0223-5234(02)01372-7. [DOI] [PubMed] [Google Scholar]
- 12.Dalla Via L, Marciani Magno S, Gia O, Marini AM, Da Settimo F, Salerno S, La Motta C, Simorini F, Taliani S, Lavecchia A, Di Giovanni C, Brancato G, Barone V, Novellino E. Benzothiopyranoindole-based antiproliferative agents: synthesis, cytotoxicity, nucleic acids interaction, and topoisomerases inhibition properties. J Med Chem. 2009;52:5429–5441. doi: 10.1021/jm900627v. [DOI] [PubMed] [Google Scholar]
- 13.Vasquez TE, Jr, Nixey T, Chenera B, Gore V, Bartberger MD, Sun Y, Hulme C. One–pot microwave assisted preparation of pyrazoloquinazolinone libraries. Mol Divers. 2003;7:161–164. doi: 10.1023/b:modi.0000006917.06401.48. [DOI] [PubMed] [Google Scholar]
- 14.Penning TD, Thomas SA, Hajduk PJ, Sauer DR, Sarris K, Giranda VL. PCT Int. Appl. WO 2007149907. Preparation of 4H–pyrazolo[1,5–a]quinazolin–5–one derivatives as PARP inhibitors. 2007
- 15.Veibel S, Arnfred NH. Pyrazole Studies. II. Anhydrides of 1–(2′–carboxyphenyl)–3–phenylpyrazolone–5 and 1–(2′– carboxyphenyl)–3–methyl–4–ethyl–pyrazolone–5. Acta Chem Scand. 1948:921–926. [Google Scholar]
- 16.Michaelis IA. 1–Phenyl–5– and 3–pyrazolone–o–carboxylic Anhydrides. Justus Liebigs Annalen der Chemie. 1910;373:129–212. [Google Scholar]
- 17.Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Differential induction of topoisomerase I-DNA cleavage complexes by the indenoisoquinoline MJ–III–65 (NSC 706744) and camptothecin: base sequence analysis and activity against camptothecin-resistant topoisomerases I. Cancer Res. 2003;63:7428–7435. [PubMed] [Google Scholar]
- 18.Antony S, Kohlhagen G, Agama K, Jayaraman M, Cao S, Durrani FA, Rustum YM, Cushman M, Pommier Y. Cellular topoisomerase I inhibition and antiproliferative activity by MJ–III–65 (NSC 706744), an indenoisoquinoline topoisomerase I poison. Mol Pharmacol. 2005;67:523–530. doi: 10.1124/mol.104.003889. [DOI] [PubMed] [Google Scholar]
- 19.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 21.Forli S, Olson AJ. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J Med Chem. 2012;55:623–638. doi: 10.1021/jm2005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.