

Abstract
Motivation: Current advances in DNA synthesis, cloning and sequencing technologies afford high-throughput implementation of artificial sequences into living cells. However, flexible computational tools for multi-objective sequence design are lacking, limiting the potential of these technologies.
Results: We developed DNA-Tailor (D-Tailor), a fully extendable software framework, for property-based design of synthetic DNA sequences. D-Tailor permits the seamless integration of multiple sequence analysis tools into a generic Monte Carlo simulation that evolves sequences toward any combination of rationally defined properties. As proof of principle, we show that D-Tailor is capable of designing sequence libraries comprising all possible combinations among three different sequence properties influencing translation efficiency in Escherichia coli. The capacity to design artificial sequences that systematically sample any given parameter space should support the implementation of more rigorous experimental designs.
Availability: Source code is available for download at https://sourceforge.net/projects/dtailor/
Contact: aparkin@lbl.gov or cambray.guillaume@gmail.com
Supplementary information: Supplementary data are available at Bioinformatics online (D-Tailor Tutorial).
1 INTRODUCTION
The accumulation of genomic data has fueled the development of numerous computational tools that infer functional behavior from biological sequences. These algorithms essentially capture our understanding of how functional information is encoded in nucleic acid and protein sequences. As a result, molecular biologists can now access a plethora of sequence analysis tools to help them predict functional behaviors from plain sequences (Altschul et al., 1997; Bailey et al., 2009; Giardine et al., 2005; Hofacker, 2003; Kingsford et al., 2007; Markham and Zuker, 2008; Thomas-Chollier et al., 2011). Common tasks comprise the identification of sequence motifs from nucleic acid (DNA/RNA) or protein sequences (e.g. promoter or termination activity, recombination or splicing sites), as well as the computation of sequence properties that are mechanistically linked to particular phenotypes (e.g. codon usage or propensity to form transmembrane protein domains).
Such sequence-analysis tools are usually used to inform biological discovery in natural genomic sequences. However, considering recent advances in DNA technologies and the concomitant rise of synthetic biology applications (Cambray et al., 2011; Carr and Church, 2009; Czar et al., 2009; Endy, 2005; Ma et al., 2012), these same tools may also be leveraged to guide the design of artificial sequences satisfying predefined functions of interest.
Ideally, elementary biological functions should be contained within well-defined sequence parts that could be re-used with acceptable reliability in different contexts [e.g. Davis et al. (2011) and Mutalik et al. (2013)]. However, it is becoming increasingly clear that many molecular behaviors result from the combined influence of several sequence determinants that cannot be neatly encapsulated within the physical boundaries of a single part, but rather emerge at the interface between the different parts (Cambray et al., 2013; Kosuri et al., 2013; Mutalik et al., 2012; Salis et al., 2009). In this context, the multidimensional examination of DNA sequences becomes necessary to better capture the inherent complexity of biological behavior and further enable predictive design of synthetic sequence functions and activities [e.g. Allert et al. (2010), Dvir et al. (2013), Kinney et al. (2010), Na et al. (2013), Rhodius and Mutalik (2010), Rodrigo et al. (2012), Salis et al. (2009), Seelig et al. (2006), Welch et al. (2009)].
Valuable sequence-design tools implementing heuristic searches have been successfully developed for multi-objective optimization within specific applications [e.g. protein synthesis optimization (Chung and Lee, 2012; Dana and Tuller, 2012; Gaspar et al., 2012, 2013; Raab et al., 2010; Racle et al., 2012; Salis et al., 2009; Welch et al., 2011)]. However, application of such optimization procedures requires an objective function relating computed sequence properties to an expected performance score. Unfortunately, the data and models required to describe these relationships are generally not sufficient to support truly reliable functional design.
Interestingly, sequence-design tools can also be used upstream of the optimization process to produce libraries of sequences that are more suited for the development of predictive models. Although large-scale studies have mostly used random approaches to introduce variability in the synthetic sequences to be interrogated (Dvir et al., 2013; Quan et al., 2011), similar endeavours have greatly benefited from following well-established design of experiments (DoE) (Allert et al., 2010; Antony, 2003; Kosuri et al., 2013; Sharon et al., 2012; Smith et al., 2013).
DoE is a general framework that fully integrates planning and analysis phases, and comprises three major steps. The first one consists in identifying the factors of interest and defining the range of values for each factor. In the case of molecular sequences, factors are properties of the primary sequence itself and can be typically identified by reanalysing available functional genomic data and published mechanistic studies. The second step consists in implementing a particular experimental design wherein multiple combinations of factor levels are selected to create an experimental dataset providing maximal information to relate the design factors to the response variable(s). For example, one of the most informative DoE is the full-factorial design, where all possible combinations of factor levels across the different factors are performed. The resulting dataset not only permits to estimate the contribution of each factor to the measured response variable, but also robustly captures the interactions between the different factors (Antony, 2003; Mutalik et al., 2013). Last, the third step includes the collection of experimental data and definition of a model relating the multiple factors to the response variable(s). Of note, this can be an iterative process wherein models derived from the third phase can inform the design of a new set of experiments.
Although implementation of experimental designs systematically varying easily manipulated factors can be straightforward (e.g. growth medium, pH, temperature or oxygen levels), the ability to design artificial sequences whose intrinsic properties can be systematically varied is not necessarily trivial (e.g. binding site affinity or the strength of an RNA secondary structure).
Here, we present D-Tailor, an extendable framework supporting integration of multiple sequence analysis tools to mine and design biological sequences. D-Tailor uses a heuristic search algorithm to enable flexible design of synthetic sequences varying multiple properties of interest so as to satisfy complex DoE. We have validated our tool by successfully designing artificial sequence libraries conforming to full-factorial designs, which represent the upper bound of experimental design complexity. More specifically, we have designed libraries systematically varying multiple sequence properties known to impact translation efficiency in E.coli. To further demonstrate the versatility of the algorithm, we also used D-Tailor to design artificial bacterial promoter sequences varying multiple cis-regulatory properties (see Supplementary Material).
2 METHODS
D-Tailor essentially implements the two-step planning process outlined above (Fig. 1). The analysis mode computes property scores from plain biological sequences. Here, the user specifies input sequences and a predefined set of properties to be computed. The design mode integrates the analysis routines with a parameterizable Monte Carlo algorithm that evolves an input sequence (seed) so as to match the specified combinations of property scores. In a typical workflow, users can use the analysis mode to identify sequence properties and operational ranges that seem worth exploring in design mode.
Fig. 1.

D-Tailor enables multidimensional analysis and design of DNA sequences. D-Tailor provides a flexible and extendable architecture to interrogate different sequence properties (box in the middle). The left panel depicts an example of the retrieval process of two properties (RNA structure and motif prediction) from multiple input sequences that can come from either FASTA or CSV files. The resulting score profile can be used to identify general trends and further define ideal parameter ranges for the design objectives. The right panel shows the design mode of D-Tailor, wherein a seed sequence is evolved to meet a user-defined combination of sequence properties. The figure depicts a full-factorial design for two different properties of interest (RNA structure and motif scores) with three levels each (low, medium and high), which yields a total of nine different combinations (colored areas).
2.1 Sequence analyzer
D-Tailor provides a generic interface for multidimensional interrogation of DNA sequences. The software is designed with a modular architecture, so that users with basic programing skills can easily implement or extend modules for handling any sequence property of interest. Such modules can be implemented using custom Python code or scripts connecting to third party software (see the Tutorial available in the Supplementary Material). In analysis mode, D-Tailor reads a set of sequences in either delimiter separated (e.g. CSV) or FASTA format files. A property profile is then computed for each of the input sequences by successively calling the analysis modules specified by the user (Fig. 1, left panel).
D-Tailor currently comprises 14 different modules to compute various sequence properties involved in diverse mechanisms of gene regulation. This collection of sequence property evaluators includes algorithms to score promoter regions or transcription factor binding sites based on sequence logos (Thomas-Chollier et al., 2011), estimate translation initiation rates based on the Shine–Dalgarno (SD) sequence (Shine and Dalgarno, 1975), predict propensity to form RNA structures, calculate nucleotide composition or compute the codon adaptation index (CAI) for a given gene sequence (Sharp and Li, 1987). Although the implementation of the different sequence property evaluators is usually self-contained within D-Tailor, the computation of specific properties may rely on third party softwares [e.g. UNAfold (Markham and Zuker, 2008) for the prediction of RNA secondary structure]. Together, these modules illustrate diverse implementation modalities and provide useful examples to guide future extensions (see Supplementary Material). The specification of adequate analysis routines is an essential prerequisite to running the design mode.
2.2 Sequence designer
As capacities for DNA synthesis increase exponentially, the ability to computationally design artificial sequences need to become more automated and transparent. The most innovative feature of D-Tailor is to provide a generic solution of designing synthetic sequences constrained by multiple properties of interest (Fig. 1, right panel).
The design process in D-Tailor starts with the specification of a seed sequence and the desired design objective (i.e. the DoE) (Fig. 1, right panel). Seed sequences serve as a template to bootstrap the evolutionary design process. Typically, users would use a particular sequence of interest from which they want to derive a mutational series. The DoE enumerates combinations of sequence properties that need to be generated, each of which constitutes a design target. D-Tailor provides a flexible scheme for the definition of DoE, which can vary from full-factorial to entirely customized designs.
The definition a finite number of targets requires the discretization of continuous property scores into a finite number of nominal or ordinal levels. For example, Figure 1 shows the discretization of two sequence property scores into three ordinal levels (low, medium and high). This framework markedly differs from usual multi-objective optimization approaches (Chung and Lee, 2012; Raab et al., 2010; Racle et al., 2012), which operate to optimize a single continuous and integrated performance score rather than explicitly target different regions of the parameter space. As illustrated in the Section 3, natural feature profiles extracted from available genomic sequences can be used to guide the discretization processes and ensure biological relevance of the sampled space. For each sequence property, users may define as many levels as necessary to attain the desired degree of resolution in the designed sequences. However, since the number of possible combinations increases geometrically with the number of properties/levels, their definition must be mindful of downstream experimental capacities.
Finding a sequence that conforms to an arbitrary combination of property levels is often computationally infeasible using a brute force approach. Indeed, the sequence space to be searched is gigantic (4N where N is the number of nucleotides in the sequence to be designed, more if indels are allowed). To optimize the search process, D-Tailor uses a Monte Carlo algorithm to evolve a given seed sequence towards the set of design targets (Fig. 2).
Fig. 2.
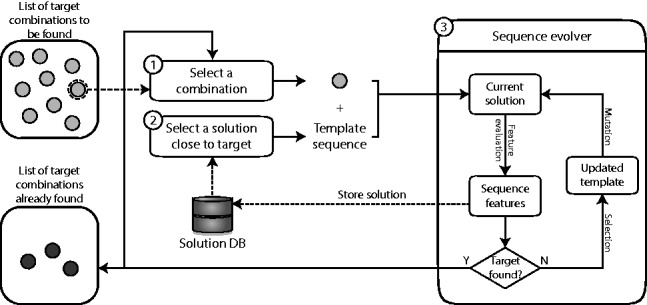
Sequence designer algorithm comprised by three different steps described in the main text. Initially, a target combination of features is selected and then a sequence that is close (i.e. short Euclidean distance) to the desired target is chosen to serve as the template in the sequence evolution step. This last step applies successive mutations until it finds a sequence matching the target combination of features
More specifically, the algorithm loops through cycles of evolution until all target combinations of property levels specified by the DoE are found. Each cycle consists in three consecutive steps: (i) a target combination of property levels is randomly selected; (ii) a template sequence is chosen from the repository of previously generated sequences using fitness proportionate selection (only seed sequences are available at the very first iteration); and (iii) a predefined number of mutational iterations are performed until a sequence satisfying the target combination of the property level is found (Fig. 2, sequence evolver). We use the inverse of the cumulative Euclidean distance (D) between property levels, as a generic fitness measure of a sequence relative to a given design target in Equation (1)
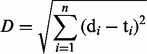 |
(1) |
where n represents the number of sequence properties; di and ti represent the levels of the i-th sequence property in the designed sequence and the desired combination, respectively.
Each iteration of the sequence evolver also comprises three steps: (i) the sequence being evolved is analyzed and a property requiring optimization (i.e. not within the target level) is randomly selected; (ii) the template sequence is then mutated following user-specified mutational rules (see below); and (iii) the feature scores of the resulting sequence are analyzed and evaluated with respect to the current design target [Equation (1)]. Every generated sequence is also screened for compliance to a user-defined set of rules meant to prevent the emergence of undesired properties in the final designed sequences (e.g. restriction sites, unexpected promoters or terminators). Only validated sequences are stored in the database.
Next, if the new sequence matches the target combination (D = 0), then the target is marked as completed and the evolution cycle is terminated. Otherwise, the algorithm updates the template for the next mutational iteration, choosing between retaining the current template sequence or accepting the mutant just derived. At this point, we defined three different selective regimes: (i) directional selection, where the sequence with the lower Euclidean distance to the target combination is chosen; (ii) neutral selection, where any of the two sequences is selected with predefined probabilities; or (iii) temperature selection, as inspired by simulated annealing optimization (Kirkpatrick et al., 1983), where the sequence is selected based on a temperature schedule that allows worse sequences (longer distances) to also be selected with a probability that decreases with the number of iterations performed.
At each of the mutational iterations, new sequences can be generated through random mutation of the template sequence, as usual in many sequence optimization tools (Chung and Lee, 2012; Gaspar et al., 2012; Salis et al., 2009). In addition, D-Tailor offers the possibility to implement specialized mutation operators that aim at improving the likelihood to generate desired property changes. Practically, a mutation operator randomly selects a property amongst those that are non-optimal in the current template (di – ti ≠ 0). We then distinguish between: (i) targeted operators, which restrict the mutational space to specific regions of the sequence that are therefore more likely to affect the property that needs to be evolved; and (ii) oriented operators, which further specify particular mutation patterns to bias the production of variants toward the current design target. For example, if the design goal specifies an increase in the CAI of a gene, the targeted mutation operator restricts the mutable region to the coding sequence and randomly replaces a codon by another one irrespective of its usage score. The oriented mutation operator further constrains the replacement of a randomly chosen codon with one associated with a higher usage score, thereby enforcing the required increase. For certain emergent features, the definition of oriented mutation might not be so straightforward. For example, we implemented oriented mutation operators for RNA secondary structure by specifically targeting mutations to bases that are predicted to be paired or unpaired, to, respectively, decrease or increase the strength of the mutated RNA structure. Importantly, any mutation operator targeting gene-coding sequences can be further constrained to only generate synonymous mutations, thereby preserving the encoded protein sequence while modifying the underlying DNA properties.
In some applications, it may be desirable to limit the overall divergence between sequences in the designed library, so that it provides small variations with respect to a particular reference sequence. Conversely, users might want to generate sequences that are as dissimilar as possible and, therefore, share as few confounding factors as possible. In D-Tailor, users can manipulate mutational patterns and the selective regime—two major parameters of the evolutionary design process—to indirectly control sequence diversity, and consequently impact the rate of sequence evolution, as well as the overall performance of the search algorithm (see below).
3 RESULTS AND VALIDATION
D-Tailor provides an integrated Python-scripting framework for multidimensional analysis of sequence properties and for the design of artificial sequences constrained by multiple sequence properties of interest.
As a case study, we have chosen three different previously reported sequence determinants of translation efficiency. In E.coli, two major factors have been shown to modulate the rate of translation initiation: (i) the strength and position of a SD motif upstream of the start codon (Barrick et al., 1994; Shine and Dalgarno, 1975); and (ii) the propensity of these sequence signals to engage in mRNA secondary structures (de Smit and van Duin, 1994; Hall et al., 1982; Kudla et al., 2009). Subsequent to initiation, the rate of elongation may also affect the overall translation efficiency and is mainly determined by the codon usage of the gene (Gustafsson et al., 2004; Ikemura 1985; Kane, 1995; Sharp and Li, 1987; Welch et al., 2009, 2011). We first illustrate how D-Tailor analysis module can be used to examine such sequence properties in the natural genome of E.coli. Then, we demonstrate how to use D-Tailor design module to generate artificial sequence libraries systematically varying the three properties of interest according to a full-factorial DoE.
3.1 Using D-Tailor to interrogate sequences
We used D-Tailor to re-analyze three different sequence properties across the entire E.coli W3110 genome (Fig. 3). Mechanistically, the SD motif stabilizes the initial binding of the 30S subunit of the ribosome by establishing canonical base pairing with the 3′-end of the 16S rRNA (anti-SD) (Shine and Dalgarno, 1975). We applied a sequence property evaluator that calculates the strength of the SD sequence by searching for a subsequence within the 25 nucleotides upstream of a start codon with highest affinity to the known anti-SD (Lithwick and Margalit, 2003). The presence of secondary structures in this region of the mRNA can hinder initiation by occluding the SD motif or the nearby start codon from recognition by the ribosomal subunits. For that purpose, we used an RNA-structure evaluator to compute the minimum free energy of the 60 nucleotides subsequence centered on the start codon (Kudla et al., 2009). Finally, we used a CAI calculator to score the codon usage of a gene sequence (Sharp and Li, 1987). Practically, the usage of these property evaluators and associated parameters requires a standard interface, which is provided by extending the abstract class Feature in D-Tailor (see Supplementary Material).
Fig. 3.
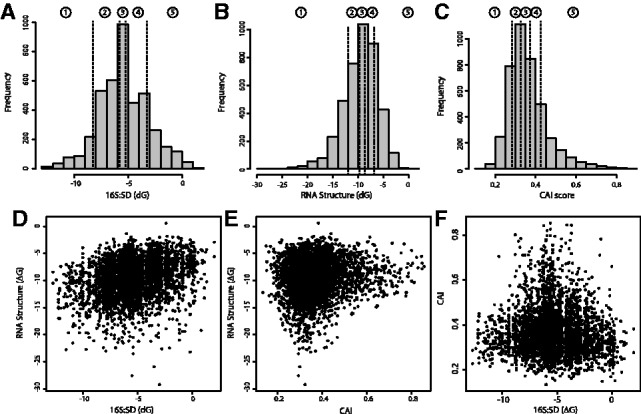
(A–C) Distribution of the three different sequence properties (hybridization energy between the 16S rRNA and SD sequence (A), minimum folding energy of RNA structure in the translation initiation region (B) and CAI of gene sequences (C)) influencing translation efficiency in E.coli. The dashed lines indicate the quintile boundaries for the scores of each property, which were later used in design mode to discretize the parameter space. (D–F) Scatter plots showing the cross-correlation between the three sequence properties of interest
The sequence property profiles resulting from a genome analysis give a solid basis to identify trends in the properties of interest, and to further determine the relevant parameter space to explore during the design step (Fig. 3A–C). Correlations amongst property scores may also provide insights onto potential functional interactions although some may be purely incidental. For example, the modest correlation between RNA structure in the translation initiation region and the affinity between ribosomes and the SD sequence (Fig. 3D) might merely reflect the thermodynamic propensity of G-rich SD motifs to engage in secondary structures. Similarly, the peculiar shape of the relationship between CAI and RNA secondary structure (Fig. 3E) might stem from the joint contributions of independent evolutionary pressures for expression levels acting on these two properties to tune expression levels [highly expressed genes being both under selection for high CAI and for low structure (Gu et al., 2010; Kudla et al., 2009; Plotkin and Kudla, 2011; Tuller et al., 2010)]. It is then up to the user to define a DoE containing combinations of sequence property scores that are more adequate to test the research hypothesis to be investigated.
3.2 Using D-Tailor to implement experimental design on sequence properties
Although recent advances in DNA synthesis, cloning and sequencing make it possible to generate and experimentally probe thousands of custom DNA/RNA sequences (Dvir et al., 2013; Kosuri et al., 2013; Patwardhan et al., 2009, 2012; Quan et al., 2011; Sharon et al., 2012; Smith et al., 2013), the availability of computational tools to aid the rational design of large sequence libraries remains very limited.
The main purpose of D-Tailor is to provide a flexible computational tool to design custom sequences satisfying complex specifications. Such task can be extremely laborious when the properties of interest physically overlap in the sequence space. For instance, in our case study, the subsequence containing the SD motif influences the formation of RNA secondary structures in that same region. Likewise, the secondary structure can be affected when modifying codon usage at the beginning of the gene. Typically, such optimization problems are best solved using a trial-and-error approach wherein sequence variants are generated using random mutations until a desired combination of property scores is found (Allert et al., 2010; Gaspar et al., 2013; Raab et al., 2010; Racle et al., 2012; Salis et al., 2009). To generalize this process, the design mode of D-Tailor provides a framework to integrate any sequence property evaluator into a parameterizable Monte Carlo algorithm that iteratively evolves sequences toward a specific set of design targets (or combinations of property levels).
We used D-Tailor to design sequences that systematically vary the three properties of interest (or factors) defined above (Fig. 3). For each of these factors, we defined five contiguous ordinal levels on the basis of the quintiles observed in the natural genome (Fig. 3A–C, dashed lines). We then instructed D-Tailor to search for sequences conforming to a full-factorial DoE based on these levels. This DoE describes a total of 125 design targets corresponding to all combinations of five levels across the three different properties (53). To validate our approach, we compared the performance of four increasingly complex evolutionary strategies available in D-Tailor at deriving full-factorial libraries for 30 different genes randomly selected in E.coli (Fig. 4A and B). In these simulations, the algorithm was run for at most 3000 generations—with a single mutational event per generation—allowing for unrestricted mutations in the 5′ UTR but only for synonymous mutations in the coding sequence.
Fig. 4.
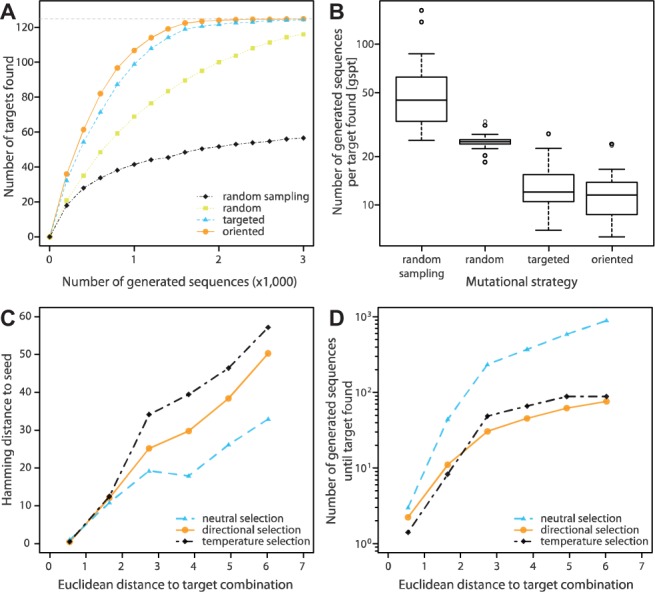
D-Tailor design simulations. (A) We performed simulations of full-factorial design using 30 different initial sequences (seeds) and four different design strategies: random sampling (black) and heuristic search using random (yellow), targeted (blue) and oriented (orange) mutations. The different lines represent the average number (across 30 simulations) of target combinations found (out of 125) as a function of the number of generated sequences (up to 3000) for the four different strategies. We observed sizeable variation between seeds (not shown for clarity, see Supplementary Material for details). (B) Number of generated sequences per target found (gspt) for the four different mutational strategies (n = 30). (C) We used the same 30 different seeds to find six different target combinations at various Euclidean distances. The different lines show the average hamming distance between the seed and the sequence matching the target combination as a function of the Euclidean distance to the target combination using neutral (light blue), directional (orange) or temperature selection (black). (D) The number of generated sequences until the desired target is found as a function of the Euclidean distance to the target combination using either neutral (light blue), directional (orange) or temperature (black) selection
We first explored the most rudimentary evolutionary strategy available in D-Tailor, random sampling, which does not implement any heuristic search and simply generates random sequences until all desired design objectives are completed. Every attempt to complete the full-factorial design before the threshold of 3000 generated sequences failed (Fig. 4A and B, black line, 54.2 generated sequences per target found [gspt] on average). The second design strategy used D-Tailor’s generic heuristic algorithm (Fig. 2 and Section 2) along with the simplest mutational method wherein new sequence variants are generated by random mutagenesis (Fig. 4A, yellow line). This strategy improved the efficiency of the search algorithm by a factor of 2 as compared to that of the random sampling method (24.8 versus 54.2 gspt on average, Mann–Whitney test P-value = 2.3 × 10−10, Fig. 4B). Still, many sequences had to be generated to meet the various design objectives. The third mutational strategy employed spatially targeted mutation operators (see Section 2) and improved the search algorithm efficiency by another factor of 2 (13.3 gspt on average, Fig. 4B). The fourth strategy used more ‘rational’ mutation operators that explicitly orient mutations toward the desired objective (see Section 2) and provided slightly faster dynamics (Fig. 4A, orange line, 11.8 versus 13.3 gspt on average, Mann–Whitney test P-value = 0.129, Fig. 4B). Since the computational time necessary to achieve a given set of design targets is dependent on the number of generated sequences per target, these results illustrate the advantage of defining specific mutation operators whenever it is possible.
When designing synthetic sequences, users may want to limit the divergence of the designed sequences with respect to the initial seed. To roughly control the spread of the generated sequences during the evolutionary process, users can manipulate the strength of selection toward the desired target(s). To better illustrate this point, we evolved each of the 30 seeds previously selected toward six different target combinations bearing different Euclidean distances from the seeds (Fig. 4C and D). We then examined the behavior and results of the algorithm in response to three contrasted selective regimes: neutral, directional and temperature selection (Section 2).
As expected, we observed that a more relaxed selection process (neutral) is able to generate sequences matching the desired target that are more similar to the seed sequence than those resulting from the directional or temperature selection approach (average hamming distance of 21 versus 31.3 and 39.2, respectively; Mann–Whitney test P-value = 0.0005 and 1.03e – 13; Fig. 4C). Nonetheless, the limitation of sequence diversity comes at the cost of longer computation time (Fig. 4D). In fact, for the 30 seed sequences, the neutral selection process requires the generation of eight and six times more sequences per target than the directional and temperature selection approach, respectively and on average. For large designs, users may have to balance the desired divergence of the designed sequences with the available computational power. A hybrid approach, wherein the algorithm is initially set with weak selection and hard constraints to limit divergence, and then progressively configured with increased selection bias and/or relaxed mutational constraints (e.g. allow non-synonymous mutations in coding sequences if it is acceptable by the user) as the rate of target discovery slows down may then be recommended. The details of such procedure are likely specific for each application, and therefore we have not sought to implement an automatic schedule to control this behavior. Since the state of a D-Tailor design mode run is permanently stored in a database, we suggest users to manually experiment with adjusting these parameters.
4 CONCLUSION
Advances in DNA-reading/writing technologies readily enable the construction and validation of complex genetic systems (Gibson et al., 2010). However, rules to successfully design synthetic sequences to functional specifications have been limited by measurements from biased natural samples and/or small scale controlled synthetic samples comprising at most hundreds of sequences (e.g. Allert et al. (2010), Amit et al. (2011), Barrick et al. (1994), Garcia et al. (2012); Mutalik et al. (2012); Na et al. (2013); Rhodius and Mutalik (2010); Rodrigo et al. (2012), Salis et al. (2009)]. This lack of knowledge strongly restrains the practical applications of ab initio design. Innovative experimental methodologies based on high-throughput technologies are scaling the characterization process up to tens of thousands of designed sequence variants, providing larger datasets to better understand sequence/activity relationships (Dvir et al., 2013; Kinney et al., 2010; Patwardhan et al., 2009, 2012; Sharon et al., 2012; Smith et al., 2013). However dramatic, this increase in throughput remains limited in comparison to the sheer immensity of the sequence space. It is therefore crucial to reduce the dimensionality of the design space to a set of sequence properties of interest that can be independently varied to facilitate estimation of their individual contribution to the measured phenotype and further support predictable design of synthetic variants (Allert et al., 2010; Sharon et al., 2012; Smith et al., 2013).
We developed D-Tailor as an extendable and flexible software platform for the multi-objective design of artificial sequences. It provides a generic interface to integrate multiple sequence analysis tools into a heuristic Monte Carlo search procedure capable of evolving sequences towards pre-defined design targets (Fig. 1). D-Tailor presents significant differences to other multi-objective sequence optimization tools (Allert et al., 2010; Chung and Lee, 2012; Dana and Tuller, 2012; Gaspar et al., 2012; Raab et al., 2010; Racle et al., 2012; Salis et al., 2009). First, it allows the definition of multiple design targets as combinations of sequence properties that embody particular DoE. A DoE can range anywhere from one specific combination of property levels to a full-factorial design, where the parameter space is fully explored. In contrast, traditional optimization tools describe design objectives in terms of desired response performances, which are linked to the sequence properties by a complex and pre-defined static objective function. Such formalization is suited for functional optimization, but do not explicitly support systematic exploration of the parameter space. Second, D-Tailor provides an evolutionary algorithm to optimize both coding and non-coding regions. Third, D-Tailor supports the implementation of advanced mutational strategies that can significantly enhance the heuristic search performance (Fig. 4B). Finally, our tool is not application-specific and provides an open source solution based on an extendable architecture, such that new sequence property evaluators can be easily implemented and integrated into the sequence design engine.
We demonstrate that D-Tailor can efficiently design artificial sequences to systematically vary any given set of properties of interest. To this end, we successfully derived full-factorial sequence libraries, starting from 30 different seed sequences, exploring the entire parameter space of three intertwined sequence properties affecting translation efficiency. Interestingly, we observed that the dynamics of target discovery varies slightly depending on the input seed (see Supplementary Material for details). This illustrates that different sequences may have distinct evolutionary landscapes; some being more amenable to generate widely variable profiles of property scores, with fewer mutational cycles than others (Cambray and Mazel, 2008; Wagner, 2008). For both targeted and oriented mutational methods, the average dynamics of target discovery revealed a relatively steady rate for the first ∼80% of targets, followed by a sharp decrease in efficiency—presumably because the remaining targets specify combinations of property levels that are harder to attain (Fig. 4A, orange and light blue lines). We also confirmed that more simplistic design approaches—such as generation of random sequences—perform poorly in comparison to a heuristic search (Fig. 4A and B).
In addition to the case study detailed here, we have used D-Tailor to systematically design synthetic bacterial promoter sequences varying multiple cis-regulatory properties (see Tutorial in Supplementary Material for details), that way demonstrating the generality and flexibility of our methods and tool.
D-Tailor permits the implementation of advanced experimental designs into artificial sequence samples that can serve as a basis to rigorously and consistently test sets of molecular hypothesis. We believe that comprehensive full-factorial libraries of sequences are needed to investigate complex biochemical activities and robustly dissect the contribution of individual factors as well as their interactions. Such libraries will aid characterizing complex multifactorial phenotypes and eventually derive quantitative relationships between sequence and activity.
Funding: Synthetic Biology Engineering Research Center under National Science Foundation (04-570/0540879); Fundação para a Ciência e a Tecnologia (FCT) (SFRH/BD/47819/2008 to J.C.G.); Human Frontiers Science Program (LT000873/2011-L to G.C.); Bettencourt Schueller foundation (to G.C.); European Regional Development Fund (ERDF) through the COMPETE Programme (to M.R.); FCT within projects COMPETE (FCOMP-01-0124-FEDER-015079, PEst-OE/EEI/ UI0752/2011 to M.R.).
Conflict of interest: none declared.
Supplementary Material
References
- Allert M, Cox JC, Hellinga HW. Multifactorial determinants of protein expression in prokaryotic open reading frames. J. Mol. Biol. 2010;402:905–918. doi: 10.1016/j.jmb.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit R, et al. Building enhancers from the ground up: a synthetic biology approach. Cell. 2011;146:105–118. doi: 10.1016/j.cell.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony J. Design of Experiments for Engineers and Scientists. Oxford: Butterworth-Heinemann; 2003. [Google Scholar]
- Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick D, et al. Quantitative analysis of ribosome binding sites in E.coli. Nucleic Acids Res. 1994;22:1287–1295. doi: 10.1093/nar/22.7.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambray G, et al. Measurement and modeling of intrinsic transcription terminators. Nucleic Acids Res. 2013;41:5139–5148. doi: 10.1093/nar/gkt163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambray G, Mazel D. Synonymous genes explore different evolutionary landscapes. PLoS Genet. 2008;4:e1000256. doi: 10.1371/journal.pgen.1000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambray G, Mutalik VK, Arkin AP. Toward rational design of bacterial genomes, Curr. Opin. Microbiol. 2011;14:624–630. doi: 10.1016/j.mib.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Carr PA, Church GM. Genome engineering. Nat. Biotechnol. 2009;27:1151–1162. doi: 10.1038/nbt.1590. [DOI] [PubMed] [Google Scholar]
- Chung BK, Lee DY. Computational codon optimization of synthetic gene for protein expression. BMC Syst. Biol. 2012;6:134. doi: 10.1186/1752-0509-6-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czar MJ, et al. Gene synthesis demystified. Trends Biotechnol. 2009;27:63–72. doi: 10.1016/j.tibtech.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Dana A, Tuller T. Efficient manipulations of synonymous mutations for controlling translation rate: an analytical approach. J. Comput. Biol. 2012;19:200–231. doi: 10.1089/cmb.2011.0275. [DOI] [PubMed] [Google Scholar]
- Davis JH, Rubin AJ, Sauer RT. Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res. 2011;39:1131–1141. doi: 10.1093/nar/gkq810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Smit MH, van Duin J. Control of translation by mRNA secondary structure in Escherichia coli. A quantitative analysis of literature data. J. Mol. Biol. 1994;244:144–150. doi: 10.1006/jmbi.1994.1714. [DOI] [PubMed] [Google Scholar]
- Dvir S, et al. Deciphering the rules by which 5'-UTR sequences affect protein expression in yeast. Proc. Natl Acad. Sci. USA. 2013;110:E2792–E2801. doi: 10.1073/pnas.1222534110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endy D. Foundations for engineering biology. Nature. 2005;438:449–453. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- Garcia HG, et al. Operator sequence alters gene expression independently of transcription factor occupancy in bacteria. Cell Rep. 2012;2:150–161. doi: 10.1016/j.celrep.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P, et al. mRNA secondary structure optimization using a correlated stem-loop prediction. Nucleic Acids Res. 2013;41:e73. doi: 10.1093/nar/gks1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P, et al. EuGene: maximizing synthetic gene design for heterologous expression. Bioinformatics. 2012;28:2683–2684. doi: 10.1093/bioinformatics/bts465. [DOI] [PubMed] [Google Scholar]
- Giardine B, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- Gu W, Zhou T, Wilke CO. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput. Biol. 2010;6:e1000664. doi: 10.1371/journal.pcbi.1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22:346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Hall MN, et al. A role for mRNA secondary structure in the control of translation initiation. Nature. 1982;295:616–618. doi: 10.1038/295616a0. [DOI] [PubMed] [Google Scholar]
- Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- Kane JF. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr. Opin. Biotechnol. 1995;6:494–500. doi: 10.1016/0958-1669(95)80082-4. [DOI] [PubMed] [Google Scholar]
- Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007;8:R22. doi: 10.1186/gb-2007-8-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JB, et al. Using deep sequencing to characterize the biophysical mechanism of a transcriptional regulatory sequence. Proc. Natl Acad. Sci. USA. 2010;107:9158–9163. doi: 10.1073/pnas.1004290107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick S, Gelatt CD, Jr, Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- Kosuri S, et al. Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proc. Natl Acad. Sci. USA. 2013;110:14024–14029. doi: 10.1073/pnas.1301301110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, et al. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lithwick G, Margalit H. Hierarchy of sequence-dependent features associated with prokaryotic translation. Genome Res. 2003;13:2665–2673. doi: 10.1101/gr.1485203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Tang N, Tian J. DNA synthesis, assembly and applications in synthetic biology. Curr. Opin. Chem. Biol. 2012;16:260–267. doi: 10.1016/j.cbpa.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol. Biol. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- Mutalik VK, et al. Quantitative estimation of activity and quality for collections of functional genetic elements. Nat. Methods. 2013;10:347–353. doi: 10.1038/nmeth.2403. [DOI] [PubMed] [Google Scholar]
- Mutalik VK, et al. Rationally designed families of orthogonal RNA regulators of translation. Nat. Chem. Biol. 2012;8:447–454. doi: 10.1038/nchembio.919. [DOI] [PubMed] [Google Scholar]
- Na D, et al. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 2013;31:170–174. doi: 10.1038/nbt.2461. [DOI] [PubMed] [Google Scholar]
- Patwardhan RP, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009;27:1173–1175. doi: 10.1038/nbt.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan J, et al. Parallel on-chip gene synthesis and application to optimization of protein expression. Nat. Biotechnol. 2011;29:449–452. doi: 10.1038/nbt.1847. [DOI] [PubMed] [Google Scholar]
- Raab D, et al. The GeneOptimizer Algorithm: using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization. Syst. Synth. Biol. 2010;4:215–225. doi: 10.1007/s11693-010-9062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racle J, Overney J, Hatzimanikatis V. A computational framework for the design of optimal protein synthesis. Biotechnol. Bioeng. 2012;109:2127–2133. doi: 10.1002/bit.24463. [DOI] [PubMed] [Google Scholar]
- Rhodius VA, Mutalik VK. Predicting strength and function for promoters of the Escherichia coli alternative sigma factor, sigmaE. Proc. Natl Acad. Sci. USA. 2010;107:2854–2859. doi: 10.1073/pnas.0915066107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo G, Landrain TE, Jaramillo A. De novo automated design of small RNA circuits for engineering synthetic riboregulation in living cells. Proc. Natl Acad. Sci. USA. 2012;109:15271–15276. doi: 10.1073/pnas.1203831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig G, et al. Enzyme-free nucleic acid logic circuits. Science. 2006;314:1585–1588. doi: 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- Sharon E, et al. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 2012;30:521–530. doi: 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shine J, Dalgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254:34–38. doi: 10.1038/254034a0. [DOI] [PubMed] [Google Scholar]
- Smith RP, et al. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 2013;45:1021–1028. doi: 10.1038/ng.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M, et al. RSAT 2011: regulatory sequence analysis tools. Nucleic Acids Res. 2011;39:W86–W91. doi: 10.1093/nar/gkr377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, et al. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl Acad. Sci. USA. 2010;107:3645–3650. doi: 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. Robustness and evolvability: a paradox resolved. Proc. Biol. Sci. 2008;275:91–100. doi: 10.1098/rspb.2007.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M, et al. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One. 2009;4:e7002. doi: 10.1371/journal.pone.0007002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M, et al. Designing genes for successful protein expression. Methods Enzymol. 2011;498:43–66. doi: 10.1016/B978-0-12-385120-8.00003-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.