

Abstract
Motivation: Methods for detecting somatic genome rearrangements in tumours using next-generation sequencing are vital in cancer genomics. Available algorithms use one or more sources of evidence, such as read depth, paired-end reads or split reads to predict structural variants. However, the problem remains challenging due to the significant computational burden and high false-positive or false-negative rates.
Results: In this article, we present Socrates (SOft Clip re-alignment To idEntify Structural variants), a highly efficient and effective method for detecting genomic rearrangements in tumours that uses only split-read data. Socrates has single-nucleotide resolution, identifies micro-homologies and untemplated sequence at break points, has high sensitivity and high specificity and takes advantage of parallelism for efficient use of resources. We demonstrate using simulated and real data that Socrates performs well compared with a number of existing structural variant detection tools.
Availability and implementation: Socrates is released as open source and available from http://bioinf.wehi.edu.au/socrates.
Contact: papenfuss@wehi.edu.au
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The identification of somatic rearrangements in tumour genomes using next-generation sequencing (NGS) data is an important step in characterizing cancers, including identifying genomic instability (Campbell et al., 2010), understanding tumour evolution (Greenman et al., 2012) and identifying potential fusion genes (Campbell et al., 2008). However, prediction of genomic rearrangements remains a challenging bioinformatics problem. Existing methods suffer from a variety of issues, including high false-positive (see Wang et al., 2011, for further discussion) or false-negative rates depending on the methods, data and nature of the genomic rearrangements.
To identify genomic rearrangements or structural variants (SVs), DNA from samples is extracted, fragmented, size selected and typically paired end (PE) sequenced. There are four distinct approaches to identifying genomic rearrangements using these data: read depth, paired end, split reads and de novo assembly methods. Read depth methods (RD) identify one class of structural variation—copy number variants. They provide only indirect evidence for break points and no information about genomic organization. RD methods involve counting reads in windows and segmenting the counts (see e.g. Miller et al., 2011). They may use single-end (SE) or paired-end reads. Their resolution and accuracy is dependent on the depth of coverage and the window size, but is typically of the order of kilobases. Examples of RD methods include readDepth (Miller et al., 2011) and CNVnator (Abyzov et al., 2011). Anomalous paired-end alignment methods (PE) use reads that contain a break point in the unsequenced region between the paired reads. These reads map anomalously or discordantly to the reference genome—further apart or closer together than expected based on the selected fragment size, to different chromosomes or with inverted orientation (see e.g. Medvedev et al., 2009). The signal of a rearrangement is a cluster of anomalous reads. The resolution of PE methods is related to the fragment size and coverage. In general, single-nucleotide resolution is not possible with PE methods. BreakDancer (Chen et al., 2009) is an example of a PE method. Split read methods (SR) rely on reads that span the break point (Fig. 1). Split reads may be identified from single-end or paired-end sequencing, although using paired-end data has the advantage of higher quality alignment. Most NGS aligners will not map split reads. With PE data, global NGS aligners will map the unsplit read, while local aligners may also map part of the split read. With paired-end reads, local NGS aligners are often greedy to align reads concordantly. Thus, the aligned part of a split read may be quite short [e.g. 20 nucleotides (nt)]. A straightforward approach to identifying a split read using an NGS aligner is via soft clipping. A soft clip is a special mismatch state in the alignment that is restricted to contiguous segments of the read at the 5′ or 3′ end (Li and Durbin, 2009). Sequencing errors, chimeric reads, errors in the reference genome or genomic rearrangement may cause soft clips. SR methods are capable of single-nucleotide resolution, although micro-homologies at fusion sites mean the break point may only be known to 1–10 nt accuracy and large imperfect homologies at the break point may cause lower accuracy again. Examples of SR methods include Splitread (Karakoc et al., 2011). Until recently, most methods used just one of these types of evidence. However, several methods have now appeared that make use of more of the available evidence to predict an SV. For example, DELLY (Rausch et al., 2012) and PRISM (Jiang et al., 2012) use PE evidence and incorporate SR evidence through a targeted Smith–Waterman alignment. CNVer (Medvedev et al., 2010) uses PE and RD signals to identify potential copy number changes. De novo assembly methods (DN) typically perform targeted sequence assembly following other evidence to locate the locus and reads for assembly. For example, CREST (Wang et al., 2011) uses SR then DN. However, unbiased assembly of the whole-genome data is also possible. Making better use of the available evidence to predict break points is obviously desirable, but how this is undertaken is critical.
Fig. 1.
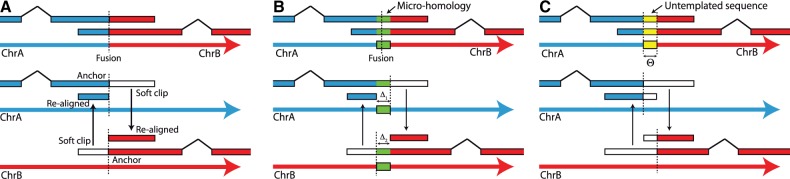
Clusters of split reads spanning a break point. Three cases are shown: (A) blunt end joining, (B) micro-homology at the break points and (C) untemplated sequence inserted between the break points
To find somatic rearrangements, evidence from a tumour and a matched normal is used. Typically, SVs are called in both samples. Variants that occur in the both genomes are most likely polymorphisms or artefacts. Somatic rearrangements are obtained by subtracting predicted normal SVs from the predicted tumour SVs. Most methods are not specifically designed for tumour genomes, and some methods that are designed for germline DNA neglect certain classes of rearrangement, which are unlikely to be present as polymorphisms (e.g. Suzuki et al., 2011), making them inappropriate for use in tumours.
The reads produced by NGS platforms are increasing dramatically in length. As a result, the capacity to create DNA libraries with fragments large enough to retain an unsequenced insert is decreasing and with it the utility of PE information. Increased length also means read mappability and the utility of SR methods is improved. Here, we describe a new method for identifying somatic rearrangements, Socrates (SOft Clip re-alignment To idEntify Structural variants), which currently relies only on SR evidence. Socrates works by efficiently identifying clusters of soft clipped alignments in the reference genome as candidate break points. The soft clipped sequences are then extracted and re-aligned to the reference genome. A genomic rearrangement is predicted and break points are identified with single-nucleotide precision when soft clipped sequences from one cluster map to another with high stringency and there is reciprocal support (Fig. 1). We have developed Socrates with Bowtie2 (Langmead and Salzberg, 2012) and BWA (Li and Durbin, 2009) in mind, but it should work with any NGS aligner that supports 5′ or 3′ soft clipping. Soft clips that are too short to align are also counted, but are handled by direct search of the cluster pair.
The idea of re-aligning soft clips has previously been used in ClipCrop (Suzuki et al., 2011), but ClipCrop’s design is inappropriate for identifying genomic rearrangements in tumours, and our approach incorporates several novel features. Socrates is designed to be fast and memory efficient. It has single-nucleotide resolution and is designed to be highly sensitive. It also has high specificity on simulated data. It identifies micro-homologies and untemplated sequence at break points and deals with promiscuous or re-used break points. Our method was originally motivated by work on the Eμ–myc transgenic mouse tumour genome and the failure of existing methods to detect known fusions using PE reads from short DNA fragments, but we have tested it extensively on simulated data and human tumours. Despite its design for somatic tissue analysis, Socrates can also be used to analyze SVs in germline DNA. We compare the algorithm’s capabilities with other established break point-detection tools using simulated and real data.
2 METHODS
Socrates is intended for PE read data with reads of at least 100 nt, but will also run on SE data. It takes aligned short read data in BAM format as input. The alignment can be performed using any aligner that supports soft clipping at the 5′ or 3′ end of reads. Currently, we find Bowtie2 to be the best choice. The aligned reads are then passed through a series of processing stages (summarized in Supplementary Fig. S1):
Preprocessing: In this stage, the input BAM file is parsed and a variety of quality filters are applied, including removal of multi-mapping reads and low-quality alignments. This stage is highly configurable via command line arguments. For further details, refer to the Supplementary Material and the software documentation. Preprocessing produces a FASTQ file of long soft clip sequences and a BAM file of reads with short soft clips. The threshold between long and short soft clips is a user-defined parameter (25 nt by default).
Re-alignment: In this stage, the algorithm uses on a short read aligner to re-map the long soft clipped parts of reads. In the output BAM file, Socrates maintains the associations between the new alignment region of the long soft clipped sequence, which we refer to as the re-aligned locus, and the original alignment region, which we term the anchor.
Split read clustering: This works on the re-aligned soft clip file and produces a data structure of split-read clusters that share the same anchor and re-aligned regions. A cluster may contain one or more reads. Clustering is the most computationally expensive stage. See Section 2.1 for details.
Cluster pairing: Clusters are parsed, and cluster pairs are formed when two clusters provide reciprocal support for a potential rearrangement. This step produces part of the algorithm’s final output. See Section 2.2 for details.
Matching short soft clips: Short soft clip sequences are used to try to find reciprocal support for the remaining unpaired clusters by directly searching the cluster anchor region using short soft clip sequences derived from the re-aligned locus. This stage adds extra cluster pairs to the output, further improving sensitivity. Further details follow in Section 2.3.
2.1 Split read clustering
The clustering stage groups split reads that support a putative genomic rearrangement involving a pair of break points from one side of the fusion. It identifies a cluster of anchored reads on one side and associated re-aligned soft clips on the other side of the fusion. Socrates parses the BAM file of the re-aligned long soft clips, creating new clusters and, if necessary, merges them with already existing clusters that support the same break point. Clusters are merged if (i) their anchor loci overlap and include the same break point, and (ii) their re-aligned loci overlap and include the same break point. However, a small degree of ‘wobble’ in the soft clip start locations is observed in a low proportion of reads (beginning too soon or too late with respect to the actual break point or the consensus of soft clip starts). This appears to be associated with sequencing errors and bases with low-quality scores. To account for this, we allow merging of two clusters even if their soft clip positions are slightly different (up to 5 nt apart by default). To perform clustering a data structure containing the anchor locus (including a specific break point—the last aligned position before the soft clipping starts), the re-aligned locus (including a break point) and a voting matrix for the re-aligned and anchor locus consensus sequence is used. The voting matrix is used to call the consensus sequence on either side of the break point. The clusters are kept in a sorted data structure to keep the search operation efficient.
2.2 Cluster pairing
Reciprocal support of break point events is at the core of the Socrates algorithm and is key to reducing false-discovery rates. A potential fusion will be predicted only if reads from both sides of the break points show soft clip evidence to support the event. This technique reduces false-positive SVs resulting from singular stochastic events such as chimeras. The cluster pairing stage of the algorithm identifies such reciprocal support. If we consider two clusters C1 and C2, which consist of anchor and re-aligned regions, there are three cases to deal with:
Blunt end joining: Blunt end joining of two loci is the most straightforward case to identify (Fig. 1A). Two clusters are paired if the re-aligned region of C1 ends at the C2 anchor region start. In other words, the re-aligned C1 soft clips map to the C2 anchor region, immediately adjacent to the C2 soft-clip site. Additionally, C2 re-aligned region must coincide with the C1 anchor region with single nucleotide stringency.
Micro-homology: Micro-homologies are short (1–10 nt) identical sequences, which may be found at either side of break points. If the true break point is in a region of micro-homology, its exact location cannot be determined. Furthermore, the reciprocal support for the break point does not identify exactly the same location. Thus, in the presence of a micro-homology, the resolution is no longer single nucleotide. The reads that contribute to C1 clip exactly at the end of the micro-homology (Fig. 1B). In C2, the reads again clip after the region of micro-homology (at its start on the negative strand). As a result, the procedure for blunt end joining described above will not suffice to pair the two clusters. The break points identified by each cluster are 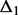 and
and 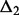 bases apart. In this case, Socrates tests whether (i) the difference in break points is consistent (
bases apart. In this case, Socrates tests whether (i) the difference in break points is consistent (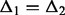 ) and (ii) the homologous sequence (the stretch between the anchor break points and re-aligned loci) is identical in both clusters (here we use the consensus sequence for comparison). If both conditions are fulfilled, the micro-homology is identified, and the two clusters are paired up.
) and (ii) the homologous sequence (the stretch between the anchor break points and re-aligned loci) is identical in both clusters (here we use the consensus sequence for comparison). If both conditions are fulfilled, the micro-homology is identified, and the two clusters are paired up.
Untemplated sequence: A third possible scenario is the presence of untemplated sequence between the two break points fusion (Fig. 1C). These are short sequences that are part of the normal genome that are generated during DNA repair. In this case, the extracted soft clip contains untemplated sequence and when re-aligned to the reference, the re-aligned soft clip sequences will themselves be soft-clipped (by Θ nt). There is an upper limit to the size of the untemplated sequence that Socrates can deal with, which depends on the read length. Socrates keeps track of this inserted sequence and includes it in the output. The insertion of untemplated sequence at a break point can also coincide with micro-homologies (Supplementary Fig. S2). In this case, the break point detection again has single-nucleotide resolution.
Finally, we point out that this scenario also applies if there are two break points in proximity to each other (fusion A to B1 and fusion B2 to C with B1 and B2 in proximity, say). In this case, the re-aligned soft clips will be placed on locus A and C omitting B as soft clips of the re-aligned locus. As a result, Socrates will detect a break point A to C with novel insertion sequence equal to B1–B2.
After successfully pairing two clusters, Socrates will parse the short soft clip input file and search directly for additional read support. This step accumulates more evidence supporting the break point and may be useful for genotyping. Every soft clip in the data that clips at the anchor loci and then matches the re-aligned consensus sequences is noted as a supporting read for the break point event.
2.3 Short soft clip cluster pairing
In the final stage of the algorithm, Socrates handles all clusters that have not been matched up as pairs in the previous stage. Socrates attempts to find supporting evidence for existing clusters by means of reciprocal short soft clip support (similar to the gathering of short soft clip support in the previous stage). This is necessary if a particular break point has long (and therefore remapped) soft clips on one side only. This may occur if there is low coverage on one side of a break point due to low mappability or high or low GC content.
In the short soft clip cluster pairing stage, the short soft clips from the re-aligned locus of unpaired clusters are extracted from the BAM file. More specifically, reads that overlap the re-aligned break point of the cluster are extracted. The algorithm then follows the same principles as the regular cluster pairing with the following difference. Because short soft clips are not remapped to the reference genome (due to the high probability of multi-mapping), they have to be compared by direct search with the cluster’s anchor locus. This is efficiently done because Socrates keeps track of the anchor locus consensus sequences. All short soft clips that match the cluster’s anchor sequence with at least 90% identity are considered as reciprocal support to the cluster. If sufficient support can be gathered this way, Socrates considers the cluster as a valid break point and includes it in the output. The level of support is included in the output for use in filtering. The same details that arise in the event of micro-homologies and untemplated sequence have to be considered in short soft clip cluster pairing as well. Short soft clip cluster pairing provides a mechanism to further increase Socrates’ sensitivity. On testing data, we found the sensitivity of the algorithm to increase by up to 15% compared with using cluster pairing only. The trade-off is a likely increase in false positives, but the minimum support parameters can be used to keep this under control. At the cost of reduced sensitivity, short soft clip pairing may be ignored by setting the short soft clip pair threshold to 0.
2.4 Implementation and algorithmic complexity
The main Socrates algorithm is implemented in java. A driver script and several utilities are implemented in python. Socrates includes a program that identifies somatic SVs from matched-tumour normal pairs, which the driver script can call. Additionally, it can also annotate whether predicted break points overlap known repeats. Socrates is designed to run efficiently on modern computing resources. The implementation supports parallelization, scaling to any number of processors on a shared memory machine and memory mapping of intermediate files, allowing for efficient usage of memory and improved speed. During preprocessing, all chromosomes are processed in parallel; in the clustering stage, chromosomal segment pairs are processed in parallel with the space distributed evenly between processors.
The most complex stages of the algorithm are the cluster pairing and short soft clip cluster pairing steps: both of them are bound by 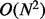 , where N is the size of the input data [comparing O(N) clusters with O(N) reads]. However, the constants for these steps are small. The most time-consuming stage of the algorithm is actually the cluster generation [which is an
, where N is the size of the input data [comparing O(N) clusters with O(N) reads]. However, the constants for these steps are small. The most time-consuming stage of the algorithm is actually the cluster generation [which is an 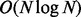 search for matches for O(N) new clusters in a sorted list]. A detailed breakdown of the theoretical complexities of Socrates’ different stages and the implementation is presented in the Supplementary Material.
search for matches for O(N) new clusters in a sorted list]. A detailed breakdown of the theoretical complexities of Socrates’ different stages and the implementation is presented in the Supplementary Material.
2.5 Testing and simulated SVs
To assess Socrates’ ability to detect structural variations and to compare it objectively with other methods, we ran Socrates on simulated data for a variety of types of genomic rearrangements. To achieve this, we developed a software tool that simulates random SVs of various types and sizes. It divides the reference genome into bins of equal sizes and creates one SV at a random location within each bin. Deletions, translocations (either moving or duplicating a random segment into the bin), inversions and tandem duplications are simulated with equal probability. The SV feature size is also chosen randomly from small (100–200 nt), medium (500–1000 nt), large (2000–10 000 nt) and extra large (20 000–100 000 nt). Additionally, tiny (5–30 nt) novel insertions were also simulated. From the resulting somatic genome, we simulated reads using SimSeq (John, 2010). These reads are then mapped to the reference genome using Bowtie2 (using the –local flag). We compared Socrates to BreakDancer (1.3) (Chen et al., 2009), CLEVER (2.0rc1) (Marschall et al., 2012), CREST (0.0.1) (Wang et al., 2011), DELLY (0.0.9) (Rausch et al., 2012), Pindel (0.2.4t) (Ye et al., 2009) and PRISM (1.1.6) (Jiang et al., 2012). These methods sample a wide range of approaches in using PE and/or SR evidence. The algorithms were typically run with default parameters (for more details see Supplementary Section S5). It should be noted that we used a consistent set of SV features to test each method. In some cases, methods did not perform well because they were not specifically designed for all SV types. We have noted this carefully where it occurs.
We also applied Socrates to several real cancer datasets: an Eμ–myc mouse lymphoma, a human melanoma and prostate cancers sequenced on an Illumina HiSeq platform using PE 100 nt reads. The Eμ–myc mice were maintained according to Peter Mac Animal Experimentation Ethics Committee protocol E352. Matched normal (buffy coat) and melanoma tissues were obtained from a consenting patient via Peter Mac Human Research Ethics Committee protocol 10/02. Experimental validation is described in the Supplementary Materials.
3 RESULTS
3.1 Evaluation on simulated SVs
To assess the performance of Socrates and several other methods, we simulated SV data and applied each tool. Simulated events were treated as genomic intervals, and we classified a predicted SV as a true positive (TP) and recalled if both ends were correct to within a small margin. For single-nucleotide resolution methods (Socrates, Pindel, CREST and PRISM), this was set to 10 bp to allow for micro-homologies around the break point sites, which unavoidably leads to loss of accuracy. For the PE methods, BreakDancer and CLEVER, which do not have single-nucleotide resolution in general, we allowed a tolerance of 250 bp. If there are redundant calls for a single event, only one was counted as a TP, the remainder were tallied (see Supplementary Table S1) and disregarded. Break points not identified are counted as false negatives (FN), and all remaining calls predicted by the algorithms as false positives (FP).
3.1.1 Escherichia coli simulations
We first simulated rearrangements in the E.coli reference genome (K12 strain). Escherichia coli was chosen for initial simulations because it has a simple and short (4.6 Mbp) genome, which should make the detection of rearrangements and other variations easy. The parameters for this simulation were 110 kbp bins (42 events in total), 300 bp DNA fragment size and a standard deviation of 30 bp. We repeated the experiment 10 times at each average coverage of 7.5, 15 and 30×.
Figure 2A (first and third panels) summarizes the precision (or positive predictive value, defined by TP/(TP + FP)) and recall (or sensitivity or true-positive rate, defined by TP/(TP + FN)) of each method on a consistent set of simulated genomic rearrangements in the E.coli genome. Increasing the coverage improves the recall for most of the methods. Pindel and PRISM particularly benefit from a higher read depth. Socrates performs well across all coverage levels. The ranking of the algorithms stays consistent across the coverage range, except for BreakDancer, which shows more robust performance at low depth of coverage. The precision of several algorithms decreased with increasing sequencing coverage. In particular, DELLY and PRISM have significantly higher false-positive rates at 30× compared with 7.5×. The precision of CREST, Pindel and Socrates is constant at about 1. We speculate that the relatively high false-positive rates for DELLY and PRISM were due to repeat events (and the misalignments that they can cause) in the genome: they tend to be clustered in focussed regions linking to various other loci in the genome. An increase in coverage is likely to produce more spurious evidence due to misalignments.
Fig. 2.

Comparison of different methods on simulated SV data. (A) Precision and recall of BreakDancer, CLEVER, CREST, DELLY, Pindel, PRISM and Socrates on simulated structural variations in E.coli and human chromosome 12. The mean precision and recall from the simulated series are plotted at 7.5, 15 and 30× coverage. (B) Detailed analysis of feature type (deletion, translocation, inversion, tandem duplication) and size [small (S), medium (M), large (L) and extra large (XL)] showing specific biases for each method on the 30× chromosome 12 data. Note that all methods are tested on a consistent set of variants, but some methods (Clever and Pindel) do not make predictions in all categories, which penalizes their performance overall in (A) and for specific classes in (B). See the text for results on novel insertions
3.1.2 Human chromosome simulations
We next tested Socrates on simulated data that are more relevant to the problem of detecting genomic rearrangements in a human cancer. A single human chromosome (Chr12) was selected as our target genome to keep runtimes short. The choice was arbitrary; however, Chr12 has a reasonably representative GC content (40%) and is interesting because it harbours a number of oncogenes such as MDM2 and CDK4, which are commonly amplified in some tumours. The main shortcoming of selecting just one chromosome is that this limits the number of paralogous and repetitive regions, especially satellite sequences, which frequently cause problems in real data. For this simulation, the bin size is 1.5 Mbp (resulting in 89 variations per simulation), the read lengths are 100 bp and the fragment length to 300 and 30 bp standard deviation. The simulation was run five times at each coverage level.
The results (Fig. 2A, second and fourth panels) show some dramatic differences to those on the E.coli genome. All algorithms, but particularly Pindel and PRISM, show lower recall, which is expected, given the higher complexity of the reference genome. A notable drop in recall is visible for the lowest coverage for CREST, Pindel and Socrates. The effect is weaker for BreakDancer, DELLY and PRISM. BreakDancer in particular holds its recall well with low coverage (and overall compared with E.coli), making it the most sensitive algorithm at 7.5×, followed by DELLY and then Socrates. CREST, Pindel and Socrates perform consistently at about 100% precision, and PRISM follows the same trend as in E.coli. DELLY, however, shows an average precision that is 40% lower than in E.coli across all coverage depths. We attribute this to the increased complexity of the genome in terms of repetitive sequence, which seems to be the main source of false-positive calls for DELLY.
3.1.3 Effect of SV type and size on false negatives
The simulations also provided us with an opportunity to investigate the effect of SV size and type on false negatives across the different methods. This provides insight into the strengths and weaknesses of each algorithm. Here, we evaluate simulations based on human Chr12 at 30×, but the results were comparable across all experiments. Figure 2B shows recall for each of the algorithms (see Supplementary Table S2 for FP, FN and total counts). We show results in all categories for CLEVER, but it is only designed for deletions and insertions. Evaluating the different variation types, we observe:
Deletions: Socrates and PRISM show the best recall for deletions with only 7 out of 117 deletions missed. CREST and Pindel show a uniform FN pattern across the entire range at roughly the same level. Deletions are CLEVER’s best category. It achieves a recall of 82%. There is a significant spike for large deletions being missed by CLEVER (it is designed to detect insertions and deletions from 20–50,000 bp). BreakDancer struggles with short deletions.
Translocations: This category consists of both randomly copied and moved genome segments. CLEVER and Pindel were not designed to detect translocations. Unsurprisingly, CLEVER finds no translocations, but Pindel does detect some as insertions. The other algorithms were effective in this category.
Inversions: DELLY is the most effective tool to predict inversions. It misses only 2 out of 180 break points. Socrates is competitive and misses only 7. PRISM has low recall for short inversions. CLEVER is not designed to predict inversions and finds none.
Tandem duplications: DELLY, PRISM and CLEVER have issues with short tandem duplications (shorter than the simulated fragment length). This would be due to the mapping distance of paired ends not being evaluated as significantly discordant. CREST and Socrates also show lower recall for short duplications (as it is difficult for the aligner to place reads within the sequence). BreakDancer shows the best performance for tandem duplications (it misses only two for the entire data)–despite having to rely exclusively on paired end information, which can be difficult to interpret for short feature sizes. Despite not explicitly handling them, CLEVER performs well on large tandem duplications, particularly
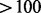 nt, treating them as insertions. Pindel does not predict any features in this category.
nt, treating them as insertions. Pindel does not predict any features in this category.
Novel insertions of tiny size were also simulated. Because they occur in only one size category (5–30 nt), we do not show these in Figure 2. PRISM performs best for novel insertions and recalls 97% of them, followed by Pindel (93%) and Socrates (65%). The other algorithms do not predict non-templated inserts at all—most of the algorithms assessed here were not designed to predict such features to begin with. We speculate that CREST fails at the BLAT alignment stage, because small novel sequences complicate the alignment results. BreakDancer is not able to detect a significant aberration in insert sizes of PE reads at this small size level, and this is just at the cusp of where CLEVER operates. Socrates only comes third in this category but is the only algorithm that actually outputs the inserted sequence, instead of just flagging a break point.
3.1.4 Mapping algorithm dependency
All the experiments discussed here are conducted with reads that were mapped with the Bowtie2 (2.0.6). The ‘–sensitive-local’ option was used (which is the default configuration for local alignments. The local alignment mode is beneficial to obtaining good soft clipping results around SVs). However, other aligners, such as BWA, can also be used. We found BWA to be less sensitive to outputting soft clipped reads when run with standard parameters. The recall of Socrates in simulations on chr12 with 15× coverage drops from >81 to 67%. Similarly, the recall achieved by CREST decreases from 68 to 61%. DELLY did not suffer from the same decrease in sensitivity—probably due to its relying more on paired end evidence than soft clipped reads. DELLY’s recall remained the same or even increased slightly when using BWA mappings. BWA can be run in the more sensitive alignment mode ‘bwasw’ that employs Smith–Waterman style alignments for increased accuracy, but we found it too slow for a whole genome scale.
3.1.5 Simulation results summary
A range of types of SV detection algorithms were tested on randomly generated variations in two organisms. Experiments were replicated to ensure reproducibility of the results, and two different sequence simulators [SimSeq and wgsim (data not shown)] were tested with consistent results. The analysed algorithms can be broadly distinguished in two classes: (i) methods that use and in some cases are primarily reliant on fragment coverage (PE information) and (ii) methods relying on sequence coverage (SR methods). Interestingly though somewhat predictably, the results reveal that the sequence coverage-dependent algorithms (CREST, Pindel, Socrates) show a larger decline in recall than the other tools as coverage decreases. However, the specificity of the SR methods is better across the experiments. We attribute this to the more stringent nature of the evidence. SR methods require multiple reads to support an event at (nearly) a single nucleotide location.
We were particularly interested in the comparison between Socrates, CREST and Pindel. While distinct in their approaches, they are most comparable in design (making use of sequence coverage and soft clipping). The three methods are similar in their overall precision and dependency of recall on coverage. Socrates shows highest sensitivity between the three methods throughout the experiments, which was a key focus of the algorithm’s design. CREST and Pindel are more conservative methods that potentially require more evidence to recall rearrangement events (for example, CREST struggles with tandem duplication and inversion events at a size level that reduces the evidence available to the algorithms).
3.2 Application to tumour genome data
We next tested Socrates on several real cancer datasets. The first was derived from the Eμ–myc transgenic mouse, which spontaneously develops lymphomas (Adams et al., 1985). The structure of the Eμ–myc transgene is well described (Adams et al., 1985; Corcoran et al., 1985). It consists of the c-myc gene in a pUC12 cloning vector with the Eμ promoter of the IgH gene inserted upstream and the  X174 bacteriophage inserted into the 3′ UTR (Fig. 3). We sequenced the genome of an Eμ–myc tumour and a DNA sample from the tail of the same mouse. The sequencing data consisted of 100 nt paired end Illumina reads with coverage of approximately 30× for each sample and average fragment size of 213 nt. The small fragment size means that about half of the fragments have overlapping PE reads, and this may be an issue for methods using PE information. We applied Socrates and several other break point-detection tools to the Eμ–myc data (Table 1). Socrates detected the three known genomic fusions (B, C and F), as did CREST and Breakdancer, while DELLY did not detect any of these. Only Socrates and CREST were able to identify the insertion site of the transgene into chr19. This is a novel finding. Socrates also identified several novel fusions that were not previously known. None of the methods detected the
X174 bacteriophage inserted into the 3′ UTR (Fig. 3). We sequenced the genome of an Eμ–myc tumour and a DNA sample from the tail of the same mouse. The sequencing data consisted of 100 nt paired end Illumina reads with coverage of approximately 30× for each sample and average fragment size of 213 nt. The small fragment size means that about half of the fragments have overlapping PE reads, and this may be an issue for methods using PE information. We applied Socrates and several other break point-detection tools to the Eμ–myc data (Table 1). Socrates detected the three known genomic fusions (B, C and F), as did CREST and Breakdancer, while DELLY did not detect any of these. Only Socrates and CREST were able to identify the insertion site of the transgene into chr19. This is a novel finding. Socrates also identified several novel fusions that were not previously known. None of the methods detected the 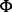 X174 insertion (Fig. 3, DE), because the
X174 insertion (Fig. 3, DE), because the 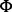 X genome was omitted from the reference sequence. Overall, Socrates dramatically outperformed the other methods on these data.
X genome was omitted from the reference sequence. Overall, Socrates dramatically outperformed the other methods on these data.
Fig. 3.
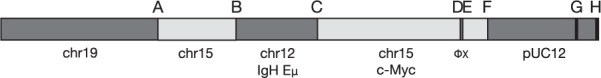
Structure of the Eμ–myc transgene. Sizes of regions are not to scale, but are indicative. A–H indicate fusions; B–F are known fusions; A and H are novel. Fusion G is the end of the pUC12 reference sequence and is due to the circular topology of the cloning vector. The break point at A is promiscuous, linking chr15 to chr9 and H
Table 1.
Detection of fusions associated with the Eμ–myc transgene by different methods
| Algorithm | A | B | C | F | G | H |
|---|---|---|---|---|---|---|
| 19–15 | 15–12 | 12–15 | 15–pUC12 | pUC12–pUC12 | pUC12–15 | |
| Socrates | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| CREST | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ |
| BreakDancer | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ |
| DELLY | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ |
Note: Fusions B–F are known (Fig. 3); others are novel or inferred.
The main reason for the poor performance of the other methods is likely to be the unfortunately small average fragment size. This means that the unsequenced region between reads is on average only 13 nt long and that about half of the reads actually overlap. This greatly reduces the usable coverage available to the PE methods such as BreakDancer and the first stage of DELLY’s alignment, thus reducing their sensitivity. DELLY in particular is not designed to detect inter-chromosomal fusions, so it can only detect the break point on the cloning vector. The complexity of break point H is particularly challenging for the methods; it is promiscuous (sharing one coordinate with the fusion to chr19), and features a 10-bp novel insertion between chr15 and pUC12. Only Socrates is equipped to recover this break point from the data.
We also applied Socrates to six prostate cancer tissue samples and six matched whole-blood samples. Each sample was sequenced to an average of 40× coverage (100 bp paired end). Tumour break point predictions were filtered using whole-blood predictions to provide an average of 4360 break points per sample using Socrates’ most sensitive settings (1 cluster containing 1 long soft clip and 1 cluster containing 1 short soft clip). In addition to DNA sequencing, RNA sequencing was performed using benign tissue as a matched normal, with an average of 120 million reads per sample (100 bp paired end). Gene fusion candidates for each sample were determined using defuse by McPherson et al.(2011) on the RNA reads. On average, 315 candidate fusions were predicted for each sample. After filtering with default parameters to enrich for high-confidence fusions (McPherson et al., 2011), defuse predicted 86 fusions per sample on average. This is expected to contain a large number of false positives. To improve performance, Socrates’ break points were used to determine true fusion events via support by matched rearrangements in the DNA. As the RNA-based fusions are determined using mature RNA transcripts, the break point resolution is at exon level. To relate DNA break points to RNA fusion points, we looked for break points between the fused exon, and the next annotated exon (unfused). This resulted in an average of three fusions per sample that were supported by both RNA and DNA. Interestingly, only 60% of these validated fusions appeared in the high-confidence enriched list produced using RNA and defuse filtering parameters. In this case, Socrates provides the ability to find fusion events that would not normally pass filtering. Furthermore, by using Socrates predictions to filter fusions, we were able to detect fusion events with as little as 4 reads support in the RNA. This highlights the ability of Socrates to improve gene fusion detection. Table 2 highlights the comparison results between Socrates and defuse.
Table 2.
Comparison of Socrates and defuse results
| Sample | All | defuse | Socrates | Overlap (%) |
|---|---|---|---|---|
| High confidence | ||||
| 1 | 340 | 84 | 6 | 5 (83%) |
| 2 | 183 | 38 | 4 | 1 (25%) |
| 3 | 431 | 134 | 3 | 3 (100%) |
| 4 | 276 | 72 | 2 | 0 (0%) |
| 5 | 220 | 68 | 1 | 1 (100%) |
| 6 | 440 | 121 | 2 | 1 (50%) |
| Average | 315 | 86 | 3 | 2 (60%) |
Note: Socrates adds increased sensitivity and single-nucleotide resolution to the RNA-seq data analysis.
Socrates is designed to be highly sensitive. We have found this to be extremely useful when integrating different sources of data (e.g. the known and inferred fusions in the Eμ–myc transgene and RNA-seq data in prostate cancer). Using simulated data, we also found it to be highly specific; however, on real tumour sequence data using its most sensitive settings without additional filtering, it generates large numbers of predicted break points and we assume these contain a large number of false positives. On the Eμ–myc tumour using the most sensitive parameter settings, Socrates called >36 000 break points. This is not an isolated problem: DELLY predicted >385 000 and BreakDancer >5000. In the absence of additional data or models, more stringent thresholding of coverage is needed.
On a human melanoma, sequenced to 60× coverage with matched normal sequenced to 30×, Socrates predicted about 105 000 SVs in the tumour when run using its most sensitive settings (in comparison Delly predicted ∼150 000; BreakDancer 7800). Increasing the stringency to two long soft clips on each side of the break point reduces the number of breakpoints to 6992, of which 678 were somatic. We randomly chose 10 break points from these somatic events for validation. Seven were validated unambiguously by PCR and sequencing (Supplementary Fig. S9); in one case the Sanger sequencing extended to just 1 bp past the break point and the result is inconclusive, and in the other two cases, further optimization to PCR conditions is required. Sanger sequencing also validated short insertions of untemplated sequence in between one of the break points that was predicted by Socrates. DELLY recovered just two of the validated break points, and BreakDancer recovered one. Increasing the threshold to five long soft clips further reduces the number of somatic events to 111, but three of the validated break points are now missed. We are thus satisfied to work with a threshold of two long soft clips. More careful control of false-positive and false-negative rates awaits large scale unbiased validation of break points. Short soft clips remain useful to more accurately estimate the absolute support and the mutant allele frequency (MAF) of break points.
It is interesting to examine the genomic context of the predicted SVs. Table 3 shows the absolute and relative abundance of different repeat classes overlapping the Socrates predictions. The labels refer to repeats that contain either of the two cluster coordinates (or are within 10 nt of these). The results show that a disproportionate number of events are predicted in repetitive regions: only 13% of the somatic break points are not affected by repeats. This number increases to 33% after basic filtering. Satellite repeats make up a large proportion of the raw output and are reduced to 10% after filtering. We also observed that break points in satellite regions and simple repeats tend to have low MAF, which are inconsistent with expected copy number ratios. This may suggest that they are false positives that arise due to stochastic events such as chimeric sequence formation in these repetitive regions and leads to the possibility of filtering based on MAF (see Supplementary Material for further discussion).
Table 3.
Summary of Socrates predictions in different classes of repetitive regions
| Repeat class | Sensitive |
Filtered |
||
|---|---|---|---|---|
| Normal | Somatic | Normal | Somatic | |
| Non-repetitive | 5509 | 10 957 (13%) | 3176 | 226 (33%) |
| LINE | 2338 | 8630 (10%) | 1114 | 123 (18%) |
| Low complexity | 195 | 1483 (2%) | 63 | 17 (3%) |
| LTR | 1052 | 2726 (3%) | 555 | 42 (6%) |
| Satellite | 8759 | 36 315 (43%) | 214 | 69 (10%) |
| Simple repeat | 2734 | 11 069 (13%) | 571 | 122 (18%) |
| SINE | 1532 | 12 461 (15%) | 621 | 79 (12%) |
Note: Sensitive means at least one long soft clip and one short soft clip supporting the fusion. Filtered means at least two long soft clips on each side.
3.3 Performance
Finally, we assessed the speed of Socrates. In the runtime experiments presented here, we used up to eight threads in parallel for the cluster-generation stage. All the experiments on Chr12 were run on different datasets to avoid caching advantages for one algorithm over the other. The other two compared algorithms do not offer ad hoc parallelization—CREST is run in parallel on all the chromosomes on the Eμ–myc tumour data, giving it 22 threads to compute. Table 4 summarizes the results. Socrates is an order of magnitude or more faster than the other methods. In terms of memory, all the algorithms are essentially bound by the same theoretic complexity—the size of the input data. Socrates uses slightly more than CREST and DELLY, but even on a whole-genome scale analysis not more than a modern desktop computer would provide.
Table 4.
Resource consumption comparison between the competing algorithms on simulated data and real sequencing reads from a cancer data set
| Algorithm | Chr12 30× simulation |
Eμ–myc |
||
|---|---|---|---|---|
| Run time (min) | Max memory (Mb) | Run time (h) | Max memory (Gb) | |
| CREST | 87 | 483 | >50a | NA |
| DELLY | 53 | 337 | 33 | 8.6 |
| Socrates | 3 | 550 | 4 | 10.1 |
aCREST failed during the soft clip extraction stage after running over 50 h, so the timing represents a lower bound. Runtimes are wall clock measures.
4 DISCUSSION
Socrates is a new break point-detection method based on split reads. It is fast and memory efficient. The main innovations in Socrates are the automatic detection of micro-homologies and untemplated sequences at fusion sites, and some of the details of its implementation leading to its high speed. On simulated data, Socrates recovers rearrangements across a broad range of types and sizes. It is specific and highly sensitive. Its high sensitivity makes it a powerful tool in the presence of additional information, and we find it is complementary to more conservative methods. On real tumour data without additional information, we find it impractical to run at its most sensitive settings, but it is easily tuned. In the future, we plan to incorporate additional types of evidence into the rearrangement prediction.
As NGS read lengths have increased, rearrangement prediction methods have evolved from PE methods towards split read and more recently hybrid methods. As this trend continues, it is becoming more difficult to generate libraries of fragments that are large enough. This is especially true with small quantities of DNA and Formalin-Fixed Parafin-Embedded (FFPE) tumour samples. Thus, we may see a resurgence of pure split read approaches. These offer higher sensitivity over the RP-guided hybrid approaches because they can analyze the smallest deviations from the reference genome on a single nucleotide level. In cancers, sensitivity is also key to detect, and make sense of, sub-clonality. High sensitivity has been a key design principle behind Socrates.
Funding: This work was supported by a National Health and Medical Research Council (NHMRC) Project Grant (637357), Victorian State Government Operational Infrastructure Support, Australian Government NHMRC IRIISS, the Melbourne Melanoma Project, the Victorian Cancer Biobank and the Victorian Cancer Agency. S.B. was supported by a Cancer Council of Victoria scholarship. G.M. and M.C. were supported by NICTA. NICTA is funded by the Australian Government as represented by the Department of Broadband, Communications and the Digital Economy and the Australian Research Council through the ICT Centre of Excellence program. R.W.J. was supported by an NHMRC Principal Research Fellowship, as well as The Cancer Council Victoria, The Leukaemia Foundation of Australia and the Victorian Cancer Agency. M.S. was supported by fellowships from Pfizer Australia and VESKI. A.T.P. was supported by an NHMRC Career Development Fellowship (1003856) and The Lorenzo and Pamela Galli Melanoma Research Fellowship.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Abyzov A, et al. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JM, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran LM, et al. Transposition of the immunoglobulin heavy chain enhancer to the myc oncogene in a murine plasmacytoma. Cell. 1985;40:71–79. doi: 10.1016/0092-8674(85)90310-1. [DOI] [PubMed] [Google Scholar]
- Greenman CD, et al. Estimation of rearrangement phylogeny for cancer genomes. Genome Res. 2012;22:346–361. doi: 10.1101/gr.118414.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, et al. PRISM: pair-read informed split-read mapping for base-pair level detection of insertion, deletion and structural variants. Bioinformatics. 2012;28:2576–2583. doi: 10.1093/bioinformatics/bts484. [DOI] [PubMed] [Google Scholar]
- John JS. Simseq. 2010 https://github.com/jstjohn/SimSeq (17 January 2014, date last accessed) [Google Scholar]
- Karakoc E, et al. Detection of structural variants and indels within exome data. Nat. Methods. 2011;9:176–178. doi: 10.1038/nmeth.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschall T, et al. Clever: clique-enumerating variant finder. Bioinformatics. 2012;28:2875–2882. doi: 10.1093/bioinformatics/bts566. [DOI] [PubMed] [Google Scholar]
- McPherson A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-seq data. PLoS Comput. Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, et al. Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods. 2009;6(Suppl. 11):S13–S20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- Medvedev P, et al. Detecting copy number variation with mated short reads. Genome Res. 2010;20:1613–1622. doi: 10.1101/gr.106344.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, et al. ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS One. 2011;6:e16327. doi: 10.1371/journal.pone.0016327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, et al. ClipCrop: a tool for detecting structural variations with single-base resolution using soft-clipping information. BMC Bioinformatics. 2011;12(Suppl. 14):S7. doi: 10.1186/1471-2105-12-S14-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods. 2011;8:652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, et al. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.