

Abstract
Motivation: Accurately identifying and eliminating allergens from biotechnology-derived products are important for human health. From a biomedical research perspective, it is also important to identify allergens in sequenced genomes. Many allergen prediction tools have been developed during the past years. Although these tools have achieved certain levels of specificity, when applied to large-scale allergen discovery (e.g. at a whole-genome scale), they still yield many false positives and thus low precision (even at low recall) due to the extreme skewness of the data (allergens are rare). Moreover, the most accurate tools are relatively slow because they use protein sequence alignment to build feature vectors for allergen classifiers. Additionally, only web server implementations of the current allergen prediction tools are publicly available and are without the capability of large batch submission. These weaknesses make large-scale allergen discovery ineffective and inefficient in the public domain.
Results: We developed Allerdictor, a fast and accurate sequence-based allergen prediction tool that models protein sequences as text documents and uses support vector machine in text classification for allergen prediction. Test results on multiple highly skewed datasets demonstrated that Allerdictor predicted allergens with high precision over high recall at fast speed. For example, Allerdictor only took ∼6 min on a single core PC to scan a whole Swiss-Prot database of ∼540 000 sequences and identified <1% of them as allergens.
Availability and implementation: Allerdictor is implemented in Python and available as standalone and web server versions at http://allerdictor.vbi.vt.edu.
Contact: lawrence@vbi.vt.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Allergy is one of the most important chronic diseases worldwide. It is also one of the main causes of asthma and asthma exacerbations, which has been an increasing health issue in developed countries (Devereux, 2006). Allergic hypersensitivity (IgE-type response) in sensitized individuals is elicited by allergens. The allergen–IgE interaction often results in mast cells and/or basophils releasing multiple inflammatory mediators such as histamine, leukotrienes, cytokines and chemokines. These mediators can cause a variety of symptoms from mild to severe including sneezing, itching, rashes, hives, difficulty in breathing and asthma attacks that can lead to death (Masoli et al., 2004; Stagg et al., 2013).
It is important to identify and eliminate potential allergens from biotechnology-derived products, such as genetically modified crops, vaccines and therapeutics, as well as identifying allergens from sequenced genomes. However, IgE-mediated allergenicity is costly and difficult to assess without human data because no single factor has been recognized as a primary identifier for allergenicity (Ladics et al., 2011; Stagg et al., 2013). Therefore, bioinformatics approaches have been widely used to prescreen novel sequences (Mari et al., 2009). The FAO/WHO guideline to assess allergenicity of genetically modified crops uses relaxed sequence similarity criteria. A protein is identified as a potential allergen if it harbors >35% identity with a known allergen over a window of 80 amino acids or has six contiguous amino acids that are also found in a known allergen (FAO/WHO, 2001; Metcalfe, 2005). These criteria are implemented in most of the allergen databases and tools (Mari et al., 2009). However, the FAO/WHO guideline focuses on sensitivity to prevent potential new allergens entering the food market rather than accurate prediction. Therefore, these criteria yield high false-positive (FP) rates such that their application is limited (Ladics et al., 2011; Stadler and Stadler, 2003). The current Codex guideline (Codex Alimentarius Commission, 2009) does not recommend the use of the six contiguous amino acid match criterion.
Many methods for allergen prediction have been developed and are more accurate than the FAO/WHO pure sequence similarity-based approach. The majority of these methods is based on supervised machine learning and differs in ways to extract useful features from amino acid sequences. Most of them rely on sequence similarity to allergen-specific peptides or motifs, including Stadler and Stadler (2003), Li et al. (2004), WebAllergen (Riaz et al., 2005), EVALLER (Barrio et al., 2007; Soeria-Atmadja et al., 2006) and SORTALLER (Zhang et al., 2012), or to known IgE epitopes, such as AlgPred (Saha and Raghava, 2006), or with known allergens and putative non-allergens, such as AllerHunter (Muh et al., 2009). Other methods use physicochemical representation of protein structure, such as APPEL (Cui et al., 2007) and the structural database of allergen proteins (SDAP) (Ivanciuc et al., 2009), or amino acid/dipeptide composition, such as AlgPred (Saha and Raghava, 2006).
Although current methods are significantly more accurate than the FAO/WHO approach, large-scale allergen prediction using these methods is still ineffective and inefficient. On large-scale data where non-allergens are naturally more abundant, the number of FP often exceeds the number of true positives (TP) that lowers the precision and thus the usefulness of the prediction. Moreover, the most accurate methods are relatively slow, as they rely on homology and use sequence alignment to construct feature vectors. Additionally, current allergen prediction methods come pretrained in the form of web servers without the capability of large batch submission making large-scale allergen prediction even more difficult.
In this article, we propose a new sequence-based allergen prediction method (Allerdictor) that can run in linear time of sequence length and is capable of producing high precision over high recall, even on highly skewed data. Allerdictor models sequences as text documents in which words are represented as overlapping k-mers generated from the sequences. We found that the k-mer approach is particularly effective in allergen prediction. Feature construction is much faster than sequence alignment-based methods and can be performed in linear time of sequence length. Allerdictor was implemented with both naive Bayes (NB) and support vector machine (SVM) classifiers. SVM outperformed NB on more difficult datasets where the level of sequence similarity between allergens and non-allergens is higher. Thus, we will mostly discuss results for the SVM-based version.
The advantages of Allerdictor make it practical for large-scale allergen prediction in applications such as whole-genome annotation, biotechnology-derived gene product screening and allergen discovery from large public sequence databases.
2 METHODS
Allerdictor represents sequences as text documents and uses NB or SVM for allergen classification.
2.1 Datasets
An initial set of allergens was built by combining sequences collected from the International Union of Immunological Societies allergen nomenclature (http://allergen.org), Allergome (Mari et al., 2009), SDAP (Ivanciuc et al., 2003), AllergenOnline (http://allergenonline.org) and AllerMatch (Fiers et al., 2004) databases. Duplicated sequences and sequences without experimental evidence, containing non-standard amino acids, or shorter than 100 amino acids were removed, and this resulted in a set of 3 907 high-quality allergen sequences. A portion of this set contains isoforms of the same allergens or allergens with similar sequences. A putative non-allergen set was created from the Swiss-Prot database (Magrane and Consortium, 2011) by removing sequences tagged with ‘predicted’ or ‘uncertain’ and sequences annotated with allergen-related keywords (‘allerg*’, ‘antigen’ or ‘atopy’) similar to other approaches (Barrio et al., 2007; Muh et al., 2009; Soeria-Atmadja et al., 2006; Zhang et al., 2012). Because many allergens have yet to be identified, this putative non-allergen set may contain some true allergens (noise). Noise was reduced by further removing sequences that were highly similar to any of the sequences collected from the allergen databases [≥90% identity and ≥90% coverage on both query and subject sequences when aligned using BLAST (Altschul et al., 1997)]. Similar to the allergen set, sequences shorter than 100 amino acids or having non-standard amino acids were also removed. This resulted in a set of 464 101 putative non-allergens. From the sets of 3907 allergens and 464 101 putative non-allergens, three datasets were derived and used in this study by the following procedures: All three datasets described later in the text were designed to contain 10 times as many non-allergens as allergens, which represents the natural imbalanced distribution of allergens and non-allergens to some degree. Sequence-based allergen prediction methods often yield low performance on datasets that include many non-allergens that share sequence similarity with allergens. Our datasets exhibited low to high levels of sequence similarity between allergens and non-allergens (Fig. 1) and thus allowed a more comprehensive evaluation of Allerdictor.
Dataset A (Allerdictor-A, 3907 allergens, 39 070 non-allergens): All allergen sequences (including isoforms) were selected and 10 times that of putative non-allergen sequences were randomly selected from the putative non-allergen set. This dataset exhibited a low level of overall sequence similarity between allergens and non-allergens. When clustered using BLASTClust (Altschul et al., 1997), only 1108 (∼3%) non-allergens together with 1293 (∼30%) allergens were grouped in 131 clusters that contained both allergens and non-allergens (allergen/non-allergen clusters) (Fig. 1). The non-allergen sequences that were clustered with allergen sequences were designated as ‘allergen-like non-allergens’.
Dataset B (Allerdictor-B, 1990 allergens, 19 900 non-allergens): All allergens were clustered using BLASTClust with ≥95% identity and ≥95% coverage on both query and subject sequences into 1990 clusters. To remove sequence redundancy, only one sequence was selected randomly from each cluster to form a set of 1990 allergens. Ten times as many non-allergen sequences were randomly selected from the putative non-allergen set. This dataset also exhibited a low level of sequence similarity between allergens and non-allergens, with only 534 (∼3%) non-allergens clustered with 473 (∼24%) allergens in 91 allergen/non-allergen clusters (Fig. 1).
Dataset C (Allerdictor-C, 1662 allergens, 16 620 non-allergens): All allergen and putative non-allergen sequences were together clustered using BLASTClust with ≥50% identity and ≥50% coverage on both query and subject and resulted in 291 allergen-only clusters, 233 allergen/non-allergen clusters and 9529 non–allergen-only clusters. From each allergen-only and allergen/non-allergen clusters, at most three allergen sequences were randomly selected. Ten times the number of non-allergen sequences were selected in similar fashion from the non–allergen-only and allergen/non-allergen clusters. The final dataset contained 1662 allergens and 16 620 non-allergens, in which a significant number of non-allergens share sequence similarity to allergens. When being re-clustered using BLASTClust, 6855 (∼41%) non-allergens were grouped together with 725 (∼44%) allergens in 232 allergen/non-allergen clusters (Fig. 1).
Fig. 1.
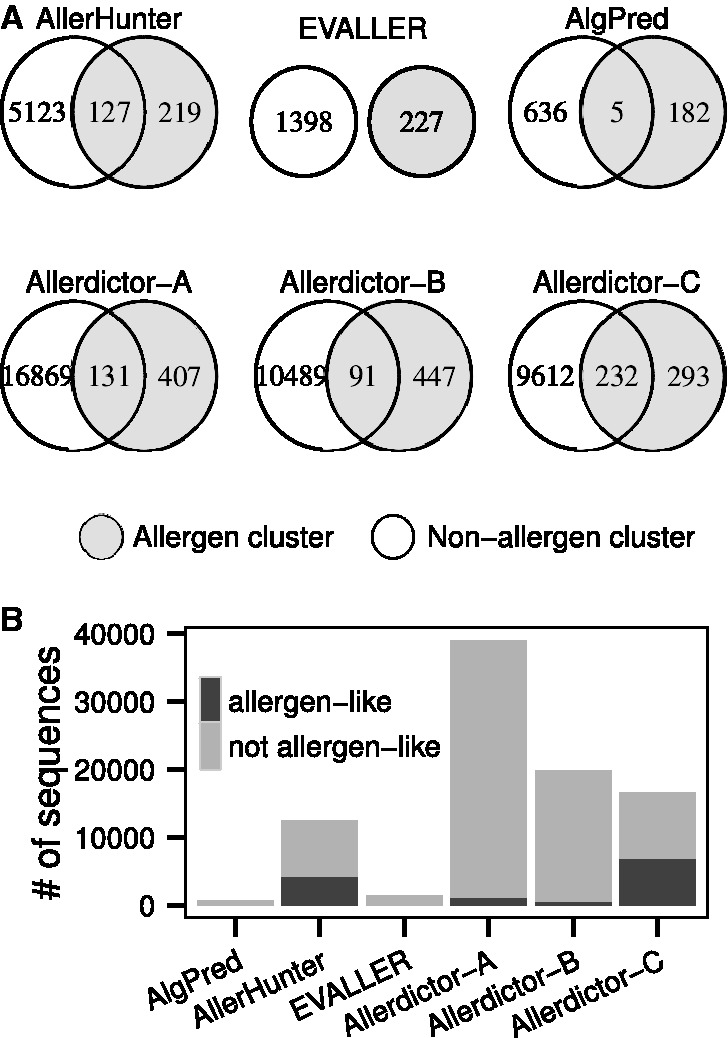
Sequence similarity between allergens and non-allergens in Allerdictor datasets and other datasets (BLASTClust cutoff ≥50% sequence identity for ≥50% query or subject coverage). The shared regions in Venn diagrams (A) are clusters that contain both allergen and non-allergen sequences. The total number of non-allergens that are allergen-like and not allergen-like are detailed in the column plot (B)
2.2 Text representation of sequences
To represent an amino acid sequence of length n as a text document, Allerdictor uses a small sliding window of size k to break the sequence into 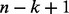 overlapping k-length peptides (k-mers). This collection of k-mers is used as a new sequence representation. If we consider a k-mer as a word, this representation is similar to the bag-of-words in document modeling (Manning et al., 2008). The set of all unique k-mers generated from training data is similar to the dictionary used in text modeling and herein called a k-mer dictionary. The feature vector for a sequence can be constructed by recording the appearance/absence of the k-mers (binary representation) or counting the frequencies of the k-mers (k-mer frequency representation). Given that N is the size of the k-mer dictionary built from training data, the k-mer frequency vector for a sequence is as follows:
overlapping k-length peptides (k-mers). This collection of k-mers is used as a new sequence representation. If we consider a k-mer as a word, this representation is similar to the bag-of-words in document modeling (Manning et al., 2008). The set of all unique k-mers generated from training data is similar to the dictionary used in text modeling and herein called a k-mer dictionary. The feature vector for a sequence can be constructed by recording the appearance/absence of the k-mers (binary representation) or counting the frequencies of the k-mers (k-mer frequency representation). Given that N is the size of the k-mer dictionary built from training data, the k-mer frequency vector for a sequence is as follows:
| (1) |
where xi is the frequency of emitting the ith k-mer of the dictionary from the sequence using the sliding window. Because only a small fraction of the k-mer dictionary can be generated from a limited length protein sequence, the k-mer feature vector is extremely sparse with the maximum of 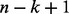 non-zero elements.
non-zero elements.
The k-mer representation of sequences also shares similar properties with the bag-of-words approach in text modeling (Joachims, 2002) such as (i) the feature space is high dimensional (the number of possible unique k-mers is 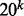 with 20 amino acid alphabet size), (ii) feature vectors are sparse and (iii) the distribution of k-mer frequencies follows Zipf’s law (Zipf, 1949) in which the number of rare k-mers is much higher than the number of frequent k-mers (data not shown).
with 20 amino acid alphabet size), (ii) feature vectors are sparse and (iii) the distribution of k-mer frequencies follows Zipf’s law (Zipf, 1949) in which the number of rare k-mers is much higher than the number of frequent k-mers (data not shown).
Many text classification methods can then be applied on k-mer sequence representation. NB and SVM were chosen for Allerdictor because they were among the best methods for text classification and fast on high dimensional sparse vectors.
2.3 Naive Bayes
NB is a simple yet effective method for text classification, especially for spam filtering (Manning et al., 2008). Using a multinomial NB model, Allerdictor-NB models the distributions of k-mer frequencies over allergen/non-allergen classes with a relaxed assumption that k-mer frequencies are independent of each other given the class. The probability of being an allergen for a sequence represented by a k-mer frequency vector X in (1) is given as follows:
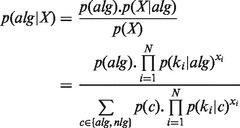 |
(2) |
where alg and nlg are allergen and non-allergen classes, respectively, and ki is the ith k-mer in the dictionary. The probability of seeing the ith k-mer in the allergen/non-allergen class 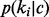 and the prior probability of the classes p(c) can be estimated from training data of known allergen and non-allergen sequences. The probability that the sequence is a non-allergen
and the prior probability of the classes p(c) can be estimated from training data of known allergen and non-allergen sequences. The probability that the sequence is a non-allergen 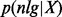 can be calculated by a similar formula.
can be calculated by a similar formula.
2.4 Support vector machine
SVM has been successfully used in numerous applications across many fields including text classification (Boser et al., 1992; Burges, 1998; Cortes and Vapnik, 1995; Joachims, 2002). Allerdictor-SVM uses a linear SVM model, and k-mer frequencies are further normalized by the total number of k-mers generated from the sequence by the sliding window. The normalized vector 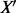 of a k-mer frequency vector X given in (1) of a sequence of length n is as follows:
of a k-mer frequency vector X given in (1) of a sequence of length n is as follows:
| (3) |
Each sequence represented by 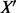 is now a point in an N-dimensional space. Given a training dataset of M sequences
is now a point in an N-dimensional space. Given a training dataset of M sequences 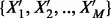 labeled with
labeled with 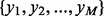 (yi = 1 if Xi is allergen, yi = -1 otherwise), a soft margin linear binary SVM classifier finds the optimal hyperplane h that separates allergens from non-allergens with the maximum margin of classification, which is equivalent to solving the following:
(yi = 1 if Xi is allergen, yi = -1 otherwise), a soft margin linear binary SVM classifier finds the optimal hyperplane h that separates allergens from non-allergens with the maximum margin of classification, which is equivalent to solving the following:
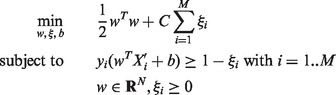 |
(4) |
where N-dimensional vector w is the normal vector of h, 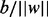 is the distance from the origin to h, slack variables
is the distance from the origin to h, slack variables 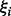 designate how far the points can pass the margin boundaries (misclassification) in cases of non-linear separable data and C is the regularization constant to control how much of the training data can be misclassified.
designate how far the points can pass the margin boundaries (misclassification) in cases of non-linear separable data and C is the regularization constant to control how much of the training data can be misclassified.
A new sequence 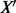 is then classified by what ‘side’ of h it lays via an SVM score (with positive score being an allergen):
is then classified by what ‘side’ of h it lays via an SVM score (with positive score being an allergen):
| (5) |
This score is then converted to a posterior probability of being an allergen by fitting a sigmoid function (Lin et al., 2007; Platt, 1999). Training and testing were conducted using SVMLight software (Joachims, 1999) with an allergen misclassification penalty weight parameter 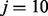 to address data imbalance. The regularization constant C was chosen by optimizing the performance via cross-validation described later in the text.
to address data imbalance. The regularization constant C was chosen by optimizing the performance via cross-validation described later in the text.
2.5 Cross-validation and dimension reduction
Nested 10-fold cross-validation was used to evaluate Allerdictor performance on three datasets A, B and C. Each dataset was randomly partitioned into 10 subsets containing roughly equal number of both allergen and non-allergen sequences. In each evaluation fold, one subset was held out (test set) and the rest nine subsets were combined and randomly partitioned into 10 other subsets for an inner 10-fold cross-validation to choose the best parameters. Mutual information (Manning et al., 2008) was used to generate feature selection scores. All k-mers were ranked by mutual information between the class variable (allergen/non-allergen) and k-mer frequency variables, and the top ranked k-mers were selected to build the prediction model. A feature abstraction technique was also used to group k-mers with the same frequency distribution in an allergen training set and in a non-allergen training set. This is a special case of distributional clustering that has been used successfully in text classification (Baker and McCallum, 1998; Pereira et al., 1993). The k-mers that were grouped together have the same frequency distribution over the allergen/non-allergen classes (observed from training data), and therefore, they received the same weights in the classification model.
3 RESULTS AND DISCUSSION
Evaluation results of Allerdictor on the three datasets built in this study (A, B and C) as well as the AllerHunter dataset demonstrated that Allerdictor is capable of obtaining high precision over high recall rates. We evaluated Allerdictor using precision/recall (PR) measures that are widely used in information retrieval (Manning et al., 2008) instead of sensitivity/specificity measures in Receiver-Operating Characteristic curve (ROC) analysis. A PR curve plots precision against recall obtained by varying the prediction score cutoff. Given the numbers of TP, FP, true negatives (TN) and false negatives (FN), precision and recall are defined as follows.
 |
(6) |
A ROC plots TP rate (also called sensitivity or recall) against FP rate (1 − specificity). Because non-allergens are much more abundant than allergens in nature, a method that predicts many more FP than TP (low precision) can still produce a good ROC as long as its sensitivity is high and thus is often misleading (Davis and Goadrich, 2006). The PR curves can reveal high FP rates and thus provide a more meaningful evaluation on naturally skewed allergen/non-allergen distributions, which are also represented in datasets A, B and C.
3.1 Length of k-mer peptides
The length of k-mer peptides is the most important parameter for Allerdictor prediction models. Allerdictor performed differently with different k values. Performance peaked at k = 5 or 6 and decreased as k moved away from the peaks (Supplementary Fig. S1). This interesting result agrees with the debatable criterion of six contiguous amino acid matches with a known allergen used by FAO/WHO guideline. The classification power of Allerdictor comes with its ability to distinguish and assign higher weight to k-mers that are more likely associated with allergens (Section 3.4). With k = 5, Allerdictor produced near perfect FP rates, whereas k = 6 allowed for better sensitivity and still maintained low FP rates. We chose k = 6 for the analyses and results reported in the following sections.
3.2 Allerdictor produces high precision over high recall
We performed nested 10-fold cross-validation to evaluate Allerdictor performance in comparison with the baseline classifiers we derived from FAO/WHO guidelines on our three datasets. The two derivatives were BLAST and MEM (maximal exact match). In the BLAST method, a protein was classified based on the best BLAST similarity score (E-value) against a database of known allergens from the training set. In MEM, the longest subsequence of contiguous amino acid matches against the allergens in the training set was chosen as the classification score. MEM was implemented using SparseMEM software (Khan et al., 2009).
For all three datasets, both Allerdictor-NB and Allerdictor-SVM performed better than BLAST and MEM with higher precision over the same recall rate as well as larger area under the PR curve (AUPRC) (Fig. 2). NB and SVM performed equally on datasets A and B, whereas with dataset C, SVM exhibited better performance. The AUPRCs for Allerdictor-SVM averaged at 0.97, 0.91 and 0.85 for datasets A, B and C, respectively. BLAST’s and MEM’s performance was acceptable on datasets exhibiting low levels of sequence similarity between allergens and non-allergens (AUPRC ≈ 0.7–0.8 for datasets A and B). However, their performance dropped dramatically when the level of sequence similarity between allergens and non-allergens increased (dataset C). BLAST appeared to be more vulnerable to a drop in performance with AUPRC ≈ 0.25 and precision rarely reaching 0.5 on dataset C. MEM was less vulnerable (AUPRC ≈ 0.63) compared with BLAST, yet failed to produce >0.6 precision over >0.6 recall. On the other hand, Allerdictor still yielded high performance on dataset C. The AUPRC was ∼0.81 for Allerdictor-NB and ∼0.85 for Allerdictor-SVM, and both still produced ∼0.8 precision over 0.8 recall. As SVM performed more robustly than NB, studies were concentrated on SVM.
Fig. 2.

PR curves for Allerdictor-SVM (A-SVM) and Allerdictor-NB (A-NB), MEM and BLAST on three datasets of increasing level of sequence similarity between allergens and non-allergens (A–C). The curves were averaged on nested 10-fold cross-validation with standard deviations as error bars
The capability of Allerdictor to produce high precision over high recall rates is due to its extremely high specificity (low FP rate). To assess Allerdictor specificity, we trained Allerdictor-SVM with each of the three datasets and predicted allergens for the whole Swiss-Prot database. The results confirmed Allerdictor had a high level of specificity with <1% of proteins in the Swiss-Prot database predicted as allergens (Table 1). Regardless of the level of similarity between allergens and non-allergens in the training datasets, Allerdictor still predicted <1% of Swiss-Prot as allergens. Homology-based methods often produce many FP when trained with datasets exhibiting low levels of sequence similarity between allergens and non-allergens. Allerdictor specificity, on the other hand, is consistent.
Table 1.
Whole Swiss-Prot (539 616 sequences) scan results for Allerdictor trained with different datasets
| Training data | Predicted allergens | Percent Swiss–Prot | Allergen-relateda |
|---|---|---|---|
| Dataset A | 3025 | 0.56% | 1069 |
| Dataset B | 4160 | 0.77% | 1109 |
| Dataset C | 2150 | 0.40% | 976 |
aPredicted allergens that are true allergens or annotated with allergen-related keywords in Swiss-Prot.
3.3 Allerdictor prediction time is linear
Sequence alignment-based approaches, which are also the most accurate current allergen prediction methods, construct features from sequence alignment. Most of the prediction time for a sequence is spent on aligning the sequence against a database of full-length allergen/non-allergen sequences and/or allergen-specific peptides. This depends on the length of both the sequence and the database. Moreover, aligning sequences requires non-linear time of the sequences’ length, which makes large-scale allergen prediction a relatively time-consuming task.
Allerdictor feature construction and prediction times are both linear of the length of the sequence. Counting frequency of k-mers from a sequence can be achieved in linear time of the length of the sequence and does not depend on training data. Prediction time for both NB and SVM has two components. The first one is the time required to look up model parameters (e.g. SVM weights) for the k-mers generated from the sequence. With proper hashing techniques, the total look up time is also linear of the number of k-mers on average. The second component is the time to compute the score of the model (NB or linear SVM) that is also linear of the number of k-mers, as it involves only non-zero elements of the sparse k-mer frequency vector. Overall, Allerdictor prediction time is linear of the length of the sequence.
The running time of Allerdictor (both web server and standalone versions) was estimated in comparison with the other methods (including EVALLER, AlgPred, AllerHunter, APPEL and SORTALLER) on a random test set of 100 protein sequences (average length of 326 amino acids) and the whole Swiss-Prot database. As only web server versions of the other methods were available, we wrote scripts to submit sequences one by one to the web servers and measured the time needed to run 100 sequences, including time for data transmission over the web. For these methods, the estimated time required to run the whole Swiss-Prot database was derived from the time used for 100 sequences. For Allerdictor web server, true running time to scan the whole Swiss-Prot database was measured using a similar submission script. The lower bound for AllerHunter feature construction was also estimated by time required to align sequences against the database of training sequences using BLAST. Allerdictor was extremely fast compared with other methods (Table 2). Allerdictor standalone version only took ∼6 min (on a single core PC), and Allerdictor web server submission took ∼34.5 h to scan the whole Swiss-Prot of 539 616 protein sequences. Web server performance depends on many factors such as web server configuration and internet connection speed, and therefore the rough estimates obtained in Table 2 were not necessarily the true performance. However, these estimates should correlate with true running time and were appropriate for comparison. The linear running time in addition to high precision over high recall makes Allerdictor more practical for large-scale allergen discovery compared with existing methods.
Table 2.
Running time for 100 random test sequences (T) and whole Swiss-Prot (SP) of 539 616 sequences
| Method | T (s) | SP (h) | Implementation note |
|---|---|---|---|
| Allerdictor | 32a | 0.1 | Standalone, implemented in Python |
| Allerdictor | 24 | 34.5 | Web server, implemented in Python, submission via Perl script |
| AlgPred-d | 114 | 174b | Web server, submission via Perl script |
| AllerHunter | 863 | 1318b | Web server, submission via Perl script |
| AllerHunter | 15c | 24c | BLAST, using AllerHunter data |
| APPEL | 3731 | 5700b | Web server, submission via Perl script |
| EVALLER | 4094 | 6255b | Web server, submission via Perl script |
| SORTALLER | 158 | 241b | Web server, submission via Perl script |
aIncluding time to read k-mer dictionary from disk. bEstimated time based on running time of 100 test sequences. cLower bound estimate (time required to run BLAST against the training sequences).
3.4 Allerdictor distinguishes allergen-related peptides
An IgE epitope is a region of an allergen that can be recognized by and interact with allergen-specific IgE antibodies. It is perhaps the most important allergenicity identification feature. However, IgE epitopes exist in both linear form (continuous amino acids) and conformational form (discontinuous amino acids brought together via protein folding) and thus are difficult to model. Sequence similarity approaches in allergen prediction such as those corresponding to the FAO/WHO guideline are centered on knowledge of IgE epitope length, which ranges from 3–71 amino acids according to the known IgE epitopes from SDAP (Ivanciuc et al., 2003). These approaches, however, cannot distinguish between sequence similarity matches in regions that are related to allergenicity such as the IgE epitopes, and those in regions that are commonly found in both allergens and non-allergens, and thus yield low performance.
Allerdictor is effective in allergen prediction because it is capable of distinguishing allergen-related short peptides (possibly but not necessarily IgE epitopes per se). For example, Allerdictor, although does not directly model IgE epitope structures, can learn and assign higher weight to k-mers that are subsequences of known IgE epitopes using a machine learning approach. We investigated this using a set of 183 known IgE epitopes (from 29 allergens) collected from SDAP (one of the most updated lists of known IgE epitopes). As given in (5), k-mers with higher weight in the linear SVM model represent more allergen predictive features (more commonly found in allergens). We ranked k-mers by their weight and investigated the distribution of the ranks of the k-mers that were subsequences of at least one known IgE epitope (IgE epitope-matched k-mers). We found >1000 IgE epitope-matched k-mers learned from each of the datasets A, B and C (Supplementary Table S1). The majority of the IgE epitope-matched k-mers (>93%) were ranked in the top 10% among ∼4.5–9.1 million k-mers obtained from the training data (or 90% of them were ranked in the top 3.4, 5.5 and 5.9% for datasets A, B and C, respectively) (Fig. 3). This result suggests that Allerdictor is capable of assigning higher weight to k-mers that are more important for allergenicity such as those found in IgE epitopes than those that are often found in both allergens and non-allergens. For datasets A and B, almost all IgE epitope-matched k-mers were ranked among the top, whereas a small number of these k-mers for dataset C had low ranks. This can be explained by the fact that dataset C only contained a fraction of the known allergens (and IgE epitopes) via the relaxed sequence clustering criteria described earlier.
Fig. 3.
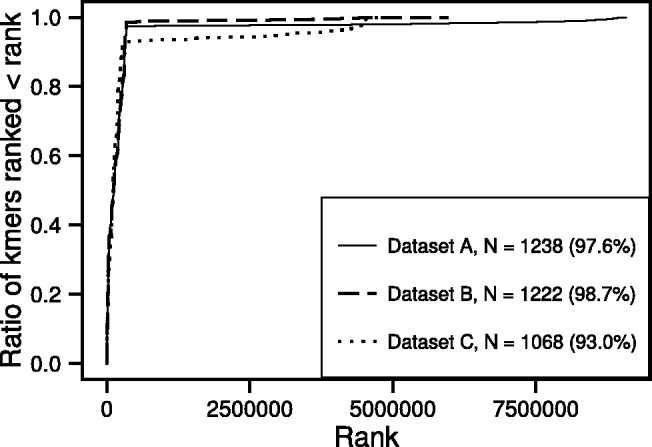
Empirical cumulative distribution of ranks of the k-mers (k = 6) that are subsequences of at least 1 of 183 known IgE epitopes from SDAP. The percentage in the brackets is the ratio of k-mers that are ranked in the top 10% of all k-mers obtained from each training set
In fact, many of the highly ranked k-mers formed continuous peptides overlapping with known IgE epitopes. We ran Allerdictor on 25 allergen proteins with IgE epitopes previously mapped (prepared from the set of 29 allergens with known IgE epitopes collected from the SDAP). The majority of the known IgE epitopes overlapped with regions formed by highly ranked k-mers, and many of them were fully covered by these regions (Supplementary Figs S2–S4). This result suggests that the regions of a protein sequence that contain highly ranked k-mers have higher probability of being part of IgE epitopes or other immunologically relevant features, and thus they are highlighted in the prediction output of Allerdictor server for further computational and/or experimental investigation by the end users.
3.5 Comparison with other methods
Current allergen prediction tools were first evaluated on a set of randomly drawn ∼10% of dataset C (167 allergens and 1663 non-allergens, test set X). For methods that produced monotonous prediction scores (AlgPred, AllerHunter, SORTALLER), the score cutoff was varied to obtain PR curves. For other methods (EVALLER, APPEL), fixed default performance measures were calculated from the number of correct and incorrect predictions. The results showed that all methods evaluated yielded low precision on the chosen test set (Supplementary Table S2 and Supplementary Fig. S5). None of the methods yielded precision >0.4 over recall >0.6 for PR curves. For default performance, only EVALLER and AllerHunter yielded Matthews correlation coefficient >0.5 with both precision and recall >0.5. Sequence similarity-based methods (AllerHunter and EVALLER) appeared to perform better in this test. As expected, performance was correlated with the time the methods were released, where later methods performed better (with the exception that SORTALLER performed poorly, although it was the latest method in this test). AllerHunter performed better than other methods, partly because it was trained on a dataset that contained many allergen-like non-allergens, a characteristic that was also exhibited by the test data.
Because performance of supervised machine learning methods depends heavily on training and testing data, we avoided comparison of Allerdictor with other methods trained with different datasets. Current allergen prediction methods were pretrained with specific datasets and only available in the form of web servers and thus prevented retraining them for comprehensive comparison with Allerdictor. Therefore, we investigated these datasets on whether they are appropriate to train and compare Allerdictor with the pretrained web servers of these methods. Among three publicly available datasets, AllerHunter was the only dataset that possessed a significant level of sequence similarity between allergens and non-allergens and had many more non-allergens than allergens (Fig. 1). The AlgPred dataset was small and sequence names were masked, whereas the EVALLER non-allergen sequences that were used to derive allergen-specific peptides were not available. The level of sequence similarity between allergens and non-allergens for AlgPred and EVALLER was low as determined by BLASTClust (Fig. 1).
The AllerHunter dataset (http://tiger.dbs.nus.edu.sg/AllerHunter/) was considered the only complete dataset for the purpose of the comparison. However, this dataset was redundant and contained noise. The AllerHunter dataset was reviewed and found to contain 48 obsolete sequences [deleted from Genbank (Benson et al., 2010) and Swiss-Prot], 233 duplicated sequences and 328 short sequences (3–50 amino acids). Also, among the non-allergens, 135 were found to be true allergens, 176 were antigens and 165 contained allergen-related ambiguous annotation (from Swiss-Prot and Allergome databases). The noise possibly resulted from new annotation added to the public databases after AllerHunter data were collected. We accepted the noise for training data and trained Allerdictor on the same training set of sequences (1266 allergens and 11 229 non-allergens) that was used to train AllerHunter server (personal communication with Martti Tammi) and compared Allerdictor with AllerHunter server on two test sets: the original AllerHunter test set (139 allergens and 1245 non-allergens) that contained noise and the revised version of the test set with reduced noise (149 allergens and 1141 non-allergens). The review process moved the newly discovered allergens from the non-allergen set to the allergen set and removed duplicated, obsolete or ambiguous sequences and non-allergen sequences that had 90% identity over 90% coverage with a known allergen (similar to the procedure used to reduce noise from Allerdictor datasets).
Comparison results on AllerHunter dataset showed that Allerdictor slightly outperformed AllerHunter with slightly larger AUPRCs (Fig. 4). An interesting trend was that Allerdictor produced higher high-range precision (>0.8) at lower recall (<0.8). At recall >0.85, both Allerdictor and AllerHunter produced many FP, and thus the precision for both methods dropped below 0.6. AllerHunter performed slightly better in lower-range precision (<0.75) at a narrow recall range from ∼0.85–0.9. High-range precision is particularly useful in large-scale prediction. For example, one often chooses the top scoring candidates from computational predictions for further experimental validation, which is equivalent to lowering recall to obtain higher precision. Along with higher high-range precision, Allerdictor also runs much faster than AllerHunter (Section 3.3), which makes it the better choice for large-scale allergen prediction.
Fig. 4.
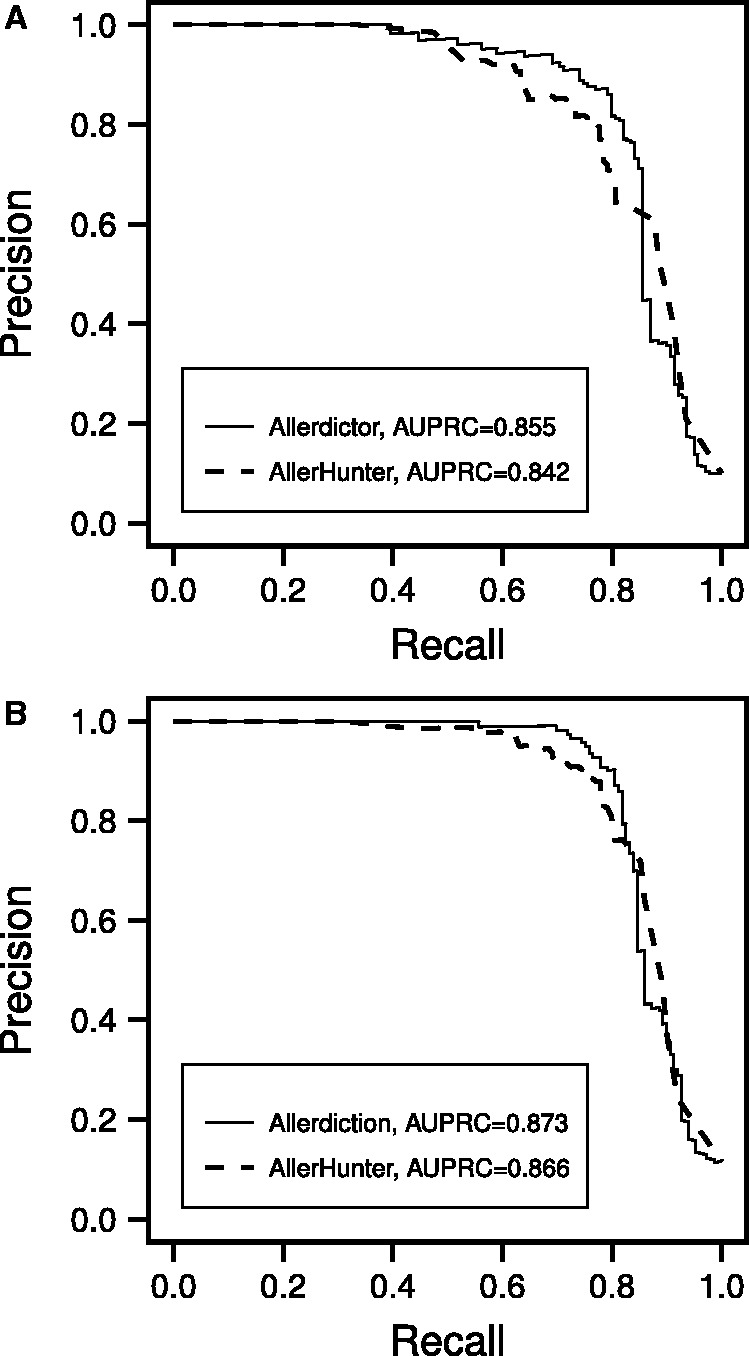
PR curves for Allerdictor and AllerHunter, both trained on the original AllerHunter training set and tested with the original AllerHunter test set (A) and the reviewed AllerHunter test set (B)
The amino acid composition and dipeptide approaches in AlgPred are special cases of k-mer approach in Allerdictor with k = 1 and 2. We found that such small values of k yielded low performance on multiple datasets, including datasets A, B and C. Also pointed out by AlgPred authors, when tested with Swiss-Prot non-allergens, AlgPred falsely predicted ∼40% of them to be allergens (Saha and Raghava, 2006).
3.6 Allerdictor prefers larger number of k-mers
The size of the k-mer dictionary (also the feature vector size) is exponential of k (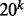 ) and therefore many machine learning approaches are prevented from using k-mer sequence representation. In reality, the size of the k-mer dictionary depends on training datasets and is much smaller than the number of possible k-mers. Allerdictor when trained with k = 6 on datasets A, B and C had feature space dimension of ∼4.6, 5.7 and 9.1 million, respectively (much smaller than 206). To test if we can reduce the number of k-mers without lowering performance, feature selection using mutual information and feature abstraction were performed with Allerdictor-SVM using k = 6.
) and therefore many machine learning approaches are prevented from using k-mer sequence representation. In reality, the size of the k-mer dictionary depends on training datasets and is much smaller than the number of possible k-mers. Allerdictor when trained with k = 6 on datasets A, B and C had feature space dimension of ∼4.6, 5.7 and 9.1 million, respectively (much smaller than 206). To test if we can reduce the number of k-mers without lowering performance, feature selection using mutual information and feature abstraction were performed with Allerdictor-SVM using k = 6.
The results on datasets A, B and C showed that no performance gain was achieved with both feature selection and feature abstraction (Supplementary Fig. S6). Using ≥20–50% k-mers, performance was similar to that obtained with all k-mers. Allerdictor performance slightly dropped when the number of selected k-mers was ≤10–20% and dramatically dropped when ≤5–10% k-mers were selected. This result suggests that Allerdictor generally performs better with more k-mers. Feature abstraction reduced ∼4.6–9.1 million k-mers down to <1000 abstract features when trained using k = 6. Surprisingly, performance for feature abstraction was close to performance using all k-mers. This interesting result opens doors for using other classification methods that can only handle a small number of features.
3.7 Effects of allergen prevalence
Supervised machine learning-based allergen prediction methods are often available to end users as tools pretrained on some specific dataset. The predictive values including positive predictive value (PPV, also called precision) and negative predictive value (NPV) of such tools are subject to the prevalence of allergens in data. We have shown that Allerdictor produced high precision over high recall when training and testing using data that exhibited low ratios of allergen sequences.
To provide a complete picture of allergen prediction performance, we also investigated the effects of allergen prevalence (in testing data) on PPV and NPV of Allerdictor and the current allergen prediction tools. As expected, predictive values of all methods were affected by the prevalence of allergens in testing data (Supplementary Figs S7–S9). When the prevalence of allergens was low, AllerHunter, APPEL and EVALLER exhibited higher predictive values than AlgPred and SORTALLER on a random set of sequences drawn from dataset C (Supplementary Fig. S8). Allerdictor exhibited stable PPV on datasets A, B, C (Supplementary Fig. S7) and on AllerHunter dataset (Supplementary Fig. S9). When allergen ratio was ≤0.5, Allerdictor achieved both PPV and NPV ≥0.8 in all datasets. Compared with AllerHunter on AllerHunter dataset, Allerdictor PPV and NPV were better when allergen prevalence was low, but AllerHunter exhibited more balanced PPV and NPV when the ratio of allergens was higher. The NPV of all methods including Allerdictor decayed rapidly as the ratio of allergens in the test sets increased. However, this behavior does not significantly limit the application of machine learning-based allergen prediction methods because allergen prevalence is low in nature and in many applications. For example, there exist ∼20 known allergens among >9000 proteins coded by the genome of the allergenic fungus Aspergillus fumigatus (Fedorova et al., 2008). Low allergen ratio is a characteristic of large sequence sets often seen in large-scale sequence annotation, which is also Allerdictor's main application.
4 CONCLUSION
This article presented an accurate sequence-based allergen protein prediction method (Allerdictor) that is much faster than the current most accurate methods while still maintaining comparable or better predictive performance (when compared with AllerHunter). The main idea is the use of the k-mer feature representation of sequences, and thus linear prediction time is achieved for both feature construction and prediction using a linear SVM model. Moreover, the k-mer approach is particularly effective for allergen prediction because supervised machine learning methods such as SVM can learn the k-mers shared by many allergens such as the one found in IgE epitopes and assign higher weights to these k-mers.
The prevalence of asthma has been an increasing human health issue. Approximately 235–300 million people worldwide were diagnosed with asthma with annual deaths of ∼250 000 (GINA, 2012; WHO, 2013). The majority of asthmatic patients have allergic asthma in which allergic reactions (caused by allergens) exacerbate asthmatic symptoms. To facilitate our understanding and prevention of this disease, it is important to identify potential allergens from massive amounts of protein sequences produced every day via both genome sequencing and sequence synthesis. Because experimental allergenicity assessment is still expensive and difficult (especially at large scale), computational allergen identification is an alternative first step.
Allerdictor addresses the shortcomings of the current allergen prediction tools. With high precision over high recall and fast speed, Allerdictor is not only useful for general sequence allergenicity assessment in applications such as screening of novel proteins introduced to genetically modified crops but also particularly suitable for allergen discovery on a large scale in applications such as whole-genome annotation and quick screening of synthesized sequences.
Supplementary Material
ACKNOWLEDGEMENT
The authors wish to thank T. M. Murali and Lenwood Heath for helpful discussions regarding this research.
Funding: National Institute of Health, USA (1R21AI094071-01) (CBL) and the Virginia Bioinformatics Institute, Virginia Tech, Blacksburg, Virginia.
Conflict of Interest: none declared.
REFERENCES
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LD, McCallum AK. Proceedings of the 21st Annual International ACM SIGIR Conference on Research and Development in Information Retrieval. 1998. Distributional clustering of words for text classification. SIGIR’98, pp. 96–103. ACM, New York, NY. [Google Scholar]
- Barrio AM, et al. EVALLER: a web server for in silico assessment of potential protein allergenicity. Nucleic Acids Res. 2007;35(Suppl. 2):W694–W700. doi: 10.1093/nar/gkm370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, et al. GenBank. Nucleic Acids Res. 2010;38(Suppl. 1):D46–D51. doi: 10.1093/nar/gkp1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boser BE, et al. Fifth Annual Workshop on Computational Learning Theory, Pittsburg, PA. New York, NY, USA: ACM Press; 1992. A training algorithm for optimal margin classifiers; pp. 144–152. [Google Scholar]
- Burges CJC. A tutorial on support vector machines for pattern recognition. Data Min. Knowl. Discov. 1998;2:121–167. [Google Scholar]
- Codex Alimentarius. 2009. Foods derived from modern biotechnology. Codex Alimentarius Commission, Joint FAO/WHO Food Standards Programme. Rome, Italy. [Google Scholar]
- Cortes C, Vapnik V. Support-vector networks. Mach. Learn. 1995;20:273–297. [Google Scholar]
- Cui J, et al. Computer prediction of allergen proteins from sequence-derived protein structural and physicochemical properties. Mol. Immunol. 2007;44:514–520. doi: 10.1016/j.molimm.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Davis J, Goadrich M. Proceedings of the 23rd International Conference on Machine Learning. 2006. The relationship between precision-recall and ROC curves. ICML’06, pp. 233–240. ACM, New York, NY. [Google Scholar]
- Devereux G. The increase in the prevalence of asthma and allergy: food for thought. Nat. Rev. Immunol. 2006;6:869–874. doi: 10.1038/nri1958. [DOI] [PubMed] [Google Scholar]
- FAO/WHO. Rome, Italy: 2001. Evaluation of allergenicity of genetically modified foods. Report of a joint FAO/WHO expert consultation on allergenicity of foods derived from biotechnology. [Google Scholar]
- Fedorova ND, et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008;4:e1000046. doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiers MW, et al. Allermatch™, a webtool for the prediction of potential allergenicity according to current FAO/WHO codex alimentarius guidelines. BMC Bioinformatics. 2004;5:133. doi: 10.1186/1471-2105-5-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Initiative for Asthma (GINA) Global Strategy for Asthma Management and Prevention. Global Initiative for Asthma. 2012 Available from http://www.ginasthma.org (16 January 2014, date last accessed) [Google Scholar]
- Ivanciuc O, et al. SDAP: database and computational tools for allergenic proteins. Nucleic Acids Res. 2003;31:359–362. doi: 10.1093/nar/gkg010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanciuc O, et al. The property distance index PD predicts peptides that cross-react with IgE antibodies. Mol. Immunol. 2009;46:873–883. doi: 10.1016/j.molimm.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachims T. Advances in Kernel Methods. Cambridge, MA: MIT Press; 1999. pp. 169–184. [Google Scholar]
- Joachims T. Learning to Classify Text using Support Vector Machines. Boston: Kluwer Academic Publishers; 2002. [Google Scholar]
- Khan Z, et al. A practical algorithm for finding maximal exact matches in large sequence datasets using sparse suffix arrays. Bioinformatics. 2009;25:1609–1616. doi: 10.1093/bioinformatics/btp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladics GS, et al. Bioinformatics and the allergy assessment of agricultural biotechnology products: industry practices and recommendations. Regul. Toxicol. Pharmacol. 2011;60:46–53. doi: 10.1016/j.yrtph.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Li K-B, et al. Predicting allergenic proteins using wavelet transform. Bioinformatics. 2004;20:2572–2578. doi: 10.1093/bioinformatics/bth286. [DOI] [PubMed] [Google Scholar]
- Lin H-T, et al. A note on platt’s probabilistic outputs for support vector machines. Mach. Learn. 2007;68:267–276. [Google Scholar]
- Magrane M, Consortium U. UniProt knowledgebase: a hub of integrated protein data. Database. 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning CD, et al. Introduction to Information Retrieval. New York: Cambridge University Press; 2008. [Google Scholar]
- Mari A, et al. Allergen databases: current status and perspectives. Curr. Allergy Asthma Rep. 2009;9:376–383. doi: 10.1007/s11882-009-0055-9. [DOI] [PubMed] [Google Scholar]
- Masoli M, et al. The global burden of asthma: executive summary of the GINA dissemination committee report. Allergy. 2004;59:469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- Metcalfe DD. Genetically modified crops and allergenicity. Nat. Immunol. 2005;6:857–860. doi: 10.1038/ni0905-857. [DOI] [PubMed] [Google Scholar]
- Muh HC, et al. AllerHunter: a SVM-Pairwise system for assessment of allergenicity and allergic cross-reactivity in proteins. PLoS One. 2009;4:e5861. doi: 10.1371/journal.pone.0005861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira F, et al. Proceedings of the 31st annual meeting on Association for Computational Linguistics. 1993. Distributional clustering of english words. ACL’93, pp. 183–190. Association for Computational Linguistics, Stroudsburg, PA. [Google Scholar]
- Platt JC. Probabilistic outputs for support vector machines and comparisons to regularized likelihood methods. In: Smola AJ, et al., editors. Advances in Large Margin Classifiers. Cambridge, MA, USA: MIT Press; 1999. pp. 61–74. [Google Scholar]
- Riaz T, et al. WebAllergen: a web server for predicting allergenic proteins. Bioinformatics. 2005;21:2570–2571. doi: 10.1093/bioinformatics/bti356. [DOI] [PubMed] [Google Scholar]
- Saha S, Raghava GPS. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006;34:W202–W209. doi: 10.1093/nar/gkl343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeria-Atmadja D, et al. Computational detection of allergenic proteins attains a new level of accuracy with in silico variable-length peptide extraction and machine learning. Nucleic Acids Res. 2006;34:3779–3793. doi: 10.1093/nar/gkl467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Stadler BM. Allergenicity prediction by protein sequence. FASEB J. 2003;17:1141–1143. doi: 10.1096/fj.02-1052fje. [DOI] [PubMed] [Google Scholar]
- Stagg NJ, et al. Workshop proceedings challenges and opportunities in evaluating protein allergenicity across biotechnology industries. Int. J. Toxicol. 2013;32:4–10. doi: 10.1177/1091581812471864. [DOI] [PubMed] [Google Scholar]
- WHO. (2013) Asthma fact sheet no. 307.
- Zhang L-D, et al. SORTALLER: predicting allergens using substantially optimized algorithm on allergen family featured peptides. Bioinformatics. 2012;28:2178–2179. doi: 10.1093/bioinformatics/bts326. [DOI] [PubMed] [Google Scholar]
- Zipf GK. Human Behavior and the Principle of Least Effort: An Introduction to Human Ecology. Mansfield Centre, CT: Martino Pub; 1949. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.