

Summary
Foxp3+ T-regulatory cells (Tregs) are primarily generated in the thymus (tTreg), but also may be generated extrathymically at peripheral sites (pTreg), or induced in cell culture (iTreg) in the presence of transforming growth factor β (TGFβ). A major unresolved issue is how these different populations of Tregs exert their suppressive function in vivo. We have developed novel systems in which the function of Tregs can be evaluated in vivo in normal mice. Our studies demonstrate that one prominent mechanism of action of polyclonal tTregs is to inhibit T-effector cell trafficking to the target organ, while antigen-specific iTregs primarily prevent T-cell priming by acting on antigen-presenting dendritic cells (DCs). Interleukin-10 (IL-10) plays an important role in the suppressive function of antigen-specific iTregs by controlling the expression of MARCH1 and CD83 on the DC. Activated tTregs may mediate infectious tolerance by delivery of cell surface-expressed TGFβ to naive responder T cells to generate pTregs. Manipulation of Treg function will require the ability to differentiate tTregs from pTregs and iTregs. The expression of the transcription factor Helios has proven to be a useful marker for the identification of stable tTregs in both mouse and man.
Keywords: Treg cells, organ-specific autoimmunity, infectious tolerance, stability, Helios, IL-10
Introduction
Forkhead box protein 3 (Foxp3)+ regulatory T cells (Tregs) play a critical role in all aspects of immune responses and the control of immune homeostasis. Over the past several years, it has become clear that the major population of Foxp3+ Treg is generated in the thymus, and we favor the term thymus-derived Treg (tTreg) for this population. Similarly, it is also clear that an unknown percentage of Tregs can be generated from Foxp3− T-conventional (Tconv) cells in peripheral sites. It has been proposed that such cell populations be termed peripherally derived Tregs (pTregs) (1). Lastly, at least in mouse models, one can generate Foxp3+ T cells by stimulation of Tconv cells in vitro in the presence of TGFβ, and these should be termed in vitro-induced Tregs (iTregs). It remains unclear whether iTregs and pTregs are similar populations that use the same suppressor mechanisms, so caution should be used when equating these two cell populations, particularly since pTregs may represent a stable population, while iTregs may be considerably more labile (2, 3). This review addresses several important issues regarding the functions of these Treg subpopulations and focuses on a comparison of the suppressive mechanisms used in vivo by polyclonal tTregs and the mechanisms used by antigen-specific iTregs. We also discuss the use of potential markers to distinguish tTregs from pTregs, as a detailed characterization of these subpopulations has important clinical implications for their use in cellular immunotherapy.
Suppression of autoimmune disease by polyclonal tTregs
The initial studies (4) describing the existence and functional properties of CD4+CD25+ Tregs demonstrated that they could prevent autoimmune diseases induced after transfer of CD4+CD25− T cells to immunodeficient recipients and also prevented induction of organ-specific autoimmunity that developed after thymectomy on the third day of life (d3Tx). The majority of CD4+CD25+ T cells were later shown to express Foxp3 (5, 6). It has been widely assumed that the majority of freshly isolated Foxp3+ T cells are actually tTregs, although the contribution of pTregs to their activity cannot be excluded due to the lack of a reagent that would allow definitive separation of tTregs from pTregs. Nevertheless, for the purposes of this review, we assume that the majority of the suppressive activity of freshly isolated Foxp3+ T cells from normal unmanipulated mice is mediated by tTregs and not by a small percentage of autoantigen-specific pTreg cells.
Although the ability of tTregs to inhibit organ-specific autoimmune disease has been well documented, the mechanisms underlying their suppressive functions remain poorly characterized. We have used autoimmune gastritis (AIG) as the model for analyzing the suppressive properties of tTregs in vivo (7). One advantage of this model is that it is one of the few models of spontaneous organ-specific autoimmune disease in which the target antigen, the proton pump of the gastric parietal cell, the H/K ATPase, has been defined (8). We have generated T-cell receptor (TCR) transgenic (Tg) mice expressing TCRs derived from T-cell clones isolated from the gastric lymph nodes of d3Tx mice with AIG (9, 10). One of these TCR-Tg strains (TxA23) spontaneously develops AIG early in life and the effector cells express a T-helper 1 (Th1) phenotype. Low numbers of CD4+CD8−Foxp3− thymocytes from TxA23 mice will readily transfer disease to nu/nu recipients. We examined the effects of polyclonal tTregs in this cell transfer model in which the target antigen is expressed and presented at physiological levels. Although the co-transfer of polyclonal tTreg prevented the induction of AIG, it did not prevent the migration of autoantigen-specific T cells to the gastric lymph node or the stomach, nor did it prevent their expansion. Thus, the primary effect of polyclonal tTregs appeared to be inhibition of the differentiation of the autoantigen-specific T cells into pathogenic Th1 cells as reflected by a decrease in antigen-stimulated interferonγ (IFNγ) production by the transferred TxA23 cells isolated from treated recipients (7).
We have also used polyclonal populations of tTregs to address whether tTregs could suppress AIG induced by fully differentiated Th1, Th2, or interleukin-17 (IL-17)-secreting autoantigen-specific T effector cells (11). One additional goal of these studies was to determine whether polyclonal tTregs could modulate cytokine production by fully differentiated effector cells in vivo. Th1, Th2, and Th17 T-effector cells were generated in vitro from CD4+CD8−Foxp3− TxA23 thymocytes and all three T helper cell types transferred AIG into nu/nu recipients. Each type of T-effector cell induced AIG with distinct histological patterns. Th17 cells induced the most destructive disease. Polyclonal tTregs could suppress the capacity of Th1 cells, moderately suppress Th2 cells, but could only minimally suppress Th17-induced disease. The major effects of the tTregs were related to their ability to inhibit T-effector cell expansion, as tTregs significantly reduced the total number of injected T-effector cells in the gastric lymph node of recipients of Th1 and Th2 effectors, but only had minimal effects on the numbers of Th17 effectors. Although Th1 and Th2 T-effector cell expansion was markedly inhibited, it was possible to recover sufficient numbers of T-effector cells from treated animals and determine whether exposure to tTreg in vivo modified T-effector function. T-effector cells were recovered from the gastric lymph node 6 weeks after transfer and re-challenged with their cognate peptide. Effector cells isolated from protected animals were not anergic and were fully competent to proliferate and produce effector cytokines ex vivo. This result is consistent with studies in the d3Tx model of oophoritis in which the continuous presence of Tregs was required for protection from disease and depletion of the Tregs resulted in disease development (12).
It is difficult to draw clear conclusions about the mechanism of action of polyclonal tTreg from these studies. We used the same Treg-T-effector cell ratio in the studies with the naive autoantigen-specific T effectors as we used with the fully differentiated T-effector cells. The results with naive T-effector cells suggested that the primary effect of tTregs was to inhibit differentiation, while the studies with the differentiated T-effector populations demonstrated that inhibition of expansion was the best correlate of protection from destructive AIG and no effect on T-effector cytokine producing capacity was observed. Several important differences were noted between these studies. Naive T effectors could easily be detected in the gastric lymph node as early as 3 days after transfer, and significant expansion could be demonstrated by day 5. However, fully differentiated effector cells could not be detected in the gastric lymph node until 7 days after transfer. This result is not surprising, as the differentiated T-effector populations expressed only low levels of CD62L, the receptor that is required for entry into lymph nodes. It is likely that the fully differentiated effector cells were first stimulated by their target antigen in the stomach and subsequently acquired the ability to traffic to the gastric lymph node. In contrast, in both models, the co-transferred tTregs could easily be detected at early time points after transfer. One possible explanation for the ability to tTreg to inhibit expansion of fully differentiated T-effector cells is that they exerted their inhibitory effects on DC resulting in inhibition of expansion of the T-effector cell populations upon their arrival in the draining lymph node. It also remains possible that the effects on the DC were mediated by a small percentage of the polyclonal tTregs that expressed receptors for the auto-antigen. These auto-antigen-specific Tregs would then be retained and subsequently expanded in the gastric lymph node and exert their inhibitory effects on DCs in a manner similar to antigen-specific iTregs (see below). Collectively, our studies clearly demonstrate a strong inhibitory effect of polyclonal tTregs on the capacity of naive T effectors as well some types of differentiated T-effector cells to induce disease and provide an experimental basis for the potential clinical use of polyclonal tTregs in human disease.
Polyclonal tTregs modulate T-effector cell trafficking
One of the major difficulties in dissecting the mechanism of action of Treg in vivo in the setting of autoimmune disease as described above is the confounding influence of lymphopenia on both Treg and T-effector cells as these studies all involved transfer of effectors to nu/nu recipients. Very few studies have addressed the mechanism of action of polyclonal tTregs in fully immunocompetent recipients. Early studies (13) in experimental autoimmune encephalomyelitis (EAE) demonstrated that augmentation of Treg cell numbers in normal recipients markedly attenuated disease accompanied by decreased T-cell infiltration into the central nervous system (CNS).
We have re-examined the potential mechanisms of suppression by polyclonal tTregs and have performed all experiments in immunologically intact recipients and carefully monitored the fate and differentiation of the T-effector cells on a single cell basis (14). Co-transfer of polyclonal activated tTreg cells into healthy mice attenuated the induction of EAE. Suppression of disease strongly correlated with a reduced number of T-effector cells in the spinal cord. Mice that had received Treg cells had a 50% reduction in the percentage of CNS infiltrating CD4+ T cells, but on a per cell basis, the cytokine production profile of these cells was identical to cells isolated from the CNS of mice that had not received Tregs. The therapeutic effects of the tTregs strongly correlated with the reduced percentage of CD4+ T-effector cells that invaded the CNS rather than Treg cell-mediated inhibition of Th1/Th17 differentiation.
To more precisely characterize the effects of polyclonal tTreg cells on the expansion/differentiation of T-effector cells in vivo, we developed a model in which congenically marked CFSE-labeled TCR Tg Foxp3− T cells were adoptively transferred to normal recipients in the presence or absence of congenically distinct pre-activated polyclonal Treg cells (13). The following day, the mice were immunized with the peptide recognized by the Tg T cells in adjuvant, and the numbers and activation status of the transferred T-effector cells were analyzed on day 4–7 after transfer. The surprising result of these studies was that Treg cells had no effect on T-effector cell proliferation as measured by carboxyfluorescein succinimidyl ester (CFSE) dilution, and this was accompanied by a twofold increase in the percentage and absolute number of T-effector cells present in the draining lymph node (Fig. 1). Furthermore, differentiation also appeared to be unimpaired in the presence of tTregs, as the percentage of cells differentiating into IFNγ or IL-17 producers was the same in the presence of absence of tTregs.
Fig. 1. Polyclonal tTregs and antigen-specific iTregs differentially modulate T-effector cell expansion in vivo.

Naive antigen-specific TCR Tg T cells were co-transferred with polyclonal tTregs (left panel) or iTregs specific for the same antigen (right panel) into normal mice. One day later, the mice were immunized with the cognate peptide, and the number of antigen-specific T effectors in the draining lymph node quantitated 4 days later. Polyclonal tTregs augmented the number of T effectors compared to non-treated mice, while antigen-specific iTregs markedly suppressed the number of T effectors recovered.
The increase in the number of T-effector cells in the draining lymph node is in apparent conflict with our studies in the EAE model that demonstrated a decreased number of T-effector cells in the target organ in the presence of an excess of Tregs. However, the total number of T cells in the lymph node is determined not only by in situ proliferation but also by the relative contribution of entry and exit from the lymph node. When we examined peripheral blood from the immunized mice, we detected fewer T-effector cells in the blood of mice that had received polyclonal tTregs than in blood of untreated controls. We also observed a markedly decreased number of T-effector cells at the site of skin testing when mice were immunized in the presence of Tregs. Taken together, these observations are consistent with the hypothesis that in the presence of polyclonal tTreg cells priming and expansion are unimpaired in the draining lymph node, but fewer cells can leave the lymph node to enter the circulation, and fewer cells are therefore available to respond to antigen at a distant site.
The mechanisms utilized by tTregs to maintain T-effector cells at the site of immunization remain to be fully elucidated. We did observe that T-effector cells recovered from lymph nodes in the presence of Treg cells expressed decreased levels of CXCR4, syndecan, and the sphinogosine phosphate receptor 1 (S1P1). S1P1 levels are rapidly downregulated on T cells following entry into lymph nodes. As T cells are primed and differentiate, they upregulate S1P1, allowing them to respond to high levels of S1P in the circulation, and exit the lymph node in response to the concentration gradient (15). By altering the expression of S1P1 on T-effector cells, Tregs would affect the ability of the effectors to migrate out of the lymph node and into the circulation to reach their target organ. One important conclusion to be drawn from these studies is polyclonal tTreg cells do not always influence the immune response by mediating immune suppression and completely shutting down T-cell activation. Thus, the therapeutic administration of polyclonal tTreg cells would not necessarily lead to global immunosuppression or the inhibition of responses to all antigens or pathogens. In contrast, polyclonal tTregs appear to specifically target trafficking pathways, thus allowing immunity to develop in lymphoid organs but limiting the number of potentially autoaggressive T cells that are allowed to migrate and enter tissues.
We have performed almost all the polyclonal tTreg transfer studies into normal mice using polyclonal T-cell populations that had been activated via their TCR and expanded in IL-2 ex vivo. The primary reason for this approach was to obtain sufficient numbers of Treg cells to use in the cell transfer model. One major issue that remains to be resolved is whether polyclonal tTregs must be activated through their TCR to exert their suppressive effects. The source of the TCR signal during in vivo activation of polyclonal populations of tTregs is not known. It is widely assumed that tTregs preferentially recognize self-antigens, and it remains possible that tTregs are continuously recognizing and being activated by ubiquitous self-peptides bound to major histocompatibility complex (MHC) class II antigens. It is also very difficult to rule out the possibility discussed above that one component of polyclonal tTreg suppression in vivo is mediated by activation of a low percentage of autoantigen-specific tTregs present within the polyclonal population.
Mechanism of action of antigen-specific iTregs in vivo
One of the major factors limiting the analysis of the mechanism of action of antigen-specific Tregs in vivo is that it is difficult, if not impossible, to isolate antigen-specific tTregs from normal, unmanipulated or even from mice that have been primed with antigen. A certain percentage of cells in TCR Tg mice do express the transgene and are Foxp3+ and cells have been used immediately after removal from the donor or after expansion in vitro. Very few studies have compared directly the in vivo effectiveness of polyclonal and monoclonal populations of tTreg. Hori et al. (16) demonstrated equivalent effectiveness of polyclonal and monoclonal populations of tTregs in preventing EAE that develops spontaneously in mice expressing a Tg anti-myelin basic protein (MBP) TCR on a RAG deficient (−/−) background. In contrast, Tang et al. (17) demonstrated that an expanded population of tTregs from a TCR Tg mouse specific for an antigen expressed by pancreatic islet cells was much more efficient than polyclonal tTregs in their ability to inhibit the induction of diabetes when diabetogenic T cells were transferred into NOD mice on a RAG−/− background. One assumption driving the use of tTregs expressing a TCR Tg receptor specific for an organ-specific peptide is that they would preferentially home to the target organ, be activated by their target autoantigen, and be capable of mediating bystander suppression even if the target autoantigen of the T effectors was not known.
Generation of iTregs in vitro
Numerous studies over the past 10 years have demonstrated that Foxp3− T cells can be induced to express Foxp3 in the presence of TGFβ (18). As these cells are induced in vitro, they will be referred to as iTregs. We have demonstrated (19) that IL-2 plays a non-redundant role in TGFβ induced Foxp3 expression. Other cytokines utilizing the common-γ-chain were unable to induce Foxp3 expression in IL-2-deficient cells. This result should be contrasted to the lack of an absolute requirement for IL-2 for the development of tTregs, as other common-γ-chain cytokines could substitute for the intrathymic development of tTregs (20). In our hands, the role of CD28 signaling in the induction of Foxp3 expression was solely related to its capacity to enhance the endogenous production of IL-2. In the absence of IL-2, TGFβ was unable to induce Foxp3 expression, even in the presence of anti-CD28. This result should be compared to the absolute requirement for CD28 signaling in the development of tTregs (21). We also evaluated the role of the strength of the TCR signal to upregulate Foxp3 in the presence of TGF-β. In the absence of CD28 costimulation, Foxp3 expression was maximal at 2μg/ml of anti-CD3 and markedly lower levels of Foxp3 expression were detected at lower concentration of anti-CD3. In contrast, in the presence of CD28 costimulation, maximal Foxp3 expression was seen at 25-fold lower levels of anti-CD3. TGFβ-iTregs were capable of suppressing the proliferation of naïve populations of T-effector cells stimulated with anti-CD3.
We have also explored the optimal requirements for the generation of antigen-specific iTregs in vitro. In most studies, we utilize CD4+Foxp3− from TCR Tg mice on a RAG−/− background. We stimulate these cells with plate-bound anti-CD3, soluble anti-CD28 in the presence of IL-2 and TGFβ (5ng/ml). These conditions routinely yield a population consisting of greater than 90% Foxp3+ T cells after 96 h of stimulation. Although Foxp3 can be induced with soluble anti-CD3 in the presence of DCs, the percentage of Foxp3+ T cells induced is usually only in the range of 50–60%. It is likely that the production of pro-inflammatory cytokines such as IL-6 by the DCs inhibits the optimal induction of Foxp3 expression.
Suppressive activity of polyclonal iTregs in vivo
Although iTregs were as efficient as tTregs in their in vitro suppressive activity, it was important to document that iTregs could manifest suppressive function in vivo. One of the most rigorous tests of the in vivo suppressive capacity of tTregs is their ability to prevent the development of disease upon transfer into neonatal scurfy mice (6). Some studies have suggested that iTregs rapidly lose Foxp3 expression following transfer in vivo (22). The use of polyclonal iTregs is further complicated by potential differences between tTregs and iTregs in their homing capabilities and in their T-cell repertoires. tTregs express a TCR repertoire with a bias for self and there is limited overlap between the tTreg TCR repertoire and the TCR repertoire of conventional cell CD4+ T cells. Thus, iTregs derived from a polyclonal population of Foxp3− T cells may be restricted in their ability to effectively suppress autoimmune disease secondary to a lack of T cells with anti-self TCRs. However, when polyclonal iTregs were transferred to neonatal scurfy mice, they suppressed all the pathologic manifestation of severe autoimmune disease in both lymphoid sites and in tissues (23). However, the potency of iTregs was considerably less than that seen with tTregs. One problem with iTregs is that a significant proportion loses expression of CD62L during in vitro generation and may be unable to home to lymph nodes. It also is possible that tTregs and iTregs use different mechanisms of suppression in vivo. Nevertheless, the TCR repertoire of the iTregs generated from normal adult mice appears to be sufficient to prevent autoimmune disease in the Scurfy mouse. The iTregs maintained high levels of Foxp3 expression in the inflammatory environment, whereas the same cells lost expression of Foxp3 when transferred to normal mice. This result suggests that certain factors such as the production of pro-inflammatory cytokines in the Scurfy mouse might act as survival or expansion signals for the iTregs.
Suppressive activity of antigen-specific iTregs in vivo
We also tested the capacity of antigen-specific iTregs to inhibit the development of disease in the AIG model. We generated iTregs specific for the naturally expressed autoantigen (H/K ATPase) by stimulation of CD4+Foxp3− thymocytes from the TxA23 TCR Tg mouse in vitro in the presence of TGFβ. When the antigen-specific iTregs were co-transferred with CD4+Foxp3− thymocytes from the same mouse to nu/nu recipients, all aspects of AIG were inhibited including the development of anti-parietal cell antibodies, as well as the destruction of gastric parietal cells (24). Several of the mice treated with the iTregs had normal stomach pathology, and the remainder had low to intermediate levels of inflammation. Antigen-specific iTregs were more efficient at preventing autoimmunity than polyclonal tTregs. The iTregs were long-lived in vivo, maintained expression of Foxp3, and protected mice from disease for long periods of time. We explored in depth possible mechanisms for antigen-specific iTreg-mediated disease suppression. The iTregs did not inhibit the initial migration of the T-effector cells to the gastric lymph node. The major effect of the iTregs appeared to be inhibition of the priming of the autoreactive effector cells. Most notably, a 40-fold reduction in the number of autoreactive effector cells was observed 5 days after transfer. When we isolated DCs that had been exposed to iTregs in vivo, the DCs had a reduced capacity to present the endogenous antigen compared to those from non-iTreg-treated mice consistent with the possibility that iTregs mediated their suppressive effects in vivo by modulating the antigen-presenting function of DCs. However, we could not directly analyze the mechanism of action of the iTregs on the small subset of autoantigen-bearing DC in the gastric lymph node due to the their limited number and lack of a method to isolate the subpopulation of autoantigen-bearing DCs.
We also evaluated the capacity of antigen-specific iTregs to modulate disease induction by fully activated autoantigen-specific Th17 effector cells that, as described above, were resistant to suppressive effects of polyclonal tTregs (25). The resistance of Th17-induced AIG to suppression by Tregs raised the possibility that Th17 cells are intrinsically not responsive to suppression by Tregs or that the inflammatory milieu generated by Th17 cells induced the production of cytokines (IL-6 and TNFα) that rendered T-effector cells resistant to suppression or neutralized the suppressive action of the Tregs (26). While neither polyclonal tTregs nor iTregs could prevent disease induced by Th17 cells, antigen-specific iTregs completely prevented tissue destruction in the gastric mucosa and also suppressed the expansion of the T-effector cells in the gastric lymph node. Antigen-specific iTregs were also capable of suppressing disease when transferred one week after transfer of the Th17 effectors. Marked expansion of the iTregs was observed in the gastric but not inguinal lymph nodes, suggesting that the presence of the autoantigen drove the expansion of the iTregs. As Th17 cells may play an important role in many human autoimmune diseases, the susceptibility of Th17 cells to suppression by antigen-specific iTregs has important implications regarding their use for cellular biotherapy.
Although antigen-specific iTregs appeared to be highly effective in preventing and potentially treating organ-specific autoimmunity and appeared to mediate their effects by preventing T-cell priming, detailed analysis of their mechanism of action is limited as most of these studies involved transfer of both the T effectors and the iTregs to immunoincompetent mice. Thus, lymphopenia-induced proliferation of either the T effector, the iTregs, or both may influence the observed results. We therefore used the cell transfer system we developed to analyze the mechanism of action of polyclonal Tregs to analyze the mechanism of action of iTregs in vivo (27, 28). We generated antigen-specific iTregs as described above and co-transferred them to normal recipients with CFSE-labeled naive T cells from the same donor. All recipients were subsequently immunized with the cognate peptide in adjuvant. Four to five days later, we determined the state of activation, differentiation, and expansion of the naive Foxp3− T cells in the draining lymph node. In marked contrast to the results seen using exactly the same protocol with polyclonal, expanded tTreg, co-transfer of antigen-specific iTregs resulted in inhibition of the expansion of the responder T cells by >90% as measured by CFSE dilution or when the absolute number of responder T cells was counted (Fig. 1). The small number of responder T cells recovered from the draining node failed to upregulate CD44 expression, indicating that their activation was also markedly impaired. Furthermore, when the recovered T cells were stimulated with phorbolmyristate acetate (PMA)/ionomycin to measure cytokine production, responder cells recovered from mice that had not been treated with iTregs produced both IFNγ and IL-17, while the few T cells recovered from mice treated with iTregs produced neither of these cytokines. Thus, antigen-specific iTregs profoundly inhibited all aspects of the T-cell activation cascade. Surprisingly, when we CFSE-labeled the iTregs, they diluted CFSE and expanded.
The profound inhibition of T-cell activation suggested that the iTregs were inhibiting antigen presentation by acting on DCs. We used a three cell transfer model (T naive, iTreg, and antigen-pulsed DC) and recovered the antigen-pulsed DCs 18 h after cell transfer and tested their capacity to activate fresh TCR Tg T cells specific for the same peptide. DCs from animals that did not receive either T naive or iTregs were quite efficient at activating the proliferation of naive T-responder cells and similar results were observed with DCs recovered from animals that received T naive only. However, DCs recovered from animals that received both T naive and iTregs were markedly deficient in their capacity to activate naive responder T cells. These studies strongly suggest that the iTregs disabled the capacity of the DCs to present antigen.
Antigen-specific iTregs impair DC function by modulating expression of MARCH1 and CD83
Although a number of studies have implicated the DCs as the major target of Treg-mediated suppression, the molecular mechanisms involved in this process remain unclear. Tregs constitutively express the inhibitory receptor cytotoxic T-lymphocyte antigen-4 (CTLA-4), and several studies (29, 30) have suggested that the interaction of CTLA-4 on Tregs with CD80/CD86 on DCs results in a decrease in CD80/CD86 expression resulting in defective T-effector cell activation. Strong support in favor of this mechanism was derived from the demonstration that the deletion of CTLA-4 in Foxp3+ Tregs resulted in the development of fatal T-cell mediated autoimmune disease (31). The major mechanism used by Tregs to mediate this cell extrinsic function of CTLA-4 has been termed transendocytosis in which CTLA-4 on the surface of Tregs binds CD80/CD86 on the surface of the DCs followed by their internalization and degradation within the Tregs (32). However, activated T-effector cells also express CTLA-4 on the cell surface and can mediate transendocytosis as efficiently as Foxp3+ Tregs, yet in most instances do not mediate suppression of T-cell activation.
To directly examine the effects of antigen-specific iTreg on DC function, we have developed a two-step culture system in which the iTregs are first co-cultured with antigen-pulsed DCs (33). The DCs are then purified from the co-culture by cell sorting and evaluated for their antigen-presenting capacity by re-culturing them with naive antigen-specific T cells with no further addition of antigen. This protocol avoids any confounding influence of effects of the Tregs on the responder T cell. iTreg-treated DCs were markedly defective in the activation of naive T-cell expansion as assayed by CFSE dilution (Fig. 2). We initially focused on the role of CTLA-4 in mediating downregulation of DC function. Antigen-specific iTregs generated from wildtype but not CTLA-4−/− mice induced a marked downregulation of the expression of CD80/CD86 on the DCs. However, iTregs from CTLA-4−/− mice were still capable of substantially impairing the antigen-presenting function of the DCs, raising the possibility that iTregs use mechanisms in addition to CTLA-4 to impair DC function. One potential candidate for this effect of iTregs on DC function was the immunosuppressive cytokine IL-10. Indeed, production of IL-10 by the iTregs appeared to be the major mediator of this process, as the effects of iTregs from wildtype animals on DC function were completely blocked by inclusion of anti-IL-10 to the iTreg DC co-cultures, and antigen-specific iTreg from IL-10−/− mice failed to decrease antigen presentation by the DCs.
Fig. 2. Antigen-specific iTregs disable the ability of DCs to present antigen.

Antigen-pulsed DCs were cultured for 18 h alone or in the presence of activated antigen-specific T-effector cells or antigen-specific iTregs. The DCs were then separated from the T cells by cell sorting and co-cultured for 72 h with naive antigen-specific T cells. The capacity of the DCs to activate T cells was quantitated by the extent of dilution of CFSE by the T effectors.
The upregulation of MHC class II and CD86 expression on the DC surface during DC maturation result from decreased rates of endocytosis and intracellular degradation secondary to decreased ubiquitination of a critical lysine residue in the MHC class II β-chain cytoplasmic tail by the ubiquitin ligase MARCH1 (34, 35). DC maturation is also accompanied by upregulation of CD83 and the transmembrane domain of CD83 inhibits the action of MARCH1 (36). As the effects of exogenous IL-10 are mediated via upregulation of MARCH1 (36), it was of interest to determine whether iTreg-derived IL-10 mediated downregulation of DC antigen presentation function by using a similar molecular pathway. Not only did antigen-specific iTregs promote the upregulation of MARCH1 mRNA, but they also suppressed the upregulation of CD83. Most, if not all, of these effects were mediated by iTreg-derived IL-10 (33). While the effects of iTreg on the expression of MARCH1 and CD83 are clear, it should be noted that co-culture of the iTregs with DCs produced little if any effect on MHC class II expression. This result was surprising, because MHC class II expression should have been greatly diminished by the enhancement of MARCH1 and the decrease in CD83 induced by iTreg-derived IL-10. Although we did not observe significant downregulation of total MHC class II expression in iTreg-treated DCs, we did observe a marked downregulation of peptide-MHC class II complexes at the 1:1 ratio of iTreg-DCs. The selective effects of iTregs on peptide-MHC class II complexes may reflect the localized effects of IL-10 secreted at the site of interaction of the iTreg TCR with its cognate MHC class II peptide complex (Fig. 3).
Fig. 3. Proposed model for antigen-specific iTreg-mediated downregulation of the expression of MHC class II peptide complexes.

iTregs recognize their cognate antigen via their TCR and locally deliver IL-10 resulting in ubiquitination and degradation of MHC class II- cognate peptide complexes, while leaving complexes lacking the specific peptide intact on the cell surface.
These studies raise important questions about the overall significance of IL-10, as one of the major mechanisms used by all types of Foxp3+ Tregs to suppress DC function and potentially to also suppress T-effector function. While mice with a conditional deletion of IL-10 in Tregs only exhibit defects in Treg function at mucosal surfaces (37), IL-10 production by Tregs is required for several models of autoimmune (38) and infectious diseases (39). Taken together, these studies suggest that IL-10 production by Tregs is a prominent Treg suppressor mechanism in a wide variety of conditions in addition to its critical role in Treg function at mucosal surfaces. The major target of Treg-produced IL-10 is most likely the antigen-presenting DC with resultant inhibition of T-effector cell priming.
Regulation of iTreg stability
Role of IL-2
The studies described above conclusively demonstrate that iTregs generated in vitro can efficiently suppress inflammation in Scurfy mice, and multiple studies in several different animals have shown that they can inhibit effector function in different animal models including AIG (24), EAE (22), and diabetes (40). However, epigenetic studies have raised questions about the stability of Foxp3 expression in iTregs. DNA methylation is one of the most established epigenetic mechanisms in T-cell development and is linked to stable gene expression patterns. Studies with both mouse and human tTregs have demonstrated complete demethylation within an evolutionary conserved region upstream of exon 1 of the Foxp3 locus, and this region has been named the Treg-specific demethylation region or TSDR (2). Demethylation of the TSDR region is a specific marker of tTregs, as CD4+Foxp3− cells and in vitro-generated iTregs display an almost complete methylation of the TSDR. In contrast, the induction of pTregs in vivo by targeting low concentrations of antigen to DCs resulted in antigen-specific pTregs with complete demethylation of the TSDR and stable expression of Foxp3 (3). The status of TSDR methylation is therefore critical for maintaining stable Foxp3 expression and a fully functional Treg phenotype in iTregs. The demonstration (2) that in vitro-induced iTregs have a fully methylated TSDR has raised doubts about their therapeutic usefulness, despite studies not only demonstrating their effectiveness but also their sustained expression of Foxp3 in vivo (24).
To resolve this controversy, we have re-evaluated the factors that regulate the expression of Foxp3 in iTreg both in vivo and in vitro (28). We have used pure populations of antigen-specific iTregs generated from Foxp3 reporter mice to avoid any possible confounding influence of small number of contaminating Foxp3− T cells that might be present in the induction cultures. iTregs restimulated via their TCR in vitro in the absence of TGFβ rapidly lost expression of Foxp3, while Foxp3 expression in iTreg was stable for at least 2 weeks when iTregs were recultured in IL-2 alone. We also examined in detail the fate of iTregs transferred to normal recipients. The results of our in vivo experiments were similar to our in vitro studies. In the absence of TCR stimulation, iTregs retained Foxp3 expression for periods as long as a month, but exhibited minimal proliferation. The iTregs that had retained Foxp3 expression still had a completely methylated TSDR. In contrast, upon restimulation with antigen in vivo, the iTregs rapidly lost Foxp3 expression but did not differentiate into pathogenic effector cells.
As our studies describing the beneficial therapeutic effects of polyclonal iTregs in the Scurfy model (23) suggested that factors present in the inflammatory environment of the Scurfy might potentiate Foxp3 expression in iTregs, we examined whether treatment of mice with IL-2/anti-IL-2 immune complexes (41) would result in stabilization of Foxp3 expression. Treatment of mice with the complexes alone enhanced the proliferation of the transferred iTregs and also markedly enhanced Foxp3 expression in mice that had received iTregs and were subsequently immunized. Most notably, analysis of the methylation status of the TSDR indicated that treatment of mice with both TCR stimulation and IL-2 immune complexes but not IL-2 complexes alone, resulted in significant demethylation of the TSDR. The role of IL-2 in the maintenance of Foxp3 expression was critical, as neutralization of IL-2 potentiated the loss of Foxp3 expression. Most importantly, in a co-transfer model in which antigen-specific iTregs inhibited the expansion of antigen-specific effector cells following immunization (see above), iTregs exhibited stabilization of Foxp3 expression. Both the maintenance of Foxp3 expression and T-suppressor function in vivo were reduced when endogenous IL-2 was neutralized with anti-IL-2. Taken together, these results demonstrate that IL-2 is not only critical for the development and homeostasis of tTregs but also that IL-2 produced by effector T cells is critical for the stability and function of iTregs in vivo. It remains unclear how the synergistic actions of IL-2 and TCR signals promote the induction of TSDR demethylation.
Role of the complement system in iTreg/pTreg induction and stability While IL-2 is absolutely required for iTreg induction in vitro and plays an important role in iTreg stability in vivo, other environmental factors also influence the generation of conventional T cells to iTregs in vitro or pTregs in vivo. The complement fragments C3a and C5a signal through G protein-coupled receptors (C3aR and C5aR) to induce the production of IL-6 and IL-12 (42) that suppress the induction of iTregs. Conversely, the absence of C3aR and C5aR signaling has been shown to potentiate iTreg induction (43). Most importantly, the absence of signaling via these receptors results in the induction of Foxp3+ Treg in vitro in the absence of exogenous TGFβ. iTreg induction was inhibited by anti-TGFβ or by antagonists of TGFβ signaling. Thus, in the absence of C3aR/C5aR signaling, the endogenous production of TGFβ by both CD4+ T cells as well as DCs is enhanced. Antigen-specific iTregs generated in vitro in the absence of C3aR/C5aR signaling exhibited enhanced stability of Foxp3 expression when transferred to normal recipients followed by immunization with their cognate peptide. Surprisingly, the enhanced stability of Foxp3 expression was not accompanied by demethylation of the TSDR raising the possibility that factors in addition to TSDR demethylation may control the stability of Foxp3 expression in vivo. iTregs generated in the absence of C3aR/C5aR signaling were more potent than iTregs generated in the presence of exogenous TGFβ in suppressing anti-CD3 induced proliferation of Foxp3− T cells in vitro, in inhibiting the expansion of antigen-specific T cells in vivo, and were potent inhibitors of established EAE. Following in vivo immunization with cognate peptide, a greater number of pTregs were generated from TCR Tg T cells that lacked C3aR/C5aR expression than from wildtype TCR Tg T cells. Under resting physiologic conditions, signaling via C3aR/C5aR is dormant, and the absence of C3aR/C5aR signaling may play a major role in the induction of pTreg to self-antigens or to foreign antigens at mucosal sites.
Infectious tolerance: induction of iTreg and pTreg by activated tTregs
The term ‘infectious tolerance’ is now used to describe a process where the tolerance inducing properties of one population of cells is transferred to another population. The demonstration that TGFβ is a potent inducer of Foxp3 expression and functional iTreg has raised the possibility Foxp3+ T cells may play a role in converting Tconv cells to iTregs in a TGFβ-dependent manner, thereby generating infectious tolerance. We initially demonstrated that latent TGFβ [TGFβ coupled to latency-associated peptide (LAP)] could be detected on the cell surface of activated but not resting tTreg (44). When activated tTreg were co-cultured with CD4+Foxp3− T cells and re-stimulated with anti-CD3 in the presence of IL-2, 10–30% of the Foxp3− T cells became Foxp3+. Freshly isolated tTreg cells were incapable of inducing Foxp3 expression, and this correlated with the increased expression of latent TGFβ on the surface of the activated tTregs. iTregs induced by co-culture with activated tTregs were markedly suppressive in standard in vitro suppression assays and could also inhibit the induction of IBD when co-transferred with CD4+Foxp3− T cells into RAG−/− mice. Activated tTregs were also capable of inducing pTregs in vivo.
The studies described above identified latent TGFβ+ T cells using a polyclonal antibody to LAP. A number of more recent studies have fully characterized the nature of the cell surface latent TGFβ complex (45–47). We have demonstrated that activated Foxp3+ T cells express a distinct cell surface protein termed GARP (or LRRC32), which binds latent TGFβ and is required for surface expression of latent TGFβ. Latent TGFβ does not have biologic activity and the release of active TGFβ from LAP is a critical regulatory step for TGFβ function. The biochemical pathways by which GARP-associated latent TGFβ is activated have not been completely elucidated, but it is likely that αV integrins play a role this process (48). Foxp3 is not essential for the expression of the GARP/latent TGFβ on activated human Tregs, because the GARP/latent TGFβ complex was completely normal following siRNA-mediated knockdown of Foxp3 (47).
We have recently extended our studies on the expression of the GARP/latent TGFβ to mouse Tregs (49). Resting mouse tTregs express low levels of GARP mostly in the absence of latent TGFβ. TCR stimulation induces an initial upregulation of GARP expression that is followed rapidly by detection of the GARP/latent TGFβ complex. A large proportion of this initial increase in cell surface expression of GARP may be secondary to its release from intracellular stores. Lower levels of expression of GARP can be seen by culturing tTregs in media alone, and further increases in expression can be induced by the addition of IL-2 or IL-4. Both in vivo (pTregs) and in vitro (iTregs) generated mouse Foxp3+ T cells expressed the GARP/latent TGFβ complex. This result should be contrasted with the results of our studies on human T cells, which failed to demonstrate GARP or LAP following in vitro induction of Foxp3 expression by TCR stimulation in the presence of TGFβ. Thus, the expression of GARP represents an excellent marker of functional Tregs in the mouse although it does not distinguish tTregs from pTregs (or iTregs). Expression of GARP was not dependent upon the expression of latent TGFβ, because cells from mice with a T-cell conditional deletion of TGFβ expressed levels of GARP equivalent to those expressed on wildtype Tregs both before and after T-cell activation. LAP expression can be restored on activated mouse Treg from TGFβ−/− mice by incubation with recombinant latent TGFβ. Thus, the GARP/latent TGFβ complex can be generated from intracellular stores and by an extracellular pathway.
What is the function of the GARP/latent TGFβ complex on activated Tregs? The contribution of cell surface expressed latent TGFβ in mediating Treg suppression of responder T-cell activation has been controversial. Tregs from GARP−/− mice were as suppressive as wildtype Tregs in mediating suppression in vitro. Although GARP−/− Tregs can still secrete latent TGFβ, it is unlikely that latent TGFβ plays any role in suppression in vitro, because the suppressive ability of TGFβ−/− Tregs is completely normal (50). As noted above, one potential function of the cell surface GARP/latent TGFβ complex may be to mediate infectious tolerance. Indeed, activated Treg from GARP−/− were poor inducers of Foxp3 expression compared to wildtype Tregs when co-cultured with naive responder cells. It is likely that in vivo Tregs and antigen-specific T cells are stimulated on the surface of the same DC. Stimulation of the Treg would induce the GARP/latent TGFβ complex that could then release activated TGFβ resulting in conversion of the antigen-specific Foxp3− T cells to a pTreg. However, one study (51) has also suggested that activated Tregs could induce Th17 cells in the presence of IL-6. The GARP/latent TGFβ complex also appears to play a predominant role in this process as activation of co-cultures of activated Tregs and naive responder cells in the presence of IL-6 resulted in induction of a substantial percentage of Th17 cells. The source of the activated TGFβ was primarily the GARP/latent TGFβ complex rather than secreted latent TGFβ. Thus, activated tTregs can utilize the GARP/latent TGFβ complex to induce both Tregs or in the presence of an inflammatory milieu (IL-6) induce Th17 cells. A clear explanation of why Foxp3+ Tregs would play an important role in the induction of potentially pathogenic Th17 cells is lacking. One possibility is that the Th17 cells induced by this pathway are primarily non-pathogenic Th17 cells.
Can functional iTregs be generated from naive human T cells?
As described above, iTregs that function both in vitro and in vivo can readily be generated from naive mouse T cells in the presence of TGFβ. It would be highly desirable to be able to similarly generate functional human iTregs. Considerable controversy exists as to whether functional human iTregs can be generated in vitro using culture conditions similar to those used in the mouse. One major issue complicating the analysis of human Tregs is that activated conventional T cells can express Foxp3 in the absence of Treg activity. It is therefore imperative that multiple assays be used to determine whether the induced Foxp3+ T cells are in fact bona fide Tregs. In our studies (52), TCR stimulation of naive human CD4+ Foxp3− T cells in the presence of TGFβ and IL-2 induced high levels of Foxp3 expression that was stable for several weeks when the cells were maintained in the presence of IL-2. In contrast to mouse iTregs that were both anergic and suppressive, the human iTregs proliferated when stimulated via the TCR and failed to suppress responder CD4+Foxp3− T cells. Furthermore, when these cells were re-stimulated with PMA and ionomycin and cytokine production measured at the single cell level by intracellular staining, the majority of the Foxp3+ T cells produced IL-2 and/or IFNγ. Similar results were obtained when naive CD4+ T cells from cord blood were induced to express Foxp3.
It remains unclear why it has been difficult to induce functional human iTregs in culture. One study has reported that the addition of retinoic acid to the induction regimen resulted in the development of anergic and suppressive human iTregs (53). We have been unsuccessful in reproducing these observations. An alternative approach involved the addition of rapamycin to the inductive cultures resulting in human iTregs that stably expressed Foxp3, which were suppressive in vitro and in vivo (54). Rapamycin has the unique properties of blocking the mTOR pathway as well as Smad7, a negative regulator of TGFβ signaling. Again, we have had difficulty reproducing these findings. Lastly, it has been possible to generate functional human iTregs by blocking C3aR/C5aR signaling with pharmacologic antagonists or mAbs to C3a and C5a (43). However, the Tregs induced by C3aR/C5aR antagonism have not been evaluated for suppressive function in vivo or for TSDR demethylation. Taken together, these studies indicate major differences exist between the susceptibility of naive human and mouse T cells to become iTregs in the presence of TGFβ. The purported success of agents such as retinoic acid and rapamycin, or antagonism of complement signaling require further studies by multiple laboratories. Large-scale clinical trials to test feasibility, efficacy, and safety will be required before iTreg cellular therapy for human diseases becomes routine practice.
Can tTregs be distinguished from pTregs or iTregs?
A large body of data supports the view that a proportion of the Foxp3+ Treg population is generated extrathymically from T-conventional cells. As the properties of these pTregs may differ markedly from tTregs, it would be important to develop reagents that would distinguish these two populations. As cellular biotherapy with Tregs is adopted for treatment of human disease, it would also be highly desirable to characterize the tTreg/pTreg makeup of the final product. While it is reasonable to assume that tTregs and pTregs differ in their TCR repertoires, one must also consider that the two populations may also differ in their stability and their suppressor mechanisms. We have proposed that expression of the transcription factor Helios distinguished tTregs from pTregs/iTregs (55).
Helios is a member of the Ikaros transcription factor family that is characterized by four highly conserved N-terminal zinc fingers and two highly conserved C-terminal zinc fingers (56). The Ikaros family is comprised of five DNA binding proteins that, as homodimers or heterodimers, function to regulate gene expression through chromatin remodeling. We became interested in Helios when we observed in a PCR-based cDNA subtraction library that Helios was preferentially expressed in Treg cells when compared to conventional CD4+CD25− T cells. To study the function of Helios, we generated Helios-deficient mice on a C57BL/6 background. It has been reported that in adult mice, Helios is expressed only in the thymus (57), so we were surprised to observe that Helios deficiency was lethal within the first day of life. During embryogenesis, Helios is first observed at day 8 in the blood islands of the yolk sac; however, it is then expressed in a subset of cells in the liver at day 11 and several epithelial tissues, including the olfactory epithelium and the lining of the gut and the respiratory tract at day 17, suggesting that Helios expression during embryogenesis, in tissues other than the immune system, is critical for survival immediately after birth. We have also developed mice in which Exon 8 of the Helios gene is flanked by loxP sites. When crossed to mice expressing either CD4-Cre to delete Helios in the T-cell compartment or to mice expressing Vav-Cre to delete Helios in the entire lymphoid compartment, all offspring were healthy and bred normally (55, authors’ unpublished data). Deletion of Helios had no effect on the development of Treg in the thymus or in their maintenance in the periphery. The suppressive function of Helios−/− Tregs was normal both in vitro and in vivo. Cai et al. (58) reported similar results.
Although our studies thus far have not uncovered a function for Helios in Tregs, we have developed a hamster monoclonal antibody (22F6) to the non-conserved N-terminal amino acid domain that allowed us to examine Helios expression on a single cell basis in both mouse and human Tregs (55). We demonstrated that Helios is first expressed at the DN2 stage of thymic differentiation, remains high during DN3 and declines in DN4. Helios expression then reappears at the CD4 SP stage in both Foxp3+ and Foxp3− cells. In the periphery, Helios is expressed in approximately 70% of CD4+Foxp3+ mouse Treg cells and 70–80% of human Tregs, as well as in a very small subset of CD4+Foxp3− cells. Helios+ Treg cells exhibit a more ‘Treg-like’ phenotype in that the MFI for GITR and CD25 is higher compared to Foxp3+Helios− cells and Helios+ Treg cells have higher percentages of CD103+, CD134+, CD120b+, and Ki67+ cells (authors’ unpublished data). We also observed that a small percentage of cells in the Foxp3+Helios− population were able to produce cytokines.
Our hypothesis that Helios is a marker of tTreg cells was based on three main observations. First, the earliest Foxp3+ T cells to arise in the thymus of 3 day-old mice and the first Foxp3+ T cells Tregs to populate the spleen in 4 day-old mice are exclusively Helios+. It is not until day 12 of life that Helios− splenic Tregs begin to appear and not until after weaning that the normal ratio of Helios−/Helios+ Tregs is established. Second, CD4+Foxp3− cells isolated from Foxp3-GFP reporter mice that are induced in vitro by activation in the presence of TGFβ to become Foxp3+Tregs (iTregs) do not express Helios. Lastly, pTregs induced in vivo using a model of oral tolerance also failed to express Helios. We have also shown that pTregs induced in vivo by intravenous injection of low dose antigen in the absence of adjuvant do not express Helios (authors’ unpublished observation).
Since our initial report, several other reports have subsequently demonstrated that Foxp3+ iTregs may under certain conditions also express Helios. In our initial experiments, iTregs were generated by stimulation of naive T cells with plate-bound anti-CD3 in the presence of TGFβ. However, Verhagen and Wraith (59) demonstrated that Helios was expressed in approximately 50% of iTregs generated from CD4+Foxp3− cells from RAG−/− TCR Tg T cells (Tg4 mice) when stimulated with their cognate peptide in the presence of antigen-presenting cells and TGFβ. They could confirm our results that iTregs induced from the same TCR Tg T cells by stimulation with plate-bound anti-CD3 and anti-CD28 were Helios−. Similar findings were observed by I. Van Driel’s group (personal communication) using CD4+CD25− cells from the TxA23 TCR Tg strain. However, we have observed that CD4+CD25− T cells from this strain contain a very significant percentage of Helios+ Foxp3− cells, and these cells may be the precursors of the Helios+Foxp3+ T cells that are generated in vitro. Akimova et al. (60) have also published that Helios does not identify thymic Tregs but, rather, is merely a marker of activation. However, in their studies, Helios expression was examined on responder CD4+Foxp3− cells in suppression co-cultures, and co-culture results are very difficult to interpret. Helios expression was examined on T-effector cells after 3 days, a time point in which there are very few, if any, cells remaining in the culture due to Treg-induced inhibition of IL-2 production. In the one experiment in which they directly examined Helios expression on iTregs induced with TGFβ, the cells were stimulated with plate-bound anti-CD3 and anti-CD28, conditions under which we, Verhagen and Wraith (59), and Van Driel (personal communication) do not observe Helios expression on the induced Foxp3+ population.
We have now done exhaustive experiments to examine the conditions under which Helios may be induced in vitro. We used a number of different protocols for TCR stimulation (plate bound anti-CD3, plate bound anti-CD3 and CD28, anti-CD3 and anti-CD28 coated beads, soluble anti-CD3 plus T-depleted splenocytes, or DC and TCR Tg CD4+ cells stimulated with antigen and various APCs). We were unable to see the induction of Helios in the absence of TGFβ. In the presence of TGFβ, we observed the induction of Foxp3+ Helios+ T cells only when TCR stimulation included non-T cell antigen-presenting cells. At this time, it is not clear why Helios is induced in the presence of antigen-presenting cells. The induction does not appear to be cytokine mediated, as antibodies to IFNγ, IL-4, IL-6, IL-12, and TNFα, separately or in combination, do not inhibit Helios induction. We have also observed that a high percentage of Foxp3− antigen-specific T cells can express Helios at early time points after immunization in vivo. Thus, it appears that some conventional T cells can express low levels of Helios under certain conditions, and it remains possible that Foxp3−Helios+ T cells will continue to express Helios when they are converted to pTreg in vivo or iTregs in vitro.
One major drawback to the use of Helios to distinguish tTreg subpopulations is that it is expressed intracellularly and cannot be used for cell isolation. Two recent reports (61, 62) have claimed that the cell surface antigen neuropilin 1 (Nrp1) distinguishes tTregs from pTregs or iTregs. In one study (61), Nrp1 was found on gene expression profiles to be expressed on Foxp3+ cells from unmanipulated mice but not on pTregs induced in vivo by oral tolerance, confirming our observations that Helios does not appear to be expressed by in vivo generated pTregs. However, when Helios expression was examined in Nrp1+ and Nrp1− Tregs, Nrp1 and Helios expression correlated well in that Nrp1+ cells were also Helios+, but Foxp3+Helios− T cells contained a significant fraction of Nrp1+ cells, particularly in the peripheral lymph node. We have similarly observed that only about 30–40% of the Foxp3+ Helios− T cells are Nrp1− (Fig. 4). One difficulty with the claim that pTregs and iTregs are Nrp1− is that expression Nrp1 is positively regulated by TGFβ and Nrp1 is expressed on TGFβ-induced iTregs (61). Furthermore, under inflammatory conditions, such as those encountered in the spinal cords of mice with EAE, Nrp1 is expressed at high levels on pTregs. Thus, it is difficult to accept the claim that the differential expression of Nrp1 is useful in distinguishing tTregs from pTregs and iTregs, as it can be detected at high levels on both pTregs and iTregs.
Fig. 4. A comparison of the expression of Helios and Nrp1 in Foxp3+ and Foxp3− T cells.
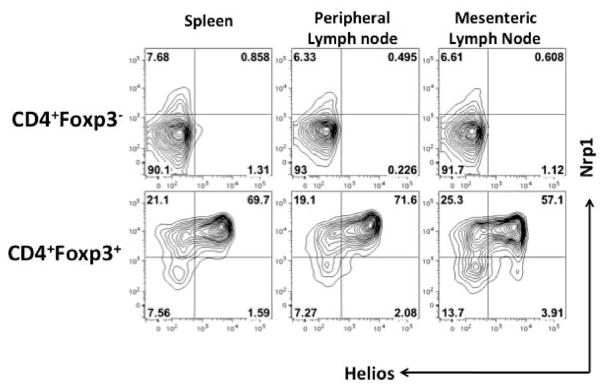
Lymphocytes from the indicated tissues of 8-week-old C57BL/6 mice were stained for CD4 and Nrp1 and then analyzed for expression of Foxp3 and Helios by intracellular staining. Helios and Nrp1 expression are shown when gated on CD4+Foxp3− cells (top) or CD4+Foxp3+ cells (bottom).
To functionally compare Foxp3+ Helios+ and Foxp3+Helios− subpopulations, we have generated Helios reporter mice and crossed them to Foxp3-RFP reporter mice. Among the various lymphoid organs, expression of the Helios reporter accurately reflected the percentage of Helios+ T cells detected by intracellular staining using the 22F6 antibody. We isolated Foxp3+Helios− and Foxp3+Helios+ cells and compared their suppressive capacity both in vitro and in vivo. Foxp3+Helios− cells are slightly, but reproducibly, less suppressive in standard in vitro assays. However, in vivo, using the CD4+CD45RBhi transfer model of IBD, we saw no differences in the suppressive properties of the two Treg subsets. The methylation status of the TSDR of the Foxp3 gene is now widely used to demonstrate stable ‘true’ Tregs. Methylation analysis of the TSDR of Foxp3+Helios− cells and Foxp3+Helios+ cells revealed less demethylation of the TSDR of the Foxp3+Helios− T (80–85%) cells compared to Foxp3+Helios+ (90%) cells (authors’ unpublished data). However, the extent of demethylation of the TSDR of the Foxp3+Helios− cells was still significantly greater than that observed with CD4+Foxp3− (10–20%) cells or Foxp3+ Helios− iTreg (10%) induced with plate-bound anti-CD3 and TGFβ. We obtained a different result when we compared the methylation status of human Foxp3+Helios+ and Foxp3+Helios− subpopulations (63). The TSDR of the human Foxp3+Helios+ subset was fully demethylated, while the TSDR region of the Foxp3+ Helios− subset was 45% demethylated. One interpretation of this finding is that the Foxp3+Helios− subset is composed of two subsets, one of which expresses a completely demethylated TSDR, while the TSDR of the second subset is completely methylated. The latter subset may, in fact, represent activated T-conventional cells that lack Treg function.
Concluding remarks
A number of studies (64–66) have reported preliminary findings regarding the use of Tregs to prevent graft versus host disease (GVHD) in human, and a phase 1 study has been initiated for the treatment of type I diabetes (67). Yet, we know very little about the proposed mechanisms of action of Tregs in vivo. In this review, we have described our studies in the mouse that have begun to dissect some of the mechanisms used by Tregs to modulate the priming of naive T cells. We have demonstrated that the effects of activated polyclonal tTregs differ markedly from the effects of antigen-specific iTregs. The former modulate T-effector trafficking, while the latter inhibit T-effector priming. We have not yet extended these studies to the analysis of the effects of these Treg subpopulations on primed T cells present in intact mice. We have also described that tTregs have the unique capacity to initiate infectious tolerance via the delivery of cell surface TGFβ bound to GARP and thereby generate antigen-specific pTregs. While pTregs are present in vivo, the ratio of tTregs to pTregs has not been completely clarified with the available markers. Future studies must resolve this issue. A clear definition of tTreg and pTreg populations will be required to determine the optimal composition of Tregs needed for the cellular biotherapy of GVHD, graft rejection, and different autoimmune diseases.
Acknowledgments
The studies described in this review were supported by funds from the Intramural Program of the National Institute of Allergy and Infectious Diseases. The authors would like to thank all members of the Cellular Immunology Section who contributed to these studies.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Abbas AK, et al. Regulatory T cells: recommendations to simplify nomenclature. Nat Immunol. 2013;14:307–308. doi: 10.1038/ni.2554. [DOI] [PubMed] [Google Scholar]
- 2.Floess S, et al. Epigenetic control of the foxp3 locus in regulatory T cells. Plos Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polansky JK, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:165–1663. doi: 10.1002/eji.200838105. [DOI] [PubMed] [Google Scholar]
- 4.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25): breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 5.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 6.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 7.DiPaolo RJ, Glass DD, Bijwaard KE, Shevach EM. CD4+CD25+ T cells prevent the development of organ-specific autoimmune disease by inhibiting the differentiation of autoreactive effector T cells. J Immunol. 2005;175:7135–7142. doi: 10.4049/jimmunol.175.11.7135. [DOI] [PubMed] [Google Scholar]
- 8.Suri-Payer E, Kehn PI, Cheever AW, Shevach EM. Pathogenesis of post-thymectomy autoimmune gastritis: identification of anti-H/K adenosine triphosphate-reactive T cells. J Immunol. 1996;157:1799–1805. [PubMed] [Google Scholar]
- 9.McHugh RS, Shevach EM, Margulies DH, Natarajan K. A T cell receptor transgenic model of severe, spontaneous organ-specific autoimmunity. Eur J Immunol. 2001;31:2094–2103. doi: 10.1002/1521-4141(200107)31:7<2094::aid-immu2094>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 10.Candon S, McHugh RS, Fourcas G, Natarajan K, Shevach EM, Margulies DH. Spontaneous organ-specific TH2-mediated autoimmunity in TCR transgenic mice. J Immunol. 2004;172:3157–3166. doi: 10.4049/jimmunol.172.5.2917. [DOI] [PubMed] [Google Scholar]
- 11.Stummvoll GH, et al. Th1, Th2 and Th17 effector T cell-induced autoimmune gastritis differs in pathological pattern and susceptibility to suppression by regulatory T cells. J Immunol. 2008;181:1908–1916. doi: 10.4049/jimmunol.181.3.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samy ET, Parker LA, Sharp CP, Tung KS. Continuous control of autoimmune disease by antigen-dependent polyclonal CD4+CD25+ regulatory T cells in regional lymph node. J Exp Med. 2005;202:771–781. doi: 10.1084/jem.20041033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting Edge: CD4+CD25high regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 14.Davidson TS, Shevach EM. Polyclonal Treg cells modulate T effector cell trafficking. Eur J Immunol. 2011;41:2862–2870. doi: 10.1002/eji.201141503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thangada S, et al. Cell surface residence of sphingosine 1-phosphae receptor 1 on lymphocytes determines lymphocyte egress kinetics. J Exp Med. 2010;207:1475–1483. doi: 10.1084/jem.20091343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hori S, Haury M, Coutinho A, Demengeot J. Specificity requirements for selection and effector function of CD25+CD4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proc Nat Acad Sci USA. 2002;99:8213–8218. doi: 10.1073/pnas.122224799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang Q, et al. In vitro expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2002;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, et al. Conversion of peripheral CD4+CD25+ naïve T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-β-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 20.Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar ML. IL-2, −7, −15, but not thymic stromal lymphopoietin, redundantly govern CD4+Foxp3+ regulatory T cell development. J Immunol. 2008;181:3285–3290. doi: 10.4049/jimmunol.181.5.3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of IL-2. Nat Immunol. 6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 22.Selveraj RK, Geiger TL. Mitigation of experimental allergic encephalomyelitis by TGF-β indudced Foxp3+ regulatory lymphocytes through the induction of anergy and infectious tolerance. J Immunl. 2008;180:6329–6337. doi: 10.4049/jimmunol.180.5.2830. [DOI] [PubMed] [Google Scholar]
- 23.Huter EN, Punkosdy GA, Glass DD, Cheng LI, Ward JM, Shevach EM. TGF-β-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur J Immunol. 2008;38:1814–1821. doi: 10.1002/eji.200838346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiPaolo RJ, Brinster C, Davidson TS, Andersson J, Glass D, Shevach EM. Autoantigen-specific TGF-β-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J Immunol. 2007;179:4685–4693. doi: 10.4049/jimmunol.179.7.4685. [DOI] [PubMed] [Google Scholar]
- 25.Huter EN, Stummvoll GH, DiPaolo RJ, Glass DD, Shevach EM. Cutting Edge: Antigen-specific TGFβ-induced regulatory T cells suppress Th17-mediated autoimmune disease. J Immunol. 2008;181:8209–8213. doi: 10.4049/jimmunol.181.12.8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korn T, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shevach EM. Biological functions of regulatory T cells. Adv Immunol. 2011;112:137–176. doi: 10.1016/B978-0-12-387827-4.00004-8. [DOI] [PubMed] [Google Scholar]
- 28.Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-β-induced Foxp3+ T cells in vivo. J Immunol. 2011;186:6329–6337. doi: 10.4049/jimmunol.1100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cederbom L, Hall H, Ivars F. CD4+ CD25+ regulatory T cells down regulate co-stimulatory molecules on antigen presenting cells. Eur J Immunol. 2000;30:1538–1543. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 30.Serra P, et al. CD40 Ligation Releases Immature Dendritic Cells from the Control of Regulatory CD4+ CD25+ T Cells. Immunity. 2003;19:877–889. doi: 10.1016/s1074-7613(03)00327-3. [DOI] [PubMed] [Google Scholar]
- 31.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 32.Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chattopadhay G, Shevach EM. Antigen-specific iTreg impair DC function via an IL-10/MARCH1-dependent mechanism. J Immunol. 2013 doi: 10.4049/jimmunol.1301693. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin JS, Ebersold M, Pypaert M, Delmarre L, Hartley A, Mellman I. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature. 2006;444:115–118. doi: 10.1038/nature05261. [DOI] [PubMed] [Google Scholar]
- 35.De Gassart A, et al. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH 1 down-regulation. Proc Natl Acad Sci USA. 2008;105:3491–3496. doi: 10.1073/pnas.0708874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tze LE, et al. CD83 increases MHC class II and CD86 on dendritic cells by opposing IL-10-driven MARCH1–mediated ubiquitination and degradation. J Exp Med. 2011;208:149–165. doi: 10.1084/jem.20092203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubtsov YP, et al. Regulatory T cell-derived IL-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, et al. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 39.Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ immunoregulatory T lymphocytes control Leishmania major persistence and the development of concomitant immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- 40.Weber SE, et al. Adaptive islet-specific regulatory CD4 T cells control autoimmune diabetes and mediate disappearance of pathogenic Th1 cells in vivo. J Immunol. 2006;176:4730–4739. doi: 10.4049/jimmunol.176.8.4730. [DOI] [PubMed] [Google Scholar]
- 41.Webster KE, et al. In vivo expansion of T reg cells with IL-2 mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med. 2009;206:751–760. doi: 10.1084/jem.20082824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strainic MG, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naïve CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4+ cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3+ regulatory T cells. Nat Immunol. 2103;14:162–171. doi: 10.1038/ni.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersson J, et al. CD4+Foxp3+ regulatory T cells confer infectious tolerance in a TGF-β-dependent manner. J Exp Med. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLos ONE. 2008;3:e2705. doi: 10.1371/journal.pone.0002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol. 2009;39:3315–3322. doi: 10.1002/eji.200939684. [DOI] [PubMed] [Google Scholar]
- 47.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent-TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Nat Acad Sci USA. 2009;106:13445–13450. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang R, Zhu J, Dong Z, Shi M, Lu C, Springer TA. GARP regulates the bioavailability and activation of TGFβ. Mol Biol Cell. 2012;23:1129–1139. doi: 10.1091/mbc.E11-12-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edwards JO, Fujii H, Zhou AX, Creemers J, Unutmaz D, Shevach EM. Regulation of the expression of GARP/Latent TGF-β1 complexes on mouse T cells and their role in regulatory T cell and Th17 differentiation. J Immunol. 2013;190:5506–5515. doi: 10.4049/jimmunol.1300199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li M, Wan YY, Flavell RA. T cell-produced transforming factor beta 1 controls T cell tolerance and regulated Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 51.Xu L, Kitani A, Fuss I, Strober W. Cutting Edge: Regulatory T cells induce CD4+CD25+Foxp3+ T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-β. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 52.Tran DQ, Ramsey H, Shevach EM. Induction of Foxp3 expression in naïve human CD4+Foxp3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ling L, Zhou X, Wang J, Zheng SG, Horwtiz DA. Characterization of protective human CD4+CD25+Foxp3+ regulatory T cells generated with IL-2, TGF-β, and retinoic acid. Plos One. 2010;5:e15150. doi: 10.1371/journal.pone.0015150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hippen KL, et al. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am J Transplantation. 2011;11:1148–1157. doi: 10.1111/j.1600-6143.2011.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thornton AM, et al. Expression of Helios, and Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;181:3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.John LB, Ward AC. The Ikaros gene family: Transcriptional regulators of hematopoiesis and immunity. Mol Immunol. 2011;48:1272–1278. doi: 10.1016/j.molimm.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 57.Kelley CM, et al. Helios, a novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Current Biol. 1998;8:508–515. doi: 10.1016/s0960-9822(98)70202-7. [DOI] [PubMed] [Google Scholar]
- 58.Cai Q, Dierich A, Oulad-Abselghani M, Chan S, Kastner P. Helios deficiency has minimal impact on T cell development and function. J Immunol. 2009;183:2303–2311. doi: 10.4049/jimmunol.0901407. [DOI] [PubMed] [Google Scholar]
- 59.Verhagen J, Wraith DC. Comment on “Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells”. J Immunol. 2010;185:7129–7129. doi: 10.4049/jimmunol.1090105. [DOI] [PubMed] [Google Scholar]
- 60.Akimova T, Beier UH, Wang L, Levine MH, Hancock WW. Helios expression is a marker of T cell activation and proliferation. Plos One. 2011;6:e24226. doi: 10.1371/journal.pone.0024226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiss JM, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012;209:1723–1742. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yadav M, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209:1713–1722. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim YC, et al. Oligodeoxynucleotides stabilize Helios-expressing Foxp3+ human T regulatory cells during in vitro expansion. Blood. 2012;119:2810–2818. doi: 10.1182/blood-2011-09-377895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trzonkowski P, et al. First in man clinical results of the treatment of patients with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin Immunol. 2009;133:22–26. doi: 10.1016/j.clim.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 65.Brunstein CG, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;20:1061–1070. doi: 10.1182/blood-2010-07-293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Ianni M, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 67.Marek-Trzonkowska N, et al. Administration of CD4+CD25highCD127− regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care. 2012;35:1817–1820. doi: 10.2337/dc12-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]