

Abstract
β-Amyloid (Aβ) concentration in the cerebrospinal fluid (CSF) of the brain may be regulated by the choroid plexus, which forms a barrier between blood and brain CSF. Aβ uptake from CSF was determined as its volume of distribution (VD) into isolated rat choroid plexus tissue. The VD of [125I]Aβ1–40 was corrected by subtraction of the VD of [14C]sucrose, a marker for extracellular space and diffusion. Aβ uptake into choroid plexus was time and temperature dependent. Uptake of [125I]Aβ was saturable. Aβ uptake was not affected by addition of transthyretin or apolipoprotein E3. In studies with primary culture monolayers of choroidal epithelial cells in Transwells, Aβ permeability across cells, corrected by [14C]sucrose, was greater from the CSF-facing membrane than from the blood-facing membrane. Similarly, cellular accumulation of [125I]Aβ was concentrative from both directions and was greater from the CSF-facing membrane, suggesting a bias for efflux. Overall, these results suggest the choroid plexus selectively cleanses Aβ from the CSF by an undetermined mechanism(s), potentially reducing Aβ from normal brains and the brains of Alzheimer’s disease patients.
Keywords: choroid plexus, β-amyloid, cerebrospinal fluid
The formation of insoluble aggregates of β-amyloid peptide (Aβ) within the brain extracellular fluid is considered to be a critical event in the etiology of Alzheimer’s disease (AD) (1, 2). Aβ accumulation in AD brains may occur by one or more processes, including overproduction in the brain, inadequate metabolic clearance within the brain, or an improper balance of import and export of Aβ or Aβ-related molecules at brain barriers. Two brain barriers separate brain cells and cerebral fluids from the blood. The blood-brain barrier (BBB), whose structural basis is the tightly connected cerebral capillary endothelia, occurs between the blood and cerebral interstitial fluid. The flux of Aβ at the BBB has been described (3). The blood–cerebrospinal fluid barrier (BCB) separates blood from the cerebrospinal fluid (CSF) circulating within the brain. The BCB occurs at the choroid plexus tissue, which transports materials into the brain by CSF secretion and serves as a primary site for efflux or clearance of brain metabolites eluted from the interstitial fluid to the ventricular CSF (4). The mechanisms by which the brain metabolizes and eliminates Aβ, specifically at the BCB, are poorly understood.
Aβ is present in the CSF of normal and AD brains. The CSF concentration ratio of two Aβ peptides (Aβ1–40 to Aβ1–42) is a suggested biomarker for AD, although the individual concentrations of the two peptides are variable among AD patients (5, 6). Aβ accumulation occurs within the choroid plexus epithelia of AD patients (7). It is unknown whether the choroid plexus plays any role in Aβ accumulation in AD; even less is known about how aging, disease, or toxicant exposure conditions that affect choroid plexus function may interfere with the homeostasis of Aβ in the CSF.
This study examined the kinetic aspect of uptake, accumulation, and transport of Aβ by the choroid plexus. We used isolated rat choroid plexus tissues to examine net Aβ1–40 uptake. Freshly isolated choroid plexus tissue maintains the primary characteristics of the BCB and is an ideal model system for obtaining the essential uptake parameters at the BCB. Aβ1–40 was selected as a model compound for all Aβ peptides because of its optimal solubility. We also used the primary culture of choroidal epithelial cells to investigate the direction of Aβ transport by the choroid plexus and the intracellular accumulation at equilibrium. Our objective was to determine whether the BCB played an important role in Aβ homeostasis within the CSF from which to understand the role of BCB in AD causation, progression, and in the design of therapeutic strategy.
Materials and Methods
Materials
Male Sprague-Dawley rats were purchased from Harlan Laboratory Animals (Indianapolis, IN). Materials were purchased from the following sources: [125I]Aβ1–40 (1700–2000 Ci/mmol) from Amersham Biosciences (Piscat-away, NJ) and Phoenix Pharmaceuticals (Belmont, CA), [14C]sucrose from Moravek (Brea, CA), cell culture reagents from Invitrogen (Carlsbad, CA), Corning Transwell-COL chambers (1 cm2 growing space, 0.4 μm pore size) from VWR (Batavia, IL), and pronase from EMD Biosciences (La Jolla, CA). All other chemicals were purchased from Sigma (St. Louis, MO). Culture medium and serum-free medium were prepared as previously described (8). In tissue experiments, stock [125I]Aβ solution was added to a MicroCon YM-3 filtration device (Millipore, Bedford, MA) with a nominal molecular weight cutoff of 3000 daltons and was spun in an Eppendorf centrifuge at 14,000 g for 30 mins. The filtrate containing free 125I was discarded, and the retentate containing [125I]Aβ1–40 was collected by centrifugation of the inverted device at 1000 g for 3 mins.
Uptake Study in Isolated Choroid Plexus
Male rats (225–275 g) were anesthetized by ip injection of pentobarbital (50 mg/kg) and euthanized by exsanguination to remove excess blood from the choroid plexus. The brains were removed immediately and washed in ice-cold PBS. The lateral ventricular choroid plexuses were removed, weighed, and immediately placed in an artificial CSF containing (in mM): Na+, 136.2; K+, 3; Ca2+, 1.1; Mg2+, 0.8; Cl−, 123.8; HPO42−, 0.6; HCO3−, 18; urea, 2; and glucose, 2, at a final pH of 7.3. For uptake studies, [125I]Aβ (0.5 μCi/ml, with unlabeled Aβ to total 10 ng/ml) and [14C]sucrose (0.5 μCi/ml) were added to each artificial CSF. Uptake inhibitors or Aβ binding agents were added to some artificial CSF solutions to measure their effects. After the time course study, an uptake time of 2 mins was selected for subsequent studies to maintain the kinetics within the linear portion of the uptake curve, where the uptake is not significantly confounded by substrate back flux from the tissue. To end the incubation, the tissue was removed from artificial CSF, dragged along a plastic slide to remove excess fluid, and solubilized. For studies of total 125I label, the tissue was solubilized in NaOH (1 M) and neutralized with equimolar HCl. In filtered [125I]Aβ uptake studies, the choroid plexuses were solubilized in artificial CSF containing 1% Triton X-100 at 4°C, followed by tissue centrifugation through a 3000 daltons mol wt cutoff membrane (14,000 g, 30 mins, 4°C). Medium aliquots and digested tissue were assayed for 125I and 14C radioactivity by liquid scintillation with corrections for crossover. All animal experiments were conducted under the guidelines of the Guide for the Care and Use of Laboratory Animals and had approval of the Purdue University Animal Care and Use Committee.
Primary Culture of Choroidal Epithelial Cells
Primary cultures of choroid plexus epithelial cells were obtained as previously described (8). Briefly, rats (150–175 g) were anesthetized and killed as described above. The brains were removed under aseptic conditions and immediately placed in ice-cold, sterile PBS. The choroid plexuses from the lateral, third, and fourth ventricles were removed. Pooled tissue was minced mechanically and digested with pronase (2 mg/ml) at 37°C for 10–15 mins. Isolated cells were washed and plated at 2 × 105 cells/Transwell insert (1 cm2) in culture medium.
AβFlux Studies
The formation of a barrier between two chambers was considered mature if it met the three following criteria: a difference in fluid height between chambers, a transepithelial electrical resistance above 80 Ω-cm2, and a [14C]sucrose permeability below 8 × 10−4 cm/ min; the latter two values were within those reported in literature (8, 9). Cells were washed three times in 37°C serum-free medium (SFM) and allowed to equilibrate for 10 mins. The final wash was replaced with SFM in the receiver chamber and SFM plus [125I]Aβ (0.5–1 μCi/ml) and [14C]sucrose (0.1–0.5 μCi/ml) in the donor chamber. For influx experiments, the outer chamber was the donor chamber; for efflux, the inner chamber was the donor chamber. Cell viability, measured by methylthiazolyldiphe-nyltetrazolium bromide (MTT) conversion to its formazan product in a parallel study, was not changed for at least 2 hrs when the cells were cultured in SFM (data not shown).
Medium aliquots (10 μL; ≤ 2% total volume) were removed from the receiver chamber at designated times and replaced with tracer-free SFM. At the end of the flux experiment, the inserts were washed 3 times in ice-cold, radioisotope-free SFM. Each filter was removed and placed in 0.2 M NaOH containing 1% SDS (0.5 ml) to solubilize the cells. The cell lysates were neutralized by equimolar HCl. Aliquots of cell lysates were disbursed to determine [125I]Aβ and [14C]sucrose uptake by liquid scintillation analysis and total protein by the bicinchoninic acid method.
Data Analysis
The volume of distribution (VD) was calculated as the ratio of tissue uptake (dpm/g tissue) to medium concentration (dpm/ml). The total 125I results were corrected for extracellular space by subtraction of the [14C]sucrose VD. For studies with the [125I]Aβ fraction, the extracellular space was removed by centrifugation, similar to the method of Teuscher et al. (9). 125I and 14C permeability values were calculated from the linear portion of the uptake curves. Apparent permeability (Papp) was calculated for cell monolayers on filters and for empty filters with the following equation:
where VReceiver is the volume of the receiver chamber; Afilter, the filter surface area; CDonor, the initial concentration in the donor chamber; and δCReceiver/δT, the change in the receiver chamber concentration over time. Monolayer permeability (PE) was determined from the Papp values of the cells and blank filters as:
Results were analyzed by linear regression or one-way ANOVA with post-hoc comparison by Dunnett’s or Bonferroni’s tests. In cases of unequal variance among treatment groups, results were analyzed by t test with correction for unequal variance.
Results
Uptake of [125I]Aβ in isolated rat choroid plexus was linear to 5 mins, after which it tapered (Fig. 1A). The total [125I]labeled species in the tissue, derived solely from [125I]Aβ added in the artificial CSF, were three times more abundant than intact [125I]Aβ in the tissue. The uptake of a constant concentration of [125I]Aβ remained steady across the first 500-fold increase in unlabeled Aβ concentration (Fig. 1B). Only at the very highest concentration (1000 ng/ ml or 0.23 μM of Aβ) did the unlabeled Aβ begin to compete with the tracer. The [125I]Aβ uptake at 1000 ng/ml was only 50% of the uptake at 500 ng/ml (0.05 ± 0.01 vs. 0.10 ± 0.01 ml/g, respectively). In contrast, adding unlabeled Aβ produced a significant, linear increase in accumulation of the total 125I label (Fig. 1B).
Figure 1.

Accumulation of β-amyloid (Aβ) in isolated rat choroid plexus. Volume of distribution values (VD) corrected by a space marker [14C]sucrose are presented as means ± SEM. Some samples were filtered to remove free iodine and small Aβ fragments and retain only [125I]Aβ (open squares). Unfiltered samples are denoted as total 125I label (filled squares). (A) Time course of Aβ uptake. The uptake was linear up to 5 mins by regression analysis for both curves (n = 3–4 per time point). (B) Concentration study of Aβ uptake (n = 3–4). The linear regression of total 125I label had a positive correlation (r = 0.96, P = 0.002). *Significant reduction (P < 0.05) versus 500 ng/ml.
Despite testing many treatments hypothesized to block energy-dependent cell functions (azide, vanadate, low temperature), to solubilize Aβ (TTR, ApoE3), to cause the precipitation of Aβ (Zn, Cu, anti-TTR), or to block putative Aβ transporters (anti-RAGE, anti-TTR), only the presence of apolipoprotein E (ApoE) and completing the experiment on ice were able to significantly change Aβ uptake (Fig. 2). ApoE3 significantly decreased the uptake of total 125I label by 49% at 14 nM and 79% at 280 nM (P < 0.05) (Fig. 2A) but had a marginal, statistically insignificant 27% decrease of [125I]Aβ uptake (Fig. 2B). Uptake of total 125I label was reduced by 90% on ice, but uptake of [125I]Aβ was reduced by only 50%.
Figure 2.

Factors affecting β-amyloid (Aβ) uptake by isolated rat choroid plexus. Values are expressed as percentage of control (mean ± SEM; n = 3–7) of the uptake of (A) total 125I label or (B) [125I]Aβ. Controls are 0.299 ± 0.046 and 0.136 ± 0.022 ml/g for total 125I label and [125I]Aβ, respectively (means ± SEM). *P < 0.05 as compared to these two controls by one-way ANOVA.
In primary cell culture studies, the flux of 125I label was linear in both directions up to 1 or 2 hrs, after which it tapered to steady-state conditions (Fig. 3). [125I]Aβ and total 125I label fluxes were not determined separately because it was not possible to distinguish if unbound 125I was released from intact [125I]Aβ that had entered the cells or if it entered directly from the medium. The [14C]sucrose permeability appears higher in influx studies (from the outer to the inner chamber) than in efflux studies (Table 1). In contrast, the difference between influx and efflux permeabilities was much less with [125I]Aβ transport. After normalizing the 125I label permeability to that of [14C]sucrose, the efflux ratio of Aβ to sucrose permeability was about 33% higher than the influx ratio. 125I label accumulation in primary cells was concentrative from both directions, with diffusion-corrected distributions (based on total protein) that were significantly higher in efflux studies than in influx studies (Fig. 4).
Figure 3.

Receiver chamber concentrations of β-amyloid (Aβ) and sucrose. [125I]Aβ and [14C]sucrose were added to the donor chamber as described. Mean values (± SEM) are shown for control treatments modeling (A) influx and (B) efflux. Flux was linear for 1–2 hrs, after which it began to approach equilibrium.
Table 1.
Permeability Constants (PE) of [125I]Aβ and [14C]Sucrose in Crossing Choroidal Epithelial Monolayer in a Transwell Devicea
| Permeability (× 10−3 cm/min) | Influx | Efflux | P value |
|---|---|---|---|
| [125I]Aβ | 1.90 ± 0.32 | 1.03 ± 0.24 | 0.10 |
| [14C]sucrose | 2.23 ± 0.60 | 0.82 ± 0.15 | 0.11 |
| Ratio Aβ/sucrose | 0.92 ± 0.09 | 1.22 ± 0.08 | 0.066 |
Values are mean ± SEM (n = 4). Comparisons between influx and efflux are shown.
Figure 4.
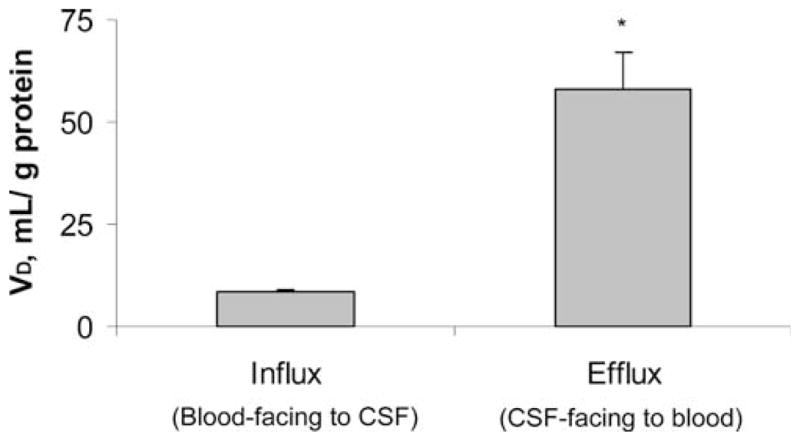
Differential accumulation of β-amyloid (Aβ) by the apical or basolateral interface of BCB in Transwell chambers. [125I]Aβ and [14C]sucrose were added into either inner (for efflux study) or outer (for influx study) chambers. VD values were corrected by a space marker, [14C]sucrose. Data represent mean ± SEM (n = 4). *P < 0.05 as compared to influx.
Discussion
This study uses two distinctive techniques to demonstrate that the choroid plexus sequesters Aβ from an artificial CSF. Freshly isolated choroid plexus provides an accurate in situ model for the intact barrier system and allows for complete control of the fluid surrounding the tissue. Primary cultures grown in Transwells produce a monolayer of polarized epithelial cells in which the cells orient in the same direction. This allows one to uniquely examine transport and uptake from either the blood-facing or the CSF-facing membranes of these choroidal cells.
The accumulation of Aβ by the BCB at the choroid plexus is demonstrated by five distinct characteristics. First, the choroid plexus takes up the intact Aβ species, but not dissociated 125I or the small fragments of [125I]Aβ present in the artificial CSF. If all uptake were attributable to free 125I that had dissociated from the Aβ, then total VD would have been much greater than observed, nearing the reported 10 ml/g (10); the time course graphs in Figure 1A would have risen to the same maximal level; and the addition of unlabeled Aβ would have very little effect on free iodine uptake, as the two molecules are likely to have very different transport mechanisms.
Second, the Aβ uptake into the choroid plexus occurs rapidly and by a nondiffusional uptake process. The Aβ uptake reached maximum within 2–5 mins and exceeded that of a reference compound, [14C]sucrose, which crosses the choroid epithelium only by diffusion (11). The reduced uptake at low temperature as observed here (Fig. 2) and in previous experiments (12) is consistent with a nondiffusional, energy-dependent mechanism. The addition of azide or vanadate, which inhibit ATP production and use, respectively, failed to inhibit Aβ uptake, suggesting that the Aβ uptake by the choroid plexus is not ATP-dependent but rather relies on another driving force. In vivo CSF clearance of Aβ following its intracerebrovascular injection into rats was reported to be rapid (13) and is consistent with nondiffusional uptake by the BBB and/or BCB.
Third, the choroid plexus has a large storage capacity for Aβ. [125I]Aβ uptake was reduced only by unlabeled Aβ at a total concentration of 0.23 μM. This accumulation may reflect storage of intact Aβ as well as metabolized Aβ fragments, as discussed below.
Fourth, the uptake of Aβ by the choroid plexus does not require several proteins with suggested roles in Aβ binding or transport, such as transthyretin (TTR), apolipoprotein E3 (ApoE3), and the receptor for advanced glycation end-products (RAGE) (Fig. 2A and B). In brain, the choroid plexus is the exclusive producer of TTR, a thyroxine transporter that also binds Aβ and prevents amyloid aggregation (14–16). In the current study, neither TTR nor an antibody against TTR significantly changed [125I]Aβ uptake by the choroid plexus. Members of the ApoE family form complexes with Aβ that cross the brain barriers, and human ApoE ε4 allele has a genetic association with AD (17–19). We hypothesized that ApoE would increase the uptake of Aβ, as reported at the BBB (17), but found an unexpected decrease. Although the underlying mechanism of Aβ inhibition by ApoE is unknown, one possibility is that the formation of ApoE-Aβ conjugates may reduce the Aβ species otherwise available for transport by ApoE-unrelated mechanisms at the BCB. RAGE appears unlikely to mediate Aβ uptake into the choroid plexus despite its established role at the human BBB (12, 20). Tissue pre- or cotreatment with antibody against RAGE had no effect on Aβ uptake by the choroid plexus. In addition, copper and zinc, two metal ions found in Aβ plaques, did not affect Aβ uptake at concentrations known to precipitate the peptide (21).
Finally, Aβ uptake by the choroid plexus favors its efflux from CSF to blood rather than its influx from blood to the CSF. The directionality of Aβ uptake was investigated in a Transwell system, in which the choroidal cells grew with the CSF-facing (apical) side oriented toward the inner chamber and the blood-facing (basolateral) side toward the outer chamber (8, 22). The [14C]sucrose permeability appears higher in influx studies than in efflux studies (Table 1), suggesting that the in vitro barrier may be more permeable to incoming sucrose than to outgoing sucrose. This unexpected difference in sucrose permeability may be a result of the presence of microvilli on the apical surface of the BCB model, which increase the surface area and provide crypt-like pockets that trap sucrose and hinder its diffusion between cells. The difference between influx and efflux permeabilities was much less with [125I]Aβ transport. Thus, in contrast to the space marker sucrose, the barrier appears to be more permeable to outgoing Aβ than to incoming Aβ. A net efflux of Aβ at the BCB is further supported by the residual Aβ accumulation studies. Total 125I label accumulation in primary cells was concentrative from both directions and was significantly higher in efflux studies than in influx studies (Fig. 4). Thus, these studies clearly demonstrate that the normal choroid plexus removes Aβ from the CSF, suggesting a novel pathway for brain to maintain Aβ homeostasis in the CSF.
Our results further suggest that the choroid plexus may metabolize Aβ into smaller fragments following initial uptake. There were three times more total [125I]labeled species in the tissue than intact [125I]Aβ following addition of [125I]Aβ to the artificial CSF. This suggests that the choroid plexus may break down or metabolize Aβ into smaller fragments that it subsequently accumulates, which is consistent with a report that Aβ remains largely intact following injection into ventricular CSF but is partly degraded during or after clearance into blood (13).
Thus, a unique mechanism(s) must exist that functions to break down or metabolize Aβ to smaller fragments in the choroid plexus. Identification and characterization of Aβ metabolism at the choroid plexus would permit a better understanding of how brain handles excess Aβ.
Although the purpose of this study was to investigate the role of the choroid plexus in removal of Aβ from the CSF and not the exact molecular species accumulated within the choroid plexus cells, the question of the specific molecule(s) that accumulates or aggregates in the choroid plexus indeed deserves further exploration. Several studies have suggested that the toxic moieties involved in Alzheimer’s disease damage are small aggregates and not the free monomers of Aβ peptides (23–25). It is unclear, however, if the small aggregates are present in the CSF and whether the aggregates found in the choroid plexus cells (7) are derived from the monomer of Aβ or directly from small aggregates in the CSF. Because Aβ is the precursor of the aggregates, the removal of it from the CSF would presumably influence the homeostasis of Aβ in the brain. If the choroid plexus accumulates only the monomer forms of Aβ and not the larger aggregates associated with AD, then perhaps the tissue plays only a limited role in AD causation and/or progression. It will be important in the future to identify the molecular species of Aβ that are taken into the choroid plexus from CSF to further elucidate the tissue’s role in AD.
In summary, the isolated choroid plexus accumulates Aβ from the CSF through an energy-dependent, nondiffusional pathway. Net flux across the choroid plexus appears to favor efflux from the CSF to blood. These results demonstrate a significant role of choroid plexus in cleansing Aβ from CSF and maintaining homeostasis of Aβ in brain extracellular fluid and should be of interest to AD researchers and clinicians.
Acknowledgments
This work was partly supported by National Institute of Environmental Health Sciences Grant ES-08146 and Johnson & Johnson Focused Giving Program.
References
- 1.Goldgaber D, Schwarzman AI, Bhasin R, Gregori L, Schmechel D, Saunders AM, Roses AD, Strittmatter WJ. Sequestration of amyloid beta-peptide. Ann N Y Acad Sci. 1993;695:139–143. doi: 10.1111/j.1749-6632.1993.tb23042.x. [DOI] [PubMed] [Google Scholar]
- 2.Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer’s disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol. 2003;29:106–117. doi: 10.1046/j.1365-2990.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- 3.Zlokovic BV. Clearing amyloid through the blood-brain barrier. J Neurochem. 2004;89:807–811. doi: 10.1111/j.1471-4159.2004.02385.x. [DOI] [PubMed] [Google Scholar]
- 4.Zheng W, Aschner M, Ghersi-Egea JF. Brain barrier systems: a new frontier in metal neurotoxicological research. Toxicol Appl Pharmacol. 2003;192:1–11. doi: 10.1016/s0041-008x(03)00251-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanai M, Matsubara E, Isoe K, Urakami K, Nakashima K, Arai H, Sasaki H, Abe K, Iwatsubo T, Kosaka T, Watanabe M, Tomidokoro Y, Shizuka M, Mizushima K, Nakamura T, Igeta Y, Ikeda Y, Amari M, Kawarabayashi T, Ishiguro K, Harigaya Y, Wakabayashi K, Okamoto K, Hirai S, Shoji M. Longitudinal study of cerebrospinal fluid levels of tau, A beta1–40, and A beta1–42(43) in Alzheimer’s disease: a study in Japan. Ann Neurol. 1998;44:17–26. doi: 10.1002/ana.410440108. [DOI] [PubMed] [Google Scholar]
- 6.Tapiola T, Pirttila T, Mikkonen M, Mehta PD, Alafuzoff I, Koivisto K, Soininen H. Three-year follow-up of cerebrospinal fluid tau, beta-amyloid 42 and 40 concentrations in Alzheimer’s disease. Neurosci Lett. 2000;280:119–122. doi: 10.1016/s0304-3940(00)00767-9. [DOI] [PubMed] [Google Scholar]
- 7.Eriksson L, Westermark P. Characterization of intracellular amyloid fibrils in the human choroid plexus epithelial cells. Acta Neuropathol (Berl) 1990;80:597–603. doi: 10.1007/BF00307626. [DOI] [PubMed] [Google Scholar]
- 8.Zheng W, Zhao Q. The blood-CSF barrier in culture. Development of a primary culture and transepithelial transport model from choroidal epithelial cells. Methods Mol Biol. 2002;188:99–114. doi: 10.1385/1-59259-185-X:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teuscher NS, Shen H, Shu C, Xiang J, Keep RF, Smith DE. Carnosine uptake in rat choroid plexus primary cell cultures and choroid plexus whole tissue from PEPT2 null mice. J Neurochem. 2004;89:375–382. doi: 10.1111/j.1471-4159.2004.02333.x. [DOI] [PubMed] [Google Scholar]
- 10.Welch K. Active transport of iodide by choroid plexus of the rabbit in vitro. Am J Physiol. 1962;202:757–760. doi: 10.1152/ajplegacy.1962.202.4.757. [DOI] [PubMed] [Google Scholar]
- 11.Dziegielewska KM, Evans CA, Malinowska DH, Mollgard K, Reynolds JM, Reynolds ML, Saunders NR. Studies of the development of brain barrier systems to lipid insoluble molecules in fetal sheep. J Physiol. 1979;292:207–231. doi: 10.1113/jphysiol.1979.sp012847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, Yan SD, Stern D, Schmidt AM, Frangione B, Zlokovic BV. Human blood-brain barrier receptors for Alzheimer’s amyloid-beta 1–40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest. 1998;102:734–743. doi: 10.1172/JCI2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghersi-Egea JF, Gorevic PD, Ghiso J, Frangione B, Patlak CS, Fenstermacher JD. Fate of cerebrospinal fluid-borne amyloid beta-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J Neurochem. 1996;67:880–883. doi: 10.1046/j.1471-4159.1996.67020880.x. [DOI] [PubMed] [Google Scholar]
- 14.Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK, Zagorski MJ, Talafous J, Eisenberg M, Saunders AM, Roses AD, Goldgaber D. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci U S A. 1994;91:8368–8372. doi: 10.1073/pnas.91.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreiber G, Aldred AR, Jaworowski A, Nilsson C, Achen MG, Segal MB. Thyroxine transport from blood to brain via transthyretin synthesis in choroid plexus. Am J Physiol. 1990;258:R338–R345. doi: 10.1152/ajpregu.1990.258.2.R338. [DOI] [PubMed] [Google Scholar]
- 16.Lignelid H, Collins VP, Jacobsson B. Cystatin C and transthyretin expression in normal and neoplastic tissues of the human brain and pituitary. Acta Neuropathol (Berl) 1997;93:494–500. doi: 10.1007/s004010050644. [DOI] [PubMed] [Google Scholar]
- 17.Martel CL, Mackic JB, Matsubara E, Governale S, Miguel C, Miao W, McComb JG, Frangione B, Ghiso J, Zlokovic BV. Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer’s amyloid beta. J Neurochem. 1997;69:1995–2004. doi: 10.1046/j.1471-4159.1997.69051995.x. [DOI] [PubMed] [Google Scholar]
- 18.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342:697–699. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 19.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 21.Bush AI. Metal complexing agents as therapies for Alzheimer’s disease. Neurobiol Aging. 2002;23:1031–1038. doi: 10.1016/s0197-4580(02)00120-3. [DOI] [PubMed] [Google Scholar]
- 22.Strazielle N, Ghersi-Egea JF. Demonstration of a coupled metabolism–efflux process at the choroid plexus as a mechanism of brain protection toward xenobiotics. J Neurosci. 1999;19:6275–6289. doi: 10.1523/JNEUROSCI.19-15-06275.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorman PM, Chakrabartty A. Alzheimer beta-amyloid peptides: structures of amyloid fibrils and alternate aggregation products. Biopolymers. 2001;60:381–394. doi: 10.1002/1097-0282(2001)60:5<381::AID-BIP10173>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 24.Dumery L, Bourdel F, Soussan Y, Fialkowsky A, Viale S, Nicolas P, Reboud-Ravaux M. beta-Amyloid protein aggregation: its implication in the physiopathology of Alzheimer’s disease. Pathol Biol (Paris) 2001;49:72–85. doi: 10.1016/s0369-8114(00)00009-2. [DOI] [PubMed] [Google Scholar]
- 25.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]