

Abstract
Alpha1‐antitrypsin (AT) deficiency is the most common genetic cause of liver disease in children and is also a cause of chronic hepatic fibrosis, cirrhosis, and hepatocellular carcinoma in adults. Recent advances in understanding how mutant AT molecules accumulate within hepatocytes and cause liver cell injury have led to a novel strategy for chemoprophylaxis of this liver disease. This strategy involves a class of drugs, which enhance the intracellular degradation of mutant AT and, because several of these drugs have been used safely in humans for other indications, the strategy can be moved immediately into clinical trials. In this review, we will also report on advances that provide a basis for several other strategies that could be used in the future for treatment of the liver disease associated with AT deficiency. Clin Trans Sci 2011; Volume 5: 289–294
Keywords: Liver disease, alpha‐1‐antitrypsin, AT deficiency
Introduction
The classic form of a1‐antitrypsin (AT) deficiency is an autosomal co‐dominant disorder that affects 1 in 2000 to 1 in 3000 live births in most populations. It causes a chronic fibrotic process in the liver that can become clinically apparent in infancy, childhood, adolescence or later in adult life with cirrhosis and/or hepatocellular carcinoma. 1 , 2 It is the most common genetic disease for which children undergo liver transplantation and a more common indication for liver transplantation in adults than previously recognized. 3 However, a prospective unbiased study of a cohort derived from nationwide newborn screening carried out in Sweden in the 1960s shows that only 8–10% of homozygotes develop clinically significant liver disease during the first 3 decades of life. 4 , 5 Thus, most individuals with this deficiency escape liver disease. There is currently no way to predict which deficient person will develop severe liver disease and the progression of liver disease in the susceptible subpopulation is quite variable. AT deficiency also predisposes to chronic obstructive pulmonary disease and people with this deficiency who also smoke cigarettes have earlier onset and more severe pulmonary disease. 6 , 7
AT is a member of the serine protease inhibitor (SERPIN) family and mainly functions as an inhibitor of neutrophil elastase and perhaps several other neutrophil proteases. 8 It is an abundant serum glycoprotein that is predominantly synthesized by the liver. In the classic form of the deficiency, homozygous for the Z allele, a point mutation renders the protein prone to misfolding, polymerization, and aggregation. The abnormal protein accumulates in the endoplasmic reticulum (ER) of liver cells as seen by periodic acid‐Schiff positive inclusions that are the histological hallmark of the disease. Serum levels of the protein are reduced to 10–15% of the levels present in normal individuals. 3 Different mechanisms account for the pathologic effects in the liver and lung: liver disease results from a gain‐of‐toxic function mechanism elicited by accumulation of mutant AT (ATZ) in the ER of liver cells; lung disease results from a loss‐of‐function mechanism involving uninhibited proteolytic destruction of the pulmonary connective tissue matrix.
In order to further understand the pathogenesis of liver disease in AT deficiency we have investigated the hypothesis that genetic and environmental modifiers determine whether a given deficient individual is susceptible to liver disease and further that these putative modifiers act at two potential levels: (1) on intracellular pathways for degradation of proteins that accumulate within the ER: and/or (2) act on stress signaling pathways designed for adaptation to ER protein accumulation. Using mammalian cell line models and recently a novel Caenorhabditis elegans model we have found that the proteasomal and autophagic pathways play important roles in intracellular degradation of ATZ. 1 These observations permitted us to conceptualize a pharmacological strategy in which drugs which enhance autophagic degradation of ATZ could ameliorate the hepatic pathology of AT deficiency. One such drug, carbamazepine (CBZ), was found to reduce hepatic fibrosis in a mouse model 9 and several additional FDA‐approved drugs which have this action are moving into preclinical trials. 10
In addition to this novel pharmacological strategy, there have been advances in developing cell transplantation and gene transfer strategies for treating liver disease due to AT deficiency. Studies in the mouse model of AT deficiency have shown that hepatocytes with massive ATZ accumulation can stimulate proliferation of hepatocytes with lesser ATZ accumulation in trans. 11 , 12 This may be a key factor in carcinogenesis but it is also the basis for a second novel therapeutic strategy involving transplantation of hepatocytes because the transplanted hepatocytes will have a selective proliferative advantage in the liver of the AT‐deficient individual. 13 Ongoing gene transfer studies have focused on inhibitory RNA (short hairpin interfering RNA) to reduce expression of the mutant gene. 14 In this review we will discuss in further detail what has been learned about this disease in recent years and the basis by which that has been translated into potential new therapies.
The Mutant ATZ Molecule
Advances in understanding the structure of the SERPIN protein family and the structural basis of SERPIN function have shed light on the structural alterations in the mutant ATZ molecule. The SERPINS have been found to fold into a metastable conformation composed of three β‐sheets, eight to nine α‐helices, and a solvent exposed reactive center loop (RCL). The mechanism by which SERPIN proteins inhibit proteases involves binding of the protease to the RCL, proteolytic cleavage of the RCL accompanied by a dramatic conformational change, analogous to a mousetrap, 15 , 16 with the RCL incorporated into the central β‐sheet‐A as an additional β‐strand (strand 4A). This conformational change from a five‐stranded to a six‐stranded β‐sheet, results in a hyperstable form of the serpin molecule.
The point mutation in ATZ, lysine for glutamate at position 342 is located at the head of strand 5A of sheet‐A and the base of the RCL. In the model originally described by Lomas et al., now known as the loop‐sheet insertion mechanism or A sheet polymerization, this substitution prevents the RCL from incorporating into the A‐sheet, opening a gap that can be filled by the RCL of an adjacent ATZ molecule (Figure 1). 16 According to this model multiple ATZ molecules undergo loop‐sheet insertion to lead to polymers and insoluble aggregates.
Figure 1.
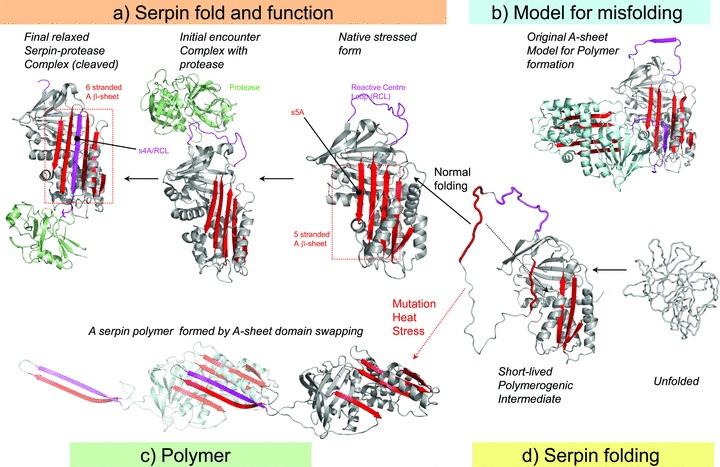
Two models for polymerization of ATZ. (A) Active serpins fold into a metastable state. Following the initial interaction with a target peptidase and reactive center loop (RCL) cleavage, the serpin undergoes a radical conformational change (RCL/s4A incorporation into b‐sheet A) that culminates in peptidase inhibition via distortion of the catalytic residues. (B) Loop‐sheet model of serpin polymerization. The RCL of one molecule is inserted into the open b‐sheet A of another. (C) Domain swapping model of serpin polymerization. This model is based on the structure of a domain‐swapped antithrombin dimer, where s5A and s4A (RCL) of a donor molecule insert into b‐sheet A of a recipient. (D) Normal serpins may fold through a polymerogenic intermediate that is stabilized by certain mutations. The black dotted arrow indicates a gap in b‐sheet A that accommodates s5A to form the s4A (RCL) exposed native molecule to A or the s5A and s4A domain‐swapped structure in C. Reproduced with permission from Figure 2 of Whisstock JC et al. J Biol Chem. 2010; 285: 24307–24312.
Yamasaki et al. have recently provided data from the crystallographic structure of a self‐terminating stable serpin dimer that has led to an alternative model for ATZ polymerization that involves a domain swapping mechanism. In contrast to the loop‐sheet insertion mechanism, domain swapping has been implicated in the formation of polymers in at least several other physiological and pathological situations. In this case a domain from the RCL (strand 4A) and strand 5A insert into the β‐sheet‐A of a neighboring ATZ molecule. 16 This model is based on the prediction that the assembly of the S5A β‐strand into β‐sheet‐A is one of the final steps of normal AT folding. The Z mutation at position 342 likely slows the final folding transition of the polymerogenic intermediate with an exposed strand 5A to the native‐state AT protein and promotes the domain swapping of S5A and S4A (RCL) into another neighboring intermediate form of the ATZ molecule (Figure 1). Furthermore, the domain swap exposes a 30‐residue hydrophobic helical linker region in the molecule that could explain the histological appearance of tangled aggregates of linear polymers seen in the ER of hepatocytes in AT deficiency. This model can explain three previously poorly understood phenomena of ATZ pathobiology: how the ATZ molecule shifts from a stable but polymerogenic intermediate into polymers and insoluble aggregates; how 10–15% of ATZ molecules are able to fold into a conformation capable of traversing the secretory pathway; and how accumulation of ATZ in the ER does not activate the unfolded protein response (UPR).
Liver Disease in AT Deficiency
Liver injury is thought to result from a gain‐of‐toxic function mechanism whereby the accumulation of mutant ATZ in the ER of hepatocytes incites a series of cytotoxic sequela. The most compelling evidence for this gain‐of‐toxic function mechanism comes from the hepatic pathology in the PiZ mouse model of AT deficiency. 17 , 18 The liver of this mouse model is characterized by many of the features of the human liver disease including hepatocellular ATZ globules, slowly progressing fibrosis with age, low‐grade inflammation and regeneration, mild steatosis, and carcinomatosis. 9 , 11 , 19 , 20 , 21 , 22 , 23 Because these mice have normal levels of the endogenous ortholog of AT, the hepatic pathology cannot be attributed to a loss‐of‐function mechanism and therefore must result from a gain‐of‐toxic function due to transgenic expression of human ATZ.
Clinically, AT deficiency is characterized by wide variability in both the expressivity and penetrance of the liver disease phenotype. Liver disease commonly presents during infancy, but AT deficiency can cause severe liver dysfunction in childhood, adolescence and adulthood 1 and predispose adults to cirrhosis and hepatocellular carcinoma. 24 Typically, liver disease is characterized by slowly progressing fibrosis and relatively low‐grade inflammation. However, only a subset of individuals homozygous for the Z mutation demonstrate clinically significant liver disease. In a unique unbiased epidemiological study carried out in Sweden over 40 years, less than 10% of the 127 infants that were identified had clinically significant liver disease over the first four decades of life. 4 , 5 This phenotypic variation has been attributed to a conceptual model in which genetic or environmental modifiers predispose a subset of homozygotes to liver disease or protect the remainder from liver disease. Furthermore, this conceptual model envisions the targets of these modifiers as being components of the pathway by which ATZ is degraded or components of the protective signaling pathways activated by ATZ accumulation. An early study by Wu et al. substantiated this conceptual by showing that disposal of ATZ was slower in genetically engineered skin fibroblast cell lines from PiZZ homozygotes with liver disease in comparison to cells taken from asymptomatic homozygotes. 25
Degradation Pathways for ATZ
The proteasome was the first system implicated in the ER degradation of ATZ and its role has been demonstrated by studies in cell‐free microsomal, mammalian cell line and yeast model systems. 26 , 27 , 28 Both the classical ubiquitin‐dependent and ubiquitin‐independent proteasomal pathways are involved. 29 We now know that mutant ATZ molecules in the ER reach the proteasome in the cytoplasm by what is known as the ERAD pathway. However, the specific ER chaperones and ER transport proteins that recognize ATZ and mediate its retrotranslocation to the cytosol and targeting to the proteasome have not been definitively elucidated.
Nevertheless, early studies indicated that proteasomal pathways could not be solely responsible for the disposal of ATZ. Autophagy, an intracellular degradation pathway in which cytosolic components are sequestered by double‐membrane vesicles (autophagosomes) and degraded upon vesicle–lysosome fusion, 30 has been implicated in the disposal of ATZ by numerous subsequent studies. First, observational studies demonstrated that ATZ expression in cell line models, in the livers of PiZ mice, and in liver biopsy specimens from human PiZZ homozygotes is associated with increased numbers of autophagosomes. 20 Second, chemical inhibitors of autophagy were found to partially attenuate the disposal of ATZ in cell line models. More conclusive genetic studies have provided the strongest evidence for the important role of the autophagic pathway in the degradation of ATZ. ATZ‐expressing murine embryonic fibroblast (MEF) cells derived from the autophagy‐deficient ATG5‐null mouse were characterized by a marked delay in the degradation of ATZ and massive accumulation of ATZ inclusions compared to ATZ‐expressing wild‐type MEFs. 31 Kruse et al., using an unrelated strategy in which they expressed ATZ in a library of yeast mutants and screened for mutants with defective ATZ degradation, corroborated the importance of autophagy in disposing of ATZ. 32 The absence of the yeast homologues for ATG6 and ATG16 resulted in a significant delay in the disposal of human ATZ. Furthermore, they demonstrated that this delay in the degradation of ATZ was only apparent at higher levels of expression of the mutant protein. This key finding has led to the concept that soluble forms of ATZ, which predominate at lower expression levels, are mainly degraded by the proteasome, while the insoluble polymers of ATZ that occur at higher levels of expression, are primarily degraded by the autophagic pathway.
Several lines of evidence also suggest that other pathways may contribute to degradation of mutant ATZ. The study by Kruse et al. 32 suggested a disposal pathway for ATZ in which it is trafficked from the trans‐Golgi network directly to the lysosome in yeast cells. To date, however, a comparable pathway has not been described in mammalian cells.
Pan et al. have recently implicated a potential genetic modifier for the hepatic phenotype in AT deficiency that could theoretically act on an intracellular degradative pathway. Infants with severe liver disease due to AT deficiency were found to have an increased prevalence of a single‐nucleotide polymorphism in the flanking region of the gene for ER mannosidase 1. 33 Because this gene product can play a role in ERAD and the polymorphic variation can suppress its translation the authors postulated that the polymorphism modifies degradation of ATZ. Further studies to confirm the cellular effects of this polymorphism and to test it in additional clinical populations are needed to validate this modifier effect.
Signaling Pathways Activated by ATZ Accumulation
Cell line and animal models with inducible expression of ATZ have been used to understand the primary signaling pathways activated when ATZ accumulates in the ER because these systems are ideally suited for investigating effects on a naive cell/tissue. We have theorized that these pathways may function to protect the cell from aggregated protein. To date, the studies have shown that ATZ accumulation is sufficient to activate the autophagic pathway, the NFκB signaling pathway, but not the UPR. For example, the Z mouse model with inducible hepatocyte‐specific expression of ATZ was been bred to the GFP‐LC3 mouse model, which generates green fluorescent autophagosomes because LC3 is an autophagosomal membrane‐specific protein. When green fluorescent autophagosomes were observed in the liver of the Z × GFP‐LC3 in the absence of starvation whereas green fluorescent autophagosomes are only seen in the liver of the GFP‐LC3 mouse after starvation, we concluded that accumulation of ATZ in the ER is sufficient to activate autophagy. 31 Similarly, ATZ expression in a cell line model, engineered for inducible expression, elicits increased LC3 conversion and that conversion is further accentuated by the presence of lysosomal protease inhibitors, 9 indicating increased autophagic flux in response to ATZ expression.
The mechanism and signaling intermediates involved in activating the autophagic system in response to ATZ accumulation are unknown. Hepatic gene expression profile analysis using the Z mouse model with hepatocyte‐specific inducible expression of ATZ identified one potential contributing mechanism, upregulation of regulator of G protein signaling 16 (RGS16). 34 Because RGS16 inhibits Gαi3 35 and Gαi3 mediates the antiautophagic effect of insulin in the liver, 36 we have hypothesized that RGS16 upregulation plays a role in activating autophagy when ATZ accumulates in the ER of hepatocytes.
The NFκB signaling pathway is also activated when ATZ accumulates in the ER. 22 Preliminary research suggests that it serves a protective role as the PiZ mouse bred onto a NFκB signaling‐deficient background has more severe liver disease and fibrosis. 1 Further elucidation of how NFκB and its downstream targets protect the liver could provide new information for development of therapeutic interventions.
Importantly, the UPR is not activated when ATZ accumulates in cells. The domain swap mechanism of ATZ polymerization, in which ATZ monomers adopt a stable polymer configuration resembling the wild‐type molecule without exposing hydrophobic residues, 16 offers a possible explanation for why the UPR is not activated. Lack of UPR signaling also has important implications for the hepatic pathobiology of AT deficiency. For instance, it could explain the relatively low rate of apoptosis seen in the liver in this disease.
Mechanisms of Hepatotoxicity and Carcinogenesis
The molecular mechanisms of hepatocellular toxicity in AT deficiency are not well understood. Studies of cell culture models of AT deficiency, the livers of PiZ mice, and the livers of AT‐deficient patients demonstrate mitochondrial injury, mitochondrial autophagy, and caspase‐3 activation. Furthermore, administration of cyclosporin A, an inhibitor of the mitochondrial permeability transition, reduced mitochondrial injury and mortality in starved PiZ mice, 19 suggesting that mitochondrial dysfunction is the final common pathway for cytotoxicity and ultimately the stereotypical fibrotic response of the liver.
The mechanisms of carcinogenesis in AT deficiency are also not well understood. The longstanding observation that only some hepatocytes in the livers of patients and PiZ mice contain ATZ globules has provided a clue. Studies of hepatocyte BrdU incorporation in the PiZ mouse showed that the globule‐devoid hepatocytes have a selective proliferative advantage when in the presence of the globule‐containing hepatocytes and are characterized by a chronic hyperproliferative state. 11 Globule‐containing hepatocytes have more aggregated ATZ, 37 activated autophagy, 31 activated NF‐κB, 22 impaired cell proliferation, 11 and enhanced levels of apoptosis. 38 These data have led to the hypothesis that carcinogenesis involves the cross‐talk between globule‐containing cells that chronically stimulate “in trans” regenerative hyperproliferation of globule‐devoid hepatocytes. The results of the BrdU studies have been recently supported by repopulation studies in PiZ mice transplanted with syngeneic normal hepatocytes. 13 Consistent with this hypothesis, the majority of carcinomas that occur in the patients with AT deficiency arise from globule‐devoid hepatocytes. 39 , 40
Novel Therapeutic Strategies for Liver Disease due to AT Deficiency
Although orthotopic liver transplantation is the only effective treatment currently available for liver failure due to AT deficiency, a recent series of observations has raised the possibility that a class of drugs which enhance autophagy could be used to ameliorate this liver disease and prevent the need for organ transplantation and chronic immunosuppressive treatment. First, Hidvegi et al. found that CBZ, a drug which has been used for years safely in humans as an anticonvulsant and mood stabilizer, enhances autophagy and perhaps other intracellular mechanisms for degradation of ATZ. 9 Further, this drug when given orally for 2 weeks to the PiZ mouse model of AT deficiency markedly reduced the hepatic load of ATZ and reduced hepatic fibrosis. Although the lowest effective dose of CBZ in mice, 200 mg/kg/day, was considerably higher than the dose range used in humans for seizures or mood stabilizing, 10–20 mg/kg/day, effective doses of drugs in mice can be 10 to 20 times as high in mice because of the higher ratio of surface area to body weight when compared to humans. This issue is being addressed in a double blinded, randomized and placebo‐controlled clinical trial for patients with severe liver disease due to AT deficiency that has just started enrolling patients. Second, Gosai et al. identified used a newly developed C. elegans model of AT deficiency and adapted it for a high‐content screening platform to identify additional therapeutic compounds. 10 The disease is modeled with exceptionally high fidelity and the screening assay provides an unbiased approach to identify additional drugs that could work by increasing degradation of mutant ATZ, increasing secretion of ATZ or decreasing synthesis of ATZ. The preliminary screening of the LOPAC drug library identified six hit compounds, four of which could be tested almost immediately in humans because they have been FDA approved for other disease indications. Despite the fact that this screen could identify drugs which act by several different mechanisms, the six hit compounds identified in the pilot screen all worked as autophagy enhancers. Furthermore two of the hit compounds, fluphenazine and pimozide, are mood stabilizers that were also identified by high throughput screening for compounds which enhance the degradation of mutant huntingtin, the aggregation‐prone protein that causes Huntington’s disease. 41 In addition to identifying fluphenazine and pimozide as candidates for human trials, these results indicate the importance of autophagy enhancer activity for potential pharmacological treatment of AT deficiency and perhaps other diseases caused by aggregation‐prone proteins (Figure 2).
Figure 2.
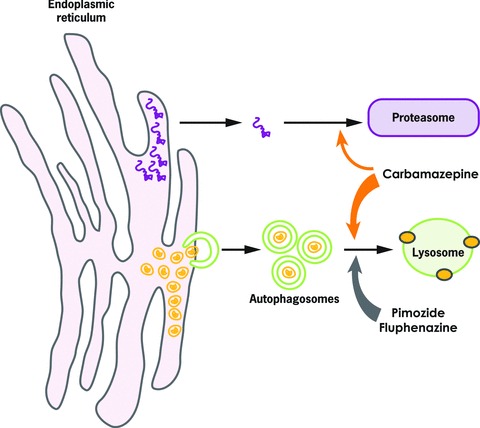
Effect of carbamazepine, fluphenazine, and pimozide on intracellular degradation of mutant ATZ. Soluble forms of ATZ (purple) are degraded by the proteasomal pathway (top right) and insoluble aggregates of ATZ (gold) are degraded by autophagy (bottom right). Carbamazepine appears to enhance autophagy and also the proteasome. Fluphenazine and pimozide enhance the autophagic degradation of ATZ.
Although these will require more preclinical work than the autophagy enhancer drugs described above, cell‐based therapy and gene therapy for AT deficiency have been the subject of several recent advances. Ding et al. have shown that hepatocyte transplantation can be engineered to almost completely repopulate the liver using the PiZ mouse model. 13 For gene therapy, Cruz et al. employed small interfering RNA (siRNA) technology to reduce ATZ hepatocellular expression. 14 siRNA sequences cloned into recombinant adeno‐associated viral vectors and packaged into viral capsids were injected into the portal vein of PiZ mice. Three weeks after injection, injected mice had a marked decrease in the hepatic load of ATZ, but without any difference in the insoluble pool of polymerized ATZ. Longer‐term studies will have to investigate whether siRNA represents a viable treatment that can attenuate ATZ globules, hepatocellular injury, and hepatic fibrosis. In addition, further research will have to establish the safety of siRNA given ongoing concerns for liver and brain toxicity. Finally, a budding new gene therapy strategy employing polymerase II‐driven micro RNAs (miRNAs) could potentially be used to target mutant ATZ expression while also simultaneously delivering wild‐type AT expression that could theoretically prevent both gain‐of‐toxic function effects in the liver and loss‐of‐function effects in the lung.
Acknowledgments
Nicholas Maurice was supported by a Howard Hughes Medical Institute Medical Research Training Fellowship. Experimental work from the authors’ laboratory was supported by NIH grants DK084512, DK076918, and HL037784.
References
- 1. Perlmutter DH. Alpha‐1‐antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease‐associated protein aggregates. Annu Rev Med. 2011; 62: 333–345. [DOI] [PubMed] [Google Scholar]
- 2. Perlmutter DH. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha‐1‐antitrypsin deficiency. Pediatr Res. 2006; 60: 233–238. [DOI] [PubMed] [Google Scholar]
- 3. Perlmutter DH. Liver injury in alpha1‐antitrypsin deficiency: an aggregated protein induces mitochondrial injury. J Clin Invest. 2002; 110: 1579–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sveger T. Liver disease in alpha1‐antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976; 294: 1316–1321. [DOI] [PubMed] [Google Scholar]
- 5. Piitulainen E, Carlson J, Ohlsson K, Sveger T. Alpha1‐antitrypsin deficiency in 26‐year‐old subjects: lung, liver, and protease/protease inhibitor studies. Chest. 2005; 128: 2076–2081. [DOI] [PubMed] [Google Scholar]
- 6. Crystal RG. Alpha 1‐antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest. 1990; 85: 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Janus ED, Phillips NT, Carrell RW. Smoking, lung function, and a1‐antitrypsin deficiency. Lancet. 1985; 1: 152–154. [DOI] [PubMed] [Google Scholar]
- 8. Silverman GA, Whisstock JC, Bottomley SP, Huntington JA, Kaiserman D, Luke CJ, Pak SC, Reichhart JM, Bird PI. Serpins flex their muscle: I. Putting the clamps on proteolysis in diverse biological systems. J Biol Chem. 2010; 285: 24,299–24,305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, et al An autophagy‐enhancing drug promotes degradation of mutant alpha1‐antitrypsin Z and reduces hepatic fibrosis. Science. 2010; 329: 229–232. [DOI] [PubMed] [Google Scholar]
- 10. Gosai SJ, Kwak JH, Luke CJ, Long OS, King DE, Kovatch KJ, Johnston PA, Shun TY, Lazo JS, Perlmutter DH, et al Automated high‐content live animal drug screening using C. elegans expressing the aggregation prone serpin a1‐antitrypsin Z. PLoS One. 2010; 12: e15460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rudnick DA, Liao Y, An JK, Muglia LJ, Perlmutter DH, Teckman JH. Analyses of hepatocellular proliferation in a mouse model of alpha‐1‐antitrypsin deficiency. Hepatology. 2004; 39: 1048–1055. [DOI] [PubMed] [Google Scholar]
- 12. Rudnick DA, Perlmutter DH. Alpha‐1 antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver diseases. Hepatology. 2005; 42: 514–521. [DOI] [PubMed] [Google Scholar]
- 13. Ding J, Yannam GR, Roy‐Chowdhury N, Hidvegi T, Basma H, Rennard SI, Wong RJ, Avsar Y, Guha C, Perlmutter DH, et al Spontaneous hepatic repopulation in transgenic mice expressing mutant human a1‐antitrypsin by wild‐type donor hepatocytes. J Clin Invest. 2011; 121: 1930–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cruz PE, Mueller C, Cossette TL, Golant A, Tang Q, Beattie SG, Brantly M, Campbell‐Thompson M, Blomenkamp KS, Teckman JH, et al In vivo post‐transcriptional gene silencing of alpha‐1 antitrypsin by adeno‐associated virus vectors expressing siRNA. Lab Invest. 2007; 87: 893–902. [DOI] [PubMed] [Google Scholar]
- 15. Huntington JA. Shape‐shifting serpins – advantages of a mobile mechanism. Trends Biochem Sci. 2006; 31: 429–435. [DOI] [PubMed] [Google Scholar]
- 16. Yamasaki M, Li W, Johnson DJ, Huntington JA. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature. 2008; 455: 1255–1258. [DOI] [PubMed] [Google Scholar]
- 17. Carlson JA, Rogers BB, Sifers RN, Hawkins HK, Finegold MJ, Woo SL. Multiple tissues express alpha 1‐antitrypsin in transgenic mice and man. J Clin Invest. 1988; 82: 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carlson JA, Rogers BB, Sifers RN, Finegold MJ, Clift SM, DeMayo FJ, Bullock DW, Woo SL. Accumulation of PiZ alpha 1‐antitrypsin causes liver damage in transgenic mice. J Clin Invest. 1989; 83: 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in alpha 1‐antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol. 2004; 286: G851–G862. [DOI] [PubMed] [Google Scholar]
- 20. Teckman JH, Perlmutter DH. Retention of mutant alpha(1)‐antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. 2000; 279: G961–G974. [DOI] [PubMed] [Google Scholar]
- 21. Teckman JH, An JK, Loethen S, Perlmutter DH. Fasting in alpha1‐antitrypsin deficient liver: constitutive activation of autophagy. Am J Physiol Gastrointest Liver Physiol. 2003; 283: G1156–G1165. [DOI] [PubMed] [Google Scholar]
- 22. Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant alpha1‐antitrypsin Z in the endoplasmic reticulum activates caspases‐4 and ‐12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem. 2005; 280: 39,002–39,015. [DOI] [PubMed] [Google Scholar]
- 23. Marcus NY, Brunt EM, Blomenkamp K, Ali F, Rudnick DA, Ahmad M, Teckman JH. Characteristics of hepatocellular carcinoma in a murine model of alpha‐1‐antitrypsin deficiency. Hepatol Res. 2010; 40: 641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha 1 antitrypsin deficiency. N Engl J Med. 1986; 314: 736–739. [DOI] [PubMed] [Google Scholar]
- 25. Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter DH. A lag in intracellular degradation of mutant alpha 1‐antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha 1‐antitrypsin deficiency. Proc Natl Acad Sci USA. 1994; 91: 9014–9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qu D, Teckman JH, Omura S, Perlmutter DH. Degradation of a mutant secretory protein, alpha1‐antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem. 1996; 271: 22,791–22,795. [DOI] [PubMed] [Google Scholar]
- 27. Werner ED, Brodsky JL, McCracken AA. Proteasome‐dependent endoplasmic reticulum‐associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci USA. 1996; 93: 13,797–13,801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Teckman JH, Burrows J, Hidvegi T, Schmidt B, Hale PD, Perlmutter DH. The proteasome participates in degradation of mutant alpha 1‐antitrypsin Z in the endoplasmic reticulum of hepatoma derived hepatocytes. J Biol Chem. 2001; 276: 44,865–44,872. [DOI] [PubMed] [Google Scholar]
- 29. Teckman JH, Gilmore R, Perlmutter DH. Role of ubiquitin in proteasomal degradation of mutant a1‐antitrypsin Z in the endoplasmic reticulum. Am J Physiol Gastrointest Liver Physiol. 2000; 278:G39–G48. [DOI] [PubMed] [Google Scholar]
- 30. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000; 290: 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T. Intracellular inclusions containing mutant alpha1‐antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006; 281: 4467–4476. [DOI] [PubMed] [Google Scholar]
- 32. Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human alpha‐1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006; 17: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pan S, Huang L, McPherson J, Muzny D, Rouhani F, Brantly M, Gibbs R, Sifers RN. Single‐nucleotide polymorhphism‐mediated translational suppression of endoplasmic reticulum mannodisase I modifies the onset of end‐stage liver disease in alpha‐1 antitrypsin deficiency. Hepatology. 2009; 50: 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hidvegi T, Mirnics K, Hale P, Ewing M, Beckett C, Perlmutter DH. Regulator of G Signaling 16 is a marker for the distinct endoplasmic reticulum stress state associated with aggregated mutant alpha1‐antitrypsin Z in the classical form of alpha1‐antitrypsin deficiency. J Biol Chem. 2007; 282: 27,769–27,780. [DOI] [PubMed] [Google Scholar]
- 35. Chen C, Zheng B, Han J, Lin SC. Characterization of a novel mammalian RGS protein that binds to Ga proteins and inhibits pheromone signaling in yeast. J Biol Chem. 1997; 272: 8679–8685. [DOI] [PubMed] [Google Scholar]
- 36. Gohla A, Klement K, Piekorz RP, Pexa K, vom Dahl S, Spicher K, Dreval V, Haussinger D, Birnbaumer L, Nurnberg B. An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc Natl Acad Sci USA. 2007; 104: 3003–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. An JK, Blomenkamp K, Lindblad D, Teckman JH. Quantitative isolation of alpha1AT mutant Z protein polymers from human and mouse livers and the effect of heat. Hepatology. 2005; 41: 160–167. [DOI] [PubMed] [Google Scholar]
- 38. Lindblad D, Blomenkamp K, Teckman J. Alpha‐1‐antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology. 2007; 46: 1228–1235. [DOI] [PubMed] [Google Scholar]
- 39. Hadzic N, Quaglia A, Mieli‐Vergani G. Hepatocellular carcinoma in a 12‐year‐old child with PiZZ a1‐antitrypsin deficiency. Hepatology. 2006; 46: 194. [DOI] [PubMed] [Google Scholar]
- 40. Zhou H, Ortiz‐Pallardo ME, Ko Y, Fischer H‐P. Is heterozygous alpha‐1‐antitrypsin deficiency type PiZ a risk factor for primary liver cancer. Cancer. 2000; 88: 2688–2676. [DOI] [PubMed] [Google Scholar]
- 41. Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, et al Small molecule regulators of autophagy identified by an image‐based high‐throughput screen. Proc Natl Acad Sci USA. 2007; 104: 19,023–19,028. [DOI] [PMC free article] [PubMed] [Google Scholar]