

Abstract
Chemical homeostasis in the extracellular fluid of the central nervous system (CNS) is maintained by two brain barrier systems, i.e., the blood–brain barrier (BBB) that separates the blood circulation from brain interstitial fluid and the blood–cerebrospinal fluid barrier (BCB) that separates the blood from the cerebrospinal fluid (CSF). The choroid plexus, where the BCB is located, is a polarized tissue, with the basolateral side of the choroidal epithelium facing the blood and the apical microvilli in direct contact with the CSF. The tissue plays a wide range of roles in brain development, aging, nutrient transport, endocrine regulation, and pathogenesis of certain neurodegenerative disorders. This chapter describes two in vitro cultures that have been well established to allow for study of the BCB structure and function. The primary choroidal epithelial cell culture can be established from rat choroid plexus tissue, and a similar immortalized murine choroidal epithelial cell culture known as Z310 cells has also been established. Both cultures display a dominant polygonal morphology, and immunochemical studies demonstrate the presence of transthyretin, a thyroxine transport protein known to be exclusively produced by the choroidal epithelia in the CNS. These cultures have been adapted for use on freely permeable Transwell® membranes sandwiched between two culture chambers, facilitating transport studies of various compounds across this barrier in vitro. These choroidal epithelia cultures with the Transwell system will perceivably assist blood–CSF barrier research.
Keywords: Choroid plexus, Cerebrospinal fluid, Transthyretin, Z310 cells
1. Introduction
The brain barrier system that separates the systemic circulation from the cerebrospinal fluid (CSF) compartment is known as the blood–CSF barrier, which is primarily located in the choroid plexus. The function of the blood–CSF barrier is to restrict the access of substances from the blood to the CSF as well as remove substances from the CSF to the blood. The choroid plexus also actively produces and secretes CSF along with critical molecules such as transthyretin (TTR) and transferrin into the brain. Under a microscope, the choroid plexus is composed of three cellular layers: (1) the apical epithelial cells facing the CSF, (2) the underlying supporting connective tissue, and (3) the inner layer of endothelial cells with immediate contact with the blood. Given that the endothelial cells of the choroid plexus are functionally leaky, the passage of substances across this barrier is essentially controlled by the apical layer of epithelial cells which are tightly connected with one another through tight junction proteins. These tight junctions constitute the structural basis for the blood–CSF barrier, through which only the selected materials may gain access to the CSF, while most of the water-soluble substances, proteins, ions, and macromolecules are impeded from the blood to the CSF.
Increasing research effort in understanding pharmacokinetics/toxicokinetics of active molecules in the CSF, a central milieu of the central nervous system (CNS), and in discovering the etiology and therapy of neurodegenerative diseases demands appropriate in vitro blood–CSF model systems that allow for characterizing the transport property of interested molecules across the blood–CSF barrier. During the past 15 years, we have successfully developed a standard protocol for primary culture of choroidal epithelial cells from rodents and routinely used the cultured cells to investigate the transport kinetics as well as molecular mechanisms of metals such as manganese (Mn), iron (Fe), and copper (Cu); chemicals such as thyroxin; and proteins such as β-amyloids (Aβ) and TTR (1–9). We have further immortalized rat choroidal epithelial cells and established a highly stable choroidal epithelial cell line known as the Z310 cell line (10, 11). Both model systems possess the essential morphology and characteristics of the blood–CSF barrier, and prove useful to study the structure and function of the blood–CSF barrier as well as examine transport properties of materials by this barrier.
In this chapter, we first describe the methods to conduct primary culture of rat choroidal epithelial cells. This is followed by the procedures to culture established Z310 cells. The methods to characterize the cells are addressed to allow the readers to typify the cells by their own hands. Finally, special notes are provided for advices learned from our own experiences and mistakes.
2. Material
2.1. Material for Primary Cell Culture
2.1.1. Coating of Culture Dishes
0.1% Type 1 Collagen (Sigma-Aldrich®) solution: Diluted to 0.01% working solution with distilled-deionized water.
Transwell® culture chambers, 12 mm in diameter, 0.4 μm pore size (Corning Costar).
35-mm tissue culture grade Petri dishes.
2.1.2. Tissue Isolation and Separation
Sprague-Dawley rats, male, 6–8 weeks old, 150–180 g (Harlan).
Ketamine/xylazine (75:10 mg/mL, 1 mL/kg body weight).
Dissection kit including scissors, bone cutter, and fine forceps.
75% Ethanol.
Phosphate-buffered saline (PBS): To 800 mL of distilled-deionized water add 8.0 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, and 0.24 g KH2PO4. Adjust the pH to 7.4 with HCl, and bring the volume up to 1,000 mL. Autoclave and store at 4°C.
2.1.3. Primary Cell Culture
Digestion solution: 4 mg/mL pronase (Calbiochem-Novobiochem). Dissolve 6 mg in 1.5 mL HBSS. The solution should then be transferred to a syringe and passed through an attached 0.22 μm low-protein-binding filter unit (Millipore). The final stock solution (4 mg/mL) should be kept on ice in a culture hood until use. The solution must be made fresh on the day of the experiment.
0.4% Trypan blue.
cis-4-Hydroxy-D-proline (Sigma-Aldrich®): Final concentration of 25 μg/mL in medium.
0.25% Trypsin, 1 mol/L EDTA (Life Technologies™).
Dulbecco’s Modified Eagle Medium (DMEM, Gibco®).
Hank’s Buffered Salt Solution (HBSS, Gibco®).
Fetal bovine serum (FBS) (heat inactivated, sterile-filtered).
Penicillin–streptomycin (10,000 units/mL penicillin Na+10,000 μg/mL streptomycin sulfate in 0.85% saline) (Gibco®).
Gentamicin (10 mg/mL) solution.
Mouse epidermal growth factor (EGF): Dissolve 0.1 mg in 1 mL of primary cell culture medium for a stock of 0.1 mg/mL.
2.2. Z310 Cell Culture
Like the culture of primary choroidal epithelial cells, the normal growth medium for Z310 cells consists of three major components in DMEM: (a) antibiotics to prevent infection, (b) FBS to provide nutrients, and (c) EGF to stimulate the growth of epithelia.
Growth medium: 450 mL DMEM supplemented with 50 mL FBS (10%), 5 mL penicillin–streptomycin (10× = 10%), penicillin–streptomycin (10,000 units/mL penicillin Na + 10,000 μg/mL streptomycin sulfate in 0.85% saline), 2 mL gentamicin solution (10 mg/mL), 50 μL of EGF (0.1 mg/mL stock) for a final concentration of 10 ng/mL.
Dexamethasone (only used in media during Transwell® studies): Dissolve in Z310 cell culture medium and dilute in medium for a final concentration of 1 μM.
Cryoprotective medium: 90–95% Culture medium supplemented with 5–10% dimethyl sulfoxide (DMSO).
2.3. Two-Chamber Transepithelial Model
Transwell® Permeable Supports (12 mm insert, Corning).
Epithelial voltohmmeter (model EVOM, World Precision Instruments).
2.4. Immuno-cytochemical Studies
Anti-TTR polyclonal antibody produced in the mouse (Sigma-Aldrich®).
4% Paraformaldehyde in PBS.
1% Permeabilization buffer: Triton™ X-100 in PBS 10 μL/mL.
1% Bovine serum albumin (BSA) in PBS.
1:250 Mouse anti-human TTR antibody, diluted in 1% BSA.
1:2,000 Fluorescein-conjugated goat anti-mouse antibody, diluted in PBS.
Confocal microscope with fluorscein isothiocyanate (FITC) and phase contrast optics.
2.5. Reverse Transcription Polymerase Chain Reaction
TRIzol® reagent (Life Technologies™).
RNeasy Mini kit (Qiagen).
Chloroform.
Ethanol.
Isopropanol.
Primers specifically selected for rat TTR (synthesized by IDT).
DEPC-treated water: Prepare by adding 1 mL of DEPC to 1,000 mL of distilled, deionized water, standing overnight, and autoclaving prior to use.
RT buffer: DEPC water, 25 mM MgCl2, 10× PCR buffer II, 10 mM dNTP mix, 50 μM Oligo dT, 20 U/μL Rnase inhibitor, MuLV reverse transcriptase (Applied Biosystems).
Master Mix: DEPC water, 25 mM MgCl2, 10 mM dNTP mix, 5× GoTaq Flexi buffer, GoTaq polymerase.
1.0% Agarose gels containing 0.5 μg/mL ethidium bromide.
3. Methods
3.1. Procedures for Cell Culture
3.1.1. Coating of Culture Dishes
To coat dishes or the membranes of Transwell® inner chambers with collagen, dilute the stock collagen (0.1%) 1:10 with DDI H2O for a 0.01% working solution (see Subheading 2.1.1).
Add an aliquot of diluted collagen to 35-mm dishes (800 μL) or Transwell® inserts (100 μL). Swirl the dishes or inserts to ensure an even distribution of the coating solution.
Incubate at room temperature in the culture hood for 4–5 h to allow the protein to bind to the surface.
Remove the excess liquid and then allow the coated dishes to air-dry in the hood under UV light to avoid contamination.
3.1.2. Tissue Isolation and Separation
Figure 1 illustrates the flowchart of the general procedures pertaining to the primary culture of choroidal epithelial cells. Please keep in mind that all procedures must be operated in sterile condition; the surgical tools must be autoclaved; the skin where the insertion made must be sterilized with 75% ethanol; and all glassware must also be autoclaved.
Fig. 1.

Flowchart of procedures in establishing primary culture of choroidal epithelial cells.
Anesthetize the rats with an i.p. injection of ketamine/xylazine.
To minimize the amount of blood present in choroid plexus tissues, draw as much blood as possible from the inferior vena cava using a syringe.
Remove the hair on the back of the head with a pair of scissors or electric trimmer.
Sterilize the exposed skin using cotton wool saturated with 75% ethanol.
Use a pair of scissors to cut the skin and to expose the skull. Use a bone cutter to remove the skull bone.
Remove the brain from the skull and place in a beaker containing PBS on ice to chill the tissue and wash off excess blood.
Once 5–8 brains have been collected, move them into a culture hood for dissection of the choroid plexuses.
Dissect the choroid plexuses from both the lateral and third ventricles and immerse them in 0.5 mL of HBSS at room temperature.
When all the choroid plexus tissues are collected, mince the plexus tissues with a pair of fine ophthalmologic scissors to roughly 1-mm cubes (~5 min of chopping).
Bring the total volume up to 1 mL by the addition of 0.5 mL HBSS.
3.2. Primary Cell Culture
3.2.1. Tissue Digestion
Add 1 mL of digestion solution (see Subheading 2.1.3, step 1) to the beaker to give a final pronase concentration of 4 mg/mL.
Swirl the beaker lightly by hand to allow a complete mixing of the digestion solution with the tissues.
Incubate at 37°C for 5–10 min.
Stop the digestion reaction by adding 4 mL of HBSS solution to the digestion mixture (see Note 1).
Centrifuge at 800 × g for 5 min at 4°C in a 15-mL sterile tube.
Discard the supernatant and wash the pellet once more with HBSS by resuspension and centrifugation. At this point the pellet should contain clumps of primary epithelial cells probably joined by tight junction proteins.
Resuspend the pellet in 2 mL of growth medium.
Mechanically dissociate the cells by 7–10 forced passages through a 20-gauge needle (see Note 1).
Remove an aliquot (0.1 mL) of cell suspension and mix with 0.1 mL of 0.4% trypan blue to count cell numbers and to assess the viability.
The procedure for cell isolation described here yields ~0.8–1×105 epithelial cells per rat.
3.2.2. Culture of Epithelial Cells
Prior to cell seeding, dilute the cell preparations with growth medium to ~1–2 × 105 cells/mL (see Note 2).
Plate the cells onto 35-mm coated Petri dishes (2–3 × 105 cells per dish) and culture in a humidified incubator with 95% air/5% CO2 at 37°C.
After 10 h in culture, remove unattached epithelial cells in culture medium and leave behind the attached fibroblast cells. This minimizes fibroblast contamination, a major problem in primary culture of epithelial cells. This “fibroblast adhering-off” method effectively leaves fibroblasts behind in the collagen-coated dishes, because fibroblasts usually attach to the collagen-coated surface much faster (6–10 h) than epithelial cells (16–24 h).
Replate the epithelial cells into new 35-mm plates and then leave cells undisturbed for at least 48 h.
Change the medium every 2–3 days thereafter for the duration of the culture.
Two days after the seeding, remove the culture medium, and replace with fresh medium containing cis-HP (see Subheading 2.1.3, step 3) to further control fibroblast contamination if necessary (see Note 3).
Usually the initial reseeding and treatment with cis-HP suffice for the purpose of inhibition of the growth of fibroblasts. Typical photographs of cultured choroidal epithelial cells under a phase contrast microscope are seen in Fig. 2.
After 3–5 days of culture with cis-HP, return the cells to normal growth medium without cis-HP, providing there are no visible fibroblasts under the microscope.
From our own experience, if the digestion procedure works well, the epithelia usually attach and grow rapidly. Therefore, the “fibroblast adhering-off” and cis-HP may not be necessary. However both methods greatly enhance the likelihood of a successful culture and are recommended.
To detach the cells for bioassays, incubate the culture with trypsin–EDTA in PBS at 37°C for 10 min.
Harvest the cells, centrifuge, and wash. They can then be used for further molecular studies or for Transwell® transport studies.
Fig. 2.
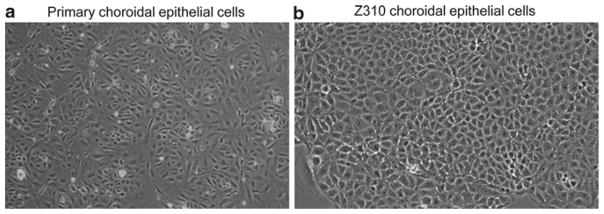
Morphology of choroidal epithelial cells in culture. (a) Primary culture of choroidal epithelial cells after 5 days in culture (10×). Note the con fluent layer of cells with a predominant polygonal cell type. The choroid plexus tissue was obtained from 6-week-old Sprague-Dawley rats. (b) Immortalized Z310 choroidal epithelial cells in culture (20×). Passage 86.
3.3. Two-Chamber Transepithelial Transport Model with Primary Choroidal Cells
The procedure for preparation of epithelial suspension is the same as described in Subheading 3.2.
Prior to seeding the cells in Transwell® chambers (inserts), coat the permeable membranes attached to the inserts with collagen as described in Subheading 3.1.1 (or purchase collagen-coated membranes).
Insert the inner chambers into the outer (basal) chambers, which should already contain 1.2 mL of growth medium (see Fig. 3).
Plate aliquots (0.8 mL) of cell suspension into the 12-mm collagen-coated culture wells (inserts) and place in incubator at 37°C.
Allow the cells to grow for 48 h.
Change the medium in both chambers every 2 days thereafter.
The formation of confluent impermeable cell monolayers is judged by three criteria: (1) the height of the culture medium in the inner chamber has to be at least 2 mm higher than that in the outer chamber for at least 24 h; (2) the appearance of a confluent monolayer on the insert under the microscope; and (3) the electrical resistance across the cell layer has to fall into the range of 65–80 Ωcm2 (see Note 4).
Transepithelial electrical resistance can be measured using an epithelial voltohmmeter after the cells have been cultured in the chambers for at least 4 days.
The net value of electrical resistance is calculated by subtracting the background (which is measured on collagen-coated cell-free chambers) from values of epithelial cell-seeded chambers.
Fig. 3.
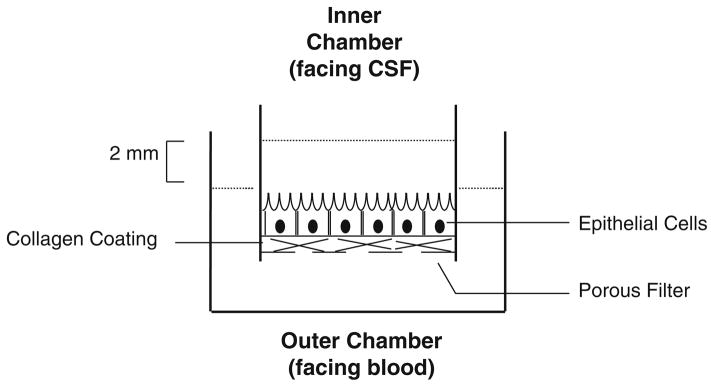
Transepithelial model of the blood–CSF barrier used to study transepithelial transport. Epithelial cells are connected by tight junctions and form a barrier between fluids in the inner and outer chambers. Fluid in the inner chamber is in contact with the apical microvilli on the surface of the cells, while the fluid in the outer chamber has access to the basal surface of the cells.
3.4. Immuno-cytochemical Studies of the Marker of Choroidal Epithelia
A reliable method for the identification of choroidal epithelial cells is visualization of TTR, a unique marker for choroidal epithelial cells (see Notes 5 and 6). TTR is a 55,000-Da protein consisting of four identical subunits in tetrahedral symmetry. Per unit of weight, rat choroid plexus contains ten times more TTR mRNA than liver, and per gram of tissue, synthesizes TTR 13 times faster than the liver; the latter is the major organ in the body producing serum TTR (12, 13).
Isolate choroid plexus tissues from the lateral ventricles as previously described in Subheading 3.1.2.
Fix the tissue in 4% paraformaldehyde in PBS.
Wash three times with PBS.
Permeabilize the tissue by incubation in 0.1% permeabilization buffer (see Subheading 2.4, step 3) for 30 min.
Wash with PBS.
Block with 1% BSA for 60 min.
Incubate the cells with anti-TTR antibody (see Subheading 2.4, step 5) overnight at 4°C.
Wash with 1% BSA.
Incubate with secondary antibody for 1 h at 37°C.
Wash three times with PBS.
Examine the tissue using confocal microscopy (see Fig. 4).
Fig. 4.
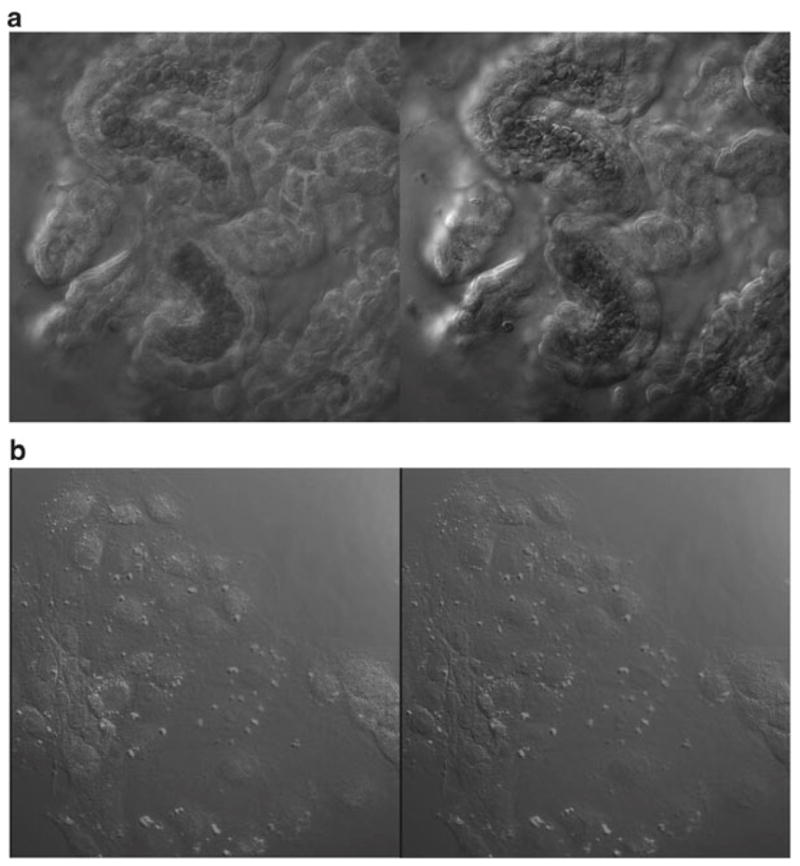
Confocal microscopic depicting TTR staining in the choroid plexus tissue (a) and cultured Z310 cells (b). Tissue was treated with anti-TTR primary antibody followed by secondary antibody conjugated with fluorescein. Note the positive staining primarily along the basolateral side of the choroid plexus tissue. A Nikon C1 series modular confocal microscope was used to view the tissue through a 60× oil immersion objective with a 488 nm laser line for excitation.
3.5. RT-PCR Analysis
While immunohistochemical staining of TTR proteins is regarded as an acceptable approach to identify choroidal epithelial cells, it may also be used in conjunction with reverse transcription polymerase chain reaction (RT-PCR) to validate expression of the mRNA encoding the protein.
Extract total RNA from the cultured cells or rat liver (as a positive control) using the TRIzol® method followed by RNA cleanup using an RNeasy mini kit.
Carry out the RT on 1 μg of total RNA, using MuLV reverse transcriptase with oligo dT primers.
The reaction should be carried out as follows: 25°C 10 min, 48°C 60 min, 95°C 5 min, and 4°C hold.
For PCR amplification, one set of specific oligonucleotide pairs should be incubated with the new cDNA synthesized in the above reaction mixture.
Add 80 μL Master Mix (see Subheading 2.5, step 9), containing 2.5 U Taq DNA polymerase, giving rise to a total volume of 100 μL.
The cycle parameters are 5 min at 94°C for initial denaturation, 0.5 min at 57°C for annealing, and 0.5 min at 72°C for extension. The subsequent cycle is as follows: 1 min at 94°C, 0.5 min at 57°C, and 0.5 min at 72°C for 40 cycles. Follow this with a 5-min incubation at 72°C.
An aliquot (5 μL) of each reaction mixture should be analyzed by electrophoresis on 1.0% agarose gels containing 0.5 μg/mL ethidium bromide (see Fig. 5).
-
The primers (custom-synthesized) designed by us specifically for rat TTR consist of:
Primer sense: 5′-TTCCCTTCGCCTGTTCTTCTT-3′.
Primer antisense: 5′-TTCTGGGGGTTAACTGACGACA-3′.
This amplifies a product of 443 bp covering the mature TTR peptide from rats.
Fig. 5.
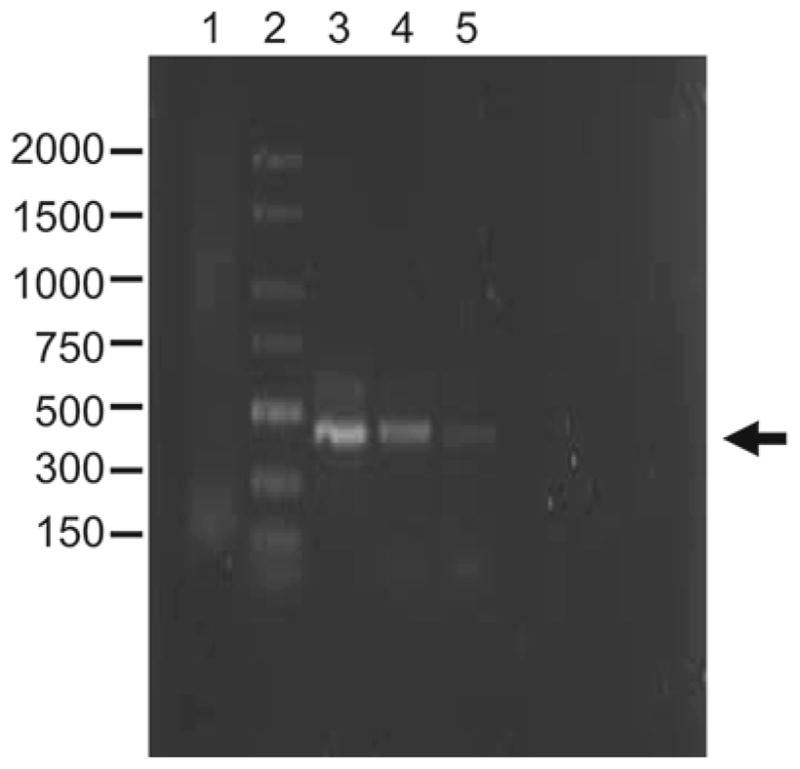
Expression of TTR mRNA in primary choroidal epithelial cells and in choroidal Z310 cells by RT-PCR analysis. All samples underwent RT-PCR unless otherwise stated. Arrow indicates bands corresponding to TTR mRNA. Lane ID: Lane 1 total RNA for PCR without RT, lane 2 base pair ladder, lane 3 liver mRNA with selected primer, lane 4 cultured primary plexus cells mRNA with selected primer, lane 5 Z310 cells mRNA with selected marker.
3.6. Immortalized Z310 Choroidal Epithelial Cells from the Murine Choroid Plexus
To facilitate the in vitro study of the blood–CSF barrier we have used a gene transfection technique to immortalize murine choroidal epithelial cells, known as Z310 cells. The cells display the same polygonal epithelial morphology and characterization as primary choroidal epithelial cells (see Fig. 2).
3.6.1. Transfer/Passage
Pre-warm PBS and growth medium in the water bath at 37°C.
Aspirate all media from the plate(s).
Add 5 mL of PBS to the plates, swirl slightly, and then aspirate PBS (repeat 2×).
Add 200 μL of trypsin–EDTA to detach the cells.
Incubate the plate(s) at 37°C for 10 min.
Add 8–10 mL of medium to each of the new plates.
After 10-min digestion, remove the cells from the incubator and observe under the microscope. If the cells look like “round balls,” the digestion is complete and cells are detached.
Add 3 mL of cell culture medium into the old plate for a 1/12 split or 4 mL for a 1/16 split.
Mix well by using a pipette to break up all the cell clusters.
Pipette 0.25 mL of cells from the old plate into a new plate which has already had 8–10 mL of new medium.
Label the new passage and place in a 37°C incubator.
Passage cells every 2–3 days, when reaching roughly 80% confluence.
3.7. Freezing and Thawing Z310 Cells
3.7.1. Freezing Z310 Cells
After cells are digested and resuspended in growth medium (see Subheading 2.2, step 1), pellet the cells (200 × g for 5–10 min, 4°C).
Aspirate supernatant above the cell pellet.
Slowly add (drop by drop) cryoprotective medium (see Subheading 2.2, step 3) to the cell pellet on ice.
Resuspend the cell pellet thoroughly in the cryoprotective medium.
Aliquot ~1 mL of cells (density ~1–2 × 107) to the appropriate tubes for storage.
Leave the aliquots on ice for 5–10 min.
Place aliquots in an insulated box (styrofoam) and place at −80°C overnight.
The next day remove the insulated container from the freezer, and place the aliquots in appropriate container for storage in liquid nitrogen.
3.7.2. Thawing Z310 Cells
Remove the vial from liquid nitrogen.
Thaw in a water bath at 37°C for 2–5 min.
Remove the vial from the bath, and clean with 70% ethanol.
Under the hood remove the cells from the vial and place in 8–10 mL of culture medium.
Use this medium to resuspend the cells and wash.
Pellet cells and resuspend in new culture medium.
Plate the cells on uncoated dishes and place into an incubator at 37°C for 1–2 days.
Pass cells upon 85–90% confluence.
3.8. Two-Chamber Transepithelial Transport Model with Z310 Cells
The procedure for preparation of epithelial suspension is the same as described in Subheading 3.6.1.
Prior to seeding the cells in Transwell® chambers (inserts), coat the permeable membranes attached to the inserts with collagen as described in Subheading 3.1.1 (or purchase collagen-coated membranes).
Insert the inner chambers into the outer (basal) chambers, which should already contain 1.2 mL of growth medium (see Fig. 3). For Z310 cells add 1 μM dexamethasone (see Subheading 2.2, step 2) in the growth medium for Transwell® studies in order to improve the tightness of the Z310 barrier.
Plate aliquots (0.8 mL) of cell suspension into the 12-mm collagen-coated culture wells (inserts).
Typical seeding numbers of Z310 cells can range from 5.0 × 104 to 2.0×106 cells per well. Depending on the seeding density, cells can reach confluence anywhere from 2 to 6 days.
Change the medium every 2 days thereafter.
The formation of confluent impermeable cell monolayers is judged by three criteria: (1) the height of the culture medium in the inner chamber has to be at least 2 mm higher than that in the outer chamber for at least 24 h; (2) the appearance of a confluent monolayer on the insert under the microscope; and (3) the electrical resistance across the cell layer has to fall into the range of 85–100 Ωcm2.
Transepithelial electrical resistance can be measured using an epithelial voltohmmeter after the cells have been cultured in the chambers for 1 day.
The net value of electrical resistance is calculated by subtracting the background (which is measured on collagen-coated cell-free chambers) from values of epithelial cell-seeded chambers.
Footnotes
Isolation of epithelial cells from the choroid plexus. To help ensure a large yield of epithelial cells from the primary culture, the digestion procedure is critical. Both collagenase (2 mg/mL) and pronase (2 mg/mL) have been used to digest the tissues in our laboratory. We have found that pronase is the most effective at releasing the epithelial cells. It is important that the concentration and duration of pronase digestion be well monitored. The ideal digestion time with pronase varies depending on the tissue mass. The general rule of thumb is to carefully watch the color change of the medium. With a complete digestion, the medium usually changes from a light red to a yellow-orange, and from transparent to slightly cloudy. Digestion with pronase should not exceed 10 min; prolonged digestion reduces cell attachment down the line. The other vital procedure is the mechanical digestion of the cells. The epithelial clumps present after digestion normally attach to the surface of the dish, but will form fewer colonies if they are not dissociated from one another. Mechanical digestion through the needle effectively dissociates the cell clumps to produce a maximal yield of epithelia and greater plate efficiency.
Density of cells for initial seeding: An effective primary culture of choroidal epithelia requires a sufficient number of cells at the initial seeding. When cells are plated at a density of <104/mL, the cell proliferation can be very slow or virtually nonexistent. We recommend seeding the cells at a density of 2–4×105 cells/35-mm dish, which is roughly 2 mL of 1–2×105 cells/mL after digestion. One further noteworthy detail pertains to the transfer of the cells from centrifuge tubes to culture dishes. It is important to pre-wet the tips of the glass pipettes prior to transferring the cells. This minimizes the number of cells adhering to the dry tip of the glass pipette.
Control of fibroblasts: Arguably the most significant challenge of this procedure is the control of fibroblast contamination in the choroidal epithelial cell culture. Fibroblasts are visible in the culture under the light microscope and are typically elongated with their nuclei condensed. Fibroblasts usually rapidly spread between the epithelial clusters. We have tried several methods to inhibit fibroblast contamination. From our experience the most successful strategy is a two-step approach. The first step is known as the “fibroblast adhering-off” approach. This technique takes advantage of the higher affinity of the fibroblast cells to collagen-coated surfaces in the early cell selection stage. A relatively complete fibroblast adherence will occur 6–12 h after the initial seeding. At this time remove all the media carefully and plate onto freshly coated 35-mm dishes. After 48 h add new medium that contains 25 μg/mL of cis-HP, which can be withdrawn from the medium a few days (3–5) later. We have found that both concentration of cis-HP as well as time of its addition to the medium are critical to its success. The earlier the cis-HP treatment the more effective it is. The caveat for cis-HP treatment is that the chemical also kills epithelial cells. Thus, we recommend adding the cis-HP no earlier than 48 h after the initial seeding. From our experience this process effectively reduces fibroblast contamination and gives us the purest epithelial culture.
Culture on Transwell® chambers: The procedure for culturing primary cells on Transwell® membranes is identical to the standard primary culture. Cells grown on the inner chamber membranes display the same morphology as those grown in culture dishes. The epithelial cells are connected by tight junctions and once they grow to confluence they form an impermeable barrier between the medium in the inner and outer chambers. The net electrical resistance across this barrier in our studies is 65–80 Ωcm2 for the primary culture and 85–100 Ωcm2 in Z310 cells (10). It is worthy to note that many factors can influence the determination of electrical resistance such as temperature, pH (physiological solutions vs. culture media), age of tissues or cultures, and freshness of the culture medium. From our experience, a higher pH, colder culture medium, and fresher culture medium usually result in a higher resistance reading. We have used this in vitro blood–CSF barrier model to study protein and metal transport and have demonstrated that the choroid plexus can remove Aβ, a vital peptide in the etiology of Alzheimer’s disease, from the CSF (2). In addition this model has been used to show that manganese (Mn) exposure alters iron (Fe) flux across the blood–CSF barrier (7). Both Z310 and primary culture in vitro Transwell® models have been used to demonstrate the effect of lead (Pb) exposure on transport of thyroxin and Aβ at the blood–CSF barrier. Pb has been shown to hinder the transepithelial transport of thyroxine as well as decrease the blood–CSF barrier’s permeability (1, 14, 15). The decrease in thyroxine transport appears to be due to the inhibitory effect of Pb on the production and secretion of TTR by the choroid plexus (5, 6). The decrease in blood–CSF barrier permeability is the result of early Pb exposure selectively inhibiting the expression of essential tight junction proteins (15). Recently, we have also applied this model to study copper transport at the blood–CSF barrier (data not shown). From these studies we have observed that the transepithelial transport of metal ions is a slow process and may take up to 24 h to reach equilibrium. Short time course studies may not be adequate in assessing the steady-state transport of substances across the blood–CSF barrier; so patience is vital for these experiments.
Use of immortalized choroidal epithelial Z310 cells: While primary culture serves as an ideal model for the in vitro study of the blood–CSF barrier, it has many limitations making it undesirable to use on a daily basis. The disadvantages include a lack of cell abundance, reliance on a large number of animals, difficulty in obtaining pure cultures, and a short life span in culture. Therefore, the development of an immortalized choroidal epithelial line for in vitro studies has been a long, overdue summon in brain barrier transport research. Z310 cells possess many of the same phenotypic characteristics of primary choroidal cells, including a polygonal epithelial type as well as the presence of TTR. These cells have been proven an acceptable model for in vitro blood–CSF study and have been widely used (9, 16).
Characterization of primary and Z310 choroidal epithelial cells: Immunohistochemical staining with an anti-TTR antibody is a reliable way to distinguish choroidal epithelia from other types of cells. The presence of TTR has been repeatedly demonstrated in the choroidal epithelia of the brain (17, 18). In addition to immunochemical staining, RT-PCR is an acceptable methodology to validate the presence of TTR mRNA transcripts in choroidal epithelia. The sequence of TTR mRNA has been reported by Dickson et al. (13). We have designed a set of primers in order to run RT-PCR for TTR mRNA from the rat. The methods presented in this article display reliable evidence for TTR expression in our cultured Z310 cells, freshly isolated choroid plexus tissues, as well as the liver. The establishment of choroidal epithelial cell cultures from various sources has been well documented in the literature. These include cells from mouse (19, 20), rat (20–22), rabbit (12), sheep (23), and cow (24), each having its own particular set of advantages and disadvantages.
References
- 1.Zheng W, Zhao Q, Graziano JH. Primary culture of rat choroidal epithelial cells: a model for in vitro study of the blood–cerebrospinal fluid barrier. In Vitro Cell Biol Dev. 1998;34:40–45. doi: 10.1007/s11626-998-0051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crossgrove JS, Li GJ, Zheng W. The choroid plexus removes beta-amyloid from the cerebrospinal fluid. Exp Biol Med. 2005;230(10):771–776. doi: 10.1177/153537020523001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deane R, Zheng W, Zlokovic BV. Brain capillary endothelium and choroid plexus epithelium regulate transport of transferrin-bound and free iron into the rat brain. J Neurochem. 2004;88:813–820. doi: 10.1046/j.1471-4159.2003.02221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng W, Deane R, Redzic Z, Preston JE, Segal MB. Transport of L-(125I)thyroxine by in-situ perfused ovine choroid plexus: inhibition by lead exposure. J Toxicol Env Health. 2003;66:435–451. doi: 10.1080/15287390306451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng W, Perry DF, Nelson DL, Aposhian HV. Protection of cerebrospinal fluid against toxic metals by the choroid plexus. FASEB J. 1991;5:2188–2193. doi: 10.1096/fasebj.5.8.1850706. [DOI] [PubMed] [Google Scholar]
- 6.Zheng W, Shen H, Blaner WS, Zhao Q, Ren X, Graziano JH. Chronic lead exposure alters transthyretin concentration in rat cerebrospinal fluid: the role of the choroid plexus. Toxicol Appl Pharmacol. 1996;139:445–450. doi: 10.1006/taap.1996.0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang XQ, Li GJ, Zheng W. Efflux of iron from the cerebrospinal fluid to the blood at the blood–CSF barrier: effect of manganese exposure. Exp Biol Med. 2008;233:1561–1571. doi: 10.3181/0803-RM-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi BS, Zheng W. Copper transport to the brain by the blood brain barrier and blood–CSF barrier. Brain Res. 2009;1248:14–21. doi: 10.1016/j.brainres.2008.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng W. Toxicology of choroid plexus: special reference to metal-induced neurotoxicities. Microsc Res Tech. 2001;52:89–103. doi: 10.1002/1097-0029(20010101)52:1<89::AID-JEMT11>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng W, Zhao Q. Establishment and characterization of an immortalized Z310 choroidal epithelial cell line from murine choroid plexus. Brain Res. 2002;958:371–380. doi: 10.1016/s0006-8993(02)03683-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi LZ, Wang SZ, Li GJ, Zheng W. Use of Z310 cells as an in vitro blood–cerebrospinal fluid barrier model: tight junction proteins and transport properties. Toxicol In Vitro. 2008;22:190–199. doi: 10.1016/j.tiv.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayer SE, Sanders-Bush E. Sodium-dependent antiporters in choroid plexus epithelial cultures from rabbit. J Neurochem. 1993;60:1304–13. doi: 10.1111/j.1471-4159.1993.tb03291.x. [DOI] [PubMed] [Google Scholar]
- 13.Dickson PW, Howlett GJ, Schreiber G. Rat transthyretin (prealbumin): molecular cloning, nucleotide sequence, and gene expression in liver and brain. J Biol Chem. 1985;260:8214–8219. [PubMed] [Google Scholar]
- 14.Zheng W, Blaner WS, Zhao Q. Inhibition by Pb of production and secretion of transthyretin in the choroid plexus: its relationship to thyroxine transport at the blood–CSF barrier. Toxicol Appl Pharmacol. 1999;155:24–31. doi: 10.1006/taap.1998.8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi LZ, Zheng W. Early lead exposure increases the leakage of the blood–cerebrospinal fluid barrier, in-vitro. Hum Exp Toxicol. 2007;26:159–167. doi: 10.1177/0960327107070560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi LZ, Zheng W. Establishment of an in vitro brain barrier epithelial transport system for pharmacological and toxicological study. Brain Res. 2005;1057:37–48. doi: 10.1016/j.brainres.2005.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aldred AR, Brack CM, Schreiber G. The cerebral expression of plasma protein genes in different species. Comp Biochem Physiol. 1995;111B:1–15. doi: 10.1016/0305-0491(94)00229-n. [DOI] [PubMed] [Google Scholar]
- 18.Herbert J, Wilcox JN, Pham KC. Transthyretin: a choroid plexus-specific transport protein in human brain. Neurology. 1986;36:900–911. doi: 10.1212/wnl.36.7.900. [DOI] [PubMed] [Google Scholar]
- 19.Bouille C, Mesnil M, Barriere H, Gabrion J. Gap junctional intercellular communication between cultured ependymal cells, revealed by Lucifer yellow CH transfer and freeze-fracture. Glia. 1991;4:25–36. doi: 10.1002/glia.440040104. [DOI] [PubMed] [Google Scholar]
- 20.Peraldi-Roux S, Nguyen-Than Dao B, Hirn M, Gabrion J. Choroidal ependymocytes in culture: expression of markers of polarity and function. Int J Dev Neurosci. 1990;8:575–588. doi: 10.1016/0736-5748(90)90050-c. [DOI] [PubMed] [Google Scholar]
- 21.Strazielle N, Ghersi-Egea JF. Demonstration of a coupled metabolism-efflux process at the choroid plexus as a mechanism of brain protection toward xenobiotics. J Neurosci. 1999;19:6275–6289. doi: 10.1523/JNEUROSCI.19-15-06275.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsutsumi M, Skinner MK, Sanders-Bush E. Transferrin gene expression and synthesis by cultured choroid plexus epithelial cells. J Biol Chem. 1989;264:9626–9631. [PubMed] [Google Scholar]
- 23.Harter DH, Hsu KC, Rose HM. Immunofluorescence and cyto-chemical studies of visna virus in cell culture. J Virol. 1967;1:1265–1270. doi: 10.1128/jvi.1.6.1265-1270.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whittico MT, Hui AC, Giacomini KM. Preparation of brush border membrane vesicles from bovine choroid plexus. J Pharmacol Methods. 1991;25:215–227. doi: 10.1016/0160-5402(91)90012-t. [DOI] [PubMed] [Google Scholar]