

Abstract
Ankyloblepharon Ectodermal Dysplasia and Cleft Lip/Palate (AEC) or Hay-Wells Syndrome is an autosomal dominant disorder characterized by a variety of phenotypes in ectodermal derivatives, including severe skin erosions, ankyloblepharon, coarse and wiry hair, scalp dermatitis, and dystrophic nails. AEC is caused by mutations in the gene encoding the TP63 transcription factor, specifically in the Sterile Alpha Motif (SAM) domain. The exact mechanism, however, by which these specific TP63 mutations lead to the observed spectrum of phenotypes is unclear. Analysis of individual TP63 target genes provides a means to understand specific aspects of the phenotypes associated with AEC. PERP is a TP63 target critical for cell-cell adhesion due to its participation in desmosomal adhesion complexes. As PERP null mice display symptoms characteristic of ectodermal dysplasia syndromes, we hypothesized that PERP dysfunction might contribute to AEC. Using luciferase reporter assays, we demonstrate here that PERP induction is in fact compromised with some, but not all, AEC-patient derived TP63 mutants. Through analysis of skin biopsies from AEC patients, we show further that a subset of these display aberrant PERP expression, suggesting the possibility that PERP dysregulation is involved in the pathogenesis of this disease. These findings demonstrate that distinct AEC TP63 mutants can differentially compromise expression of downstream targets, providing a rationale for the variable spectra of symptoms seen in AEC patients. Elucidating how specific TP63 target genes contribute to the pathogenesis of AEC will ultimately help design novel approaches to diagnose and treat AEC.
Keywords: TP63, mutations, ectodermal dysplasia, PERP, PERP protein, TP53 apoptosis effector protein related to PMP22, skin, biopsy, transgenic mice, Western blotting, immunohistochemistry
INTRODUCTION
TP63 is a transcriptional activator critical for the development of the skin and other ectodermal derivatives. This essential role was originally revealed through studies of TP63-deficient mice, which die postnatally from a failure of proper skin development and which lack a variety of ectodermal appendages, including hair follicles, teeth and mammary glands [Mills et al. 1999; Yang et al. 1999]. TP63 mutations have also been associated with several dominantly inherited human syndromes, including Ectrodactyly-Ectodermal Dysplasia and Cleft Lip/Palate (EEC) [Celli et al. 1999], Ankyloblepharon Ectodermal Dysplasia and Cleft Lip/Palate (AEC) [McGrath et al. 2001], Acro-Dermato-Ungual-Lacrimal-Tooth Syndrome (ADULT) [Amiel et al. 2001], Rapp-Hodgkin’s Syndrome (RHS) [Kantaputra et al. 2003], and Limb-Mammary Syndrome (LMS) [van Bokhoven et al. 1999]. Each syndrome is associated with a spectrum of phenotypes affecting the skin and various ectodermal derivatives, underscoring the important role for TP63 in the development of these structures. Although each syndrome is characterized by a unique set of symptoms, there is overlap in phenotypes between the diseases. For example, while ectodermal dysplasia is a cardinal feature of both the EEC and AEC Syndromes, ankyloblepharon and severe skin erosions are distinguishing characteristics of AEC.
Some of these syndromes are associated with mutations in specific domains of the TP63 protein, which presumably accounts for the distinct spectra of symptoms seen in each disease. For example, mutations in the DNA binding domain are associated with EEC, while mutations in the sterile alpha motif, a purported protein-protein interaction domain, are linked to AEC [van Bokhoven and McKeon 2002]. However, the mechanisms by which these different mutations predispose to distinct collections of symptoms remain unclear. EEC mutants, which harbor alterations in the DNA binding domain of TP63, are likely completely defective for transactivation of TP63 target genes. In contrast, at least some of the SAM domain mutations found in AEC do not disrupt TP63 DNA binding function, and may instead affect specific protein-protein interactions, potentially leading to selective effects on particular TP63 target genes [Lo Iacono et al. 2006]. TP63 directs the development of the skin and ectodermal derivatives through the induction of a plethora of target genes involved in different subprograms critical for the genesis and function of epithelia, including those involved in proliferation, differentiation, and cell-cell adhesion [Carroll et al. 2007; Ihrie et al. 2005; Truong et al. 2006; Yang et al. 2006]. Expression of individual target genes may be differentially affected in the various syndromes, thereby explaining the different spectra of symptoms associated with each disease. In addition, different effects of distinct TP63 mutants associated with a particular syndrome on specific target genes could explain the clinical variability in symptoms within that syndrome. Understanding the contribution of individual TP63 targets to the phenotypes associated with these syndromes will help better define the basis of these diseases.
PERP, which encodes a tetraspan membrane protein, was one of the first TP63 target genes shown to play a fundamental role in the integrity of stratified epithelia [Attardi et al. 2000; Ihrie et al. 2005]. PERP-/- mice display postnatal lethality accompanied by dramatic blistering of the skin and oral mucosa, reflecting a crucial role for PERP in cell-cell adhesion through multiprotein complexes known as desmosomes. These structures confer strength on tissues exposed to mechanical stress by virtue of mediating cell-cell contact and connecting to the intermediate filament network [Kottke et al. 2006]. Mutations in genes encoding desmosomal components are found in several human diseases associated with defects in skin, hair, and other ectodermal derivatives. For example, patients with mutations in PLAKOPHILIN-1 exhibit symptoms of ectodermal dysplasias, including defects in skin, hair, teeth, nails and sweat glands [McGrath et al. 1997] and individuals with mutations in PLAKOGLOBIN or DESMOPLAKIN have woolly hair and thickened skin of the palms and soles known as palmoplantar keratoderma [Kottke et al. 2006]. These findings underscore the importance of desmosomal adhesion in the proper function of ectodermal derivatives. Consistent with this idea, we have found that while the majority of PERP null mice die postnatally, a small number of these mice survive to adulthood and develop ectodermal dysplasia phenotypes reminiscent of those seen in EEC and AEC. In particular, PERP-/- mice exhibit palmoplantar keratoderma, malformed nails, abnormally thickened skin, disorganized and greasy hair, and are prone to bacterial infections [Ihrie et al. 2006]. These observations suggest that PERP dysregulation or inactivation could be associated with human diseases, either through direct effects on PERP or through alterations in TP63 in syndromes such as AEC. Here, we examine the hypothesis that PERP may be improperly regulated in AEC patients, thereby contributing to some of the myriad phenotypes characteristic of these patients. These studies will not only provide insight into the basis of AEC phenotypes but also into the genetics of TP63-associated diseases.
MATERIALS AND METHODS
Reporter Assays
25,000 TP63-/-;TP53-/- Mouse Embryo Fibroblasts (MEFs) were plated in 24-well plates. The following day, the MEFs were transiently transfected with 250 ng of empty vector (pcDNA3) or TA-TP63-alpha expression constructs, 250 ng of the pPERPLucPS reporter construct [Ihrie et al. 2005], and 25 ng of PRLnull (Promega) using Fugene 6 (Roche). pcDNA3-TA-TP63-alpha expression constructs encoding the TA-TP63-alpha-L514F, TA-TP63-alpha-G530V and TA-TP63-alpha-Q536L AEC mutants (originally identified as L518F, G534V, Q540L, respectively) and the TA-TP63-alpha-R279H and TA-TP63-alpha-R304W EEC mutants were the kind gift of Dr. Hans van Bokhoven [McGrath et al. 2001]. After 24 hr, cells were harvested and luciferase activity was measured using the Dual-Luciferase Reporter Assay system (Promega). Firefly luciferase values were divided by Renilla luciferase values to control for transfection efficiency. Assays were performed three times, with triplicate samples in each experiment.
Western Blotting
500,000 H1299 cells were cultured on 60 mm plates and then transiently transfected with 4 μg of each cDNA-TA-TP63-alpha expression construct using Fugene 6 (Roche). Protein extracts were prepared 24 hr later using 50 mM Tris (pH 8.0), 200 mM NaCl, 10% glycerol, and 1% Nonidet P-40. 40 μg of lysate were separated by SDS-PAGE, and western blotting was performed using standard methods. Antibodies directed against MYC (clone 9E10) and GAPDH (Research Diagnostics, Inc) were utilized at 1:1000 and 1:10,000 dilutions, respectively.
PERP Immunohistochemistry
Paraffin-embedded AEC patient samples were obtained from unaffected regions of AEC patient epidermis through Dr. Maranke Koster (University of Colorado, Denver) from Dr. Alanna Bree (Baylor College of Medicine), who is principal investigator for IRB protocol #20716. Immunohistochemistry was performed as described (Ihrie et al. 2005). Polyclonal antibodies against the first extracellular loop (Abcam) or against the carboxy-terminal tail of PERP [Ihrie et al. 2005] were used at 1:50 and 1:250, respectively.
RESULTS AND DISCUSSION
To gain insight into AEC pathogenesis, we sought to define the effects of AEC patient-derived TP63 mutants on PERP expression. We first addressed this question by performing luciferase reporter assays in which expression constructs encoding either wild-type TA-TP63-alpha or any of three AEC mutants (TA-TP63-alpha-L514F, TA-TP63-alpha-G530V and TA-TP63-alpha-Q536L) were transfected along with a PERP luciferase reporter construct bearing the critical TP63-responsive site in intron 1 into TP63-/-;TP53-/- fibroblasts. TP63 exists as multiple isoforms, the TA isoforms bearing the entire transactivation domain and the ΔN isoforms carrying a truncated transactivation domain. Both the TA-TP63 and ΔN-TP63 isoforms activate PERP, and here, we chose to examine the TA-TP63 isoform because it robustly activates PERP, therefore providing a large range over which to assess the effects of AEC mutants [Ihrie et al. 2005]. The mutants we examined were selected because they were amongst the first originally associated with AEC and because they represent the two major categories of AEC mutants suggested by homology modeling of the SAM domain through comparison to the TP73 (SAM domain solution structure [McGrath et al. 2001]. Certain residues, exemplified by L514, are buried in the core of the SAM domain and upon mutation are predicted to disrupt the overall stability of the domain. In contrast, residues G530 and Q536 are predicted to be on the surface of the SAM domain and therefore to affect specific protein-protein interactions rather than causing gross structural alterations. Interestingly, we observed differences in transactivation activity with the various mutants. While TA-TP63-alpha-L514F was completely defective in activating the PERP luciferase reporter, TA-TP63-alpha-G530V showed some transactivation potential and TA-TP63-alpha-Q536L exhibited near wild-type activity (Fig. 1A). Differences in the transactivation capacity are not due to differences in TP63 expression levels, as the TA-TP63-alpha-Q536L mutant is expressed at lower levels than the other mutants, yet is the most potent activator (Fig. 1B). The differences in activity of the various mutants are in contrast to the EEC-derived TA-TP63-alpha-R304W and TA-TP63-alpha-R279H mutants, which are known to be completely defective in DNA binding, and which were both deficient for PERP activation (Fig. 1A). Together, these data suggest that distinct mutations within the SAM domain differentially compromise the ability of TP63 to transactivate a particular TP63 target gene, in this case PERP, and provide the first evidence that the various TP63 mutants associated with a particular syndrome may manifest differences in transcriptional activation potential. We hypothesize that specific protein-protein interactions needed for full PERP transactivation by TP63 are retained in the TA-TP63-alpha-Q536L mutant, but are abolished when the structure of the SAM domain is compromised, as in the TA-TP63-alpha-L514F mutant. It is also likely that this set of AEC mutants will differ in their capacity to transactivate other TP63 target genes, a notion that can be investigated in future studies. The differential activity manifested by diverse AEC mutants in the activation of PERP and possibly other key TP63 targets provides a potential rationale for the clinical variability in the phenotypes observed amongst AEC patients.
Fig. 1.
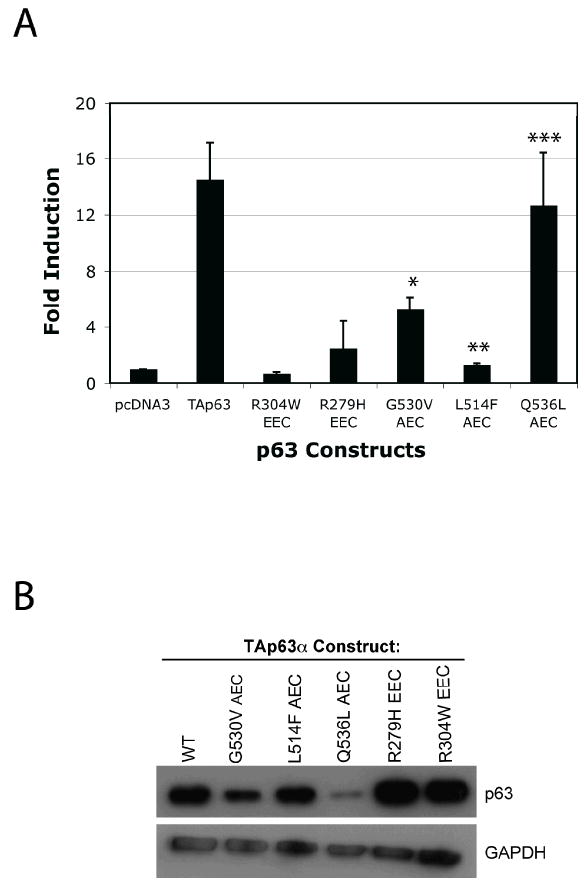
AEC-derived TP63 mutants display differential abilities to transactivate a PERP reporter construct. (A) The pPERPLucPS luciferase reporter construct was transfected into TP63-/-;TP53-/- MEFs along with constructs encoding either wild-type or any of several mutant versions of TA-TP63-alpha [McGrath et al. 2001]. The graph shows the fold induction of each TA-TP63-alpha protein relative to the empty pcDNA3 vector and represents the average of three experiments +/-SEM. P-values comparing the activity of TA-TP63-alpha mutants relative to wild-type TA-TP63-alpha are shown (*p<0.03, ** p<0.008, and ***p<0.8). (B) Western Blot analysis showing the expression levels of wild-type or mutant TA-TP63-alpha proteins transfected into H1299 cells. GAPDH serves as a loading control.
To determine whether compromised PERP expression might provide an explanation for some of the phenotypes commonly associated with AEC, we sought to investigate whether PERP levels are altered in epidermis derived from AEC patients. We obtained skin biopsies from non-lesional regions of 16 AEC patients exhibiting signs of skin erosions and performed PERP immunohistochemistry to examine any potential change in PERP protein expression. While skin from a control unaffected individual and many of the AEC patients displayed a normal pattern of PERP membrane staining throughout the cells of the epidermis (Fig. 2A-E), we identified two patient samples with anomalous staining profiles (Fig. 2G, H, J, K, Table I). Specifically, in these samples harboring either the F526L or the G561V mutation, we found that PERP membrane staining is dramatically diminished in the basal and suprabasal layers of the skin while remaining intact in the uppermost layers (Fig. 2G, H, J, K). This pattern is consistent with disrupted TP63 action, as TP63 is localized to the more basal layers of the epidermis in AEC patients, and suggests that some other factor regulates PERP in the upper layers of the epidermis. Thus, these data suggest that in a subset of AEC patients, proper PERP expression is perturbed, and that this could contribute to the symptoms of the disease. In addition, the levels of PERP may be diminished in some AEC patient samples, such as #12 (Fig. 2F), as seen with the TP63 G530V mutant in the reporter assay. This possibility can be further clarified in future through more quantitative analysis. Any observed perturbations in PERP expression in the epidermis would be expected to compromise desmosome function, increasing the likelihood of epithelial blistering. Although no overt blisters were observed in the biopsies we examined from these patients, there may still be desmosome abnormalities, which likely would not result in blisters until the tissue was subjected to mechanical stress. Indeed, acantholysis, a sign of defective cell-cell adhesion, has been observed in some cases of AEC [Payne et al. 2005], and loss of PERP may contribute to this phenotype.
Fig. 2.
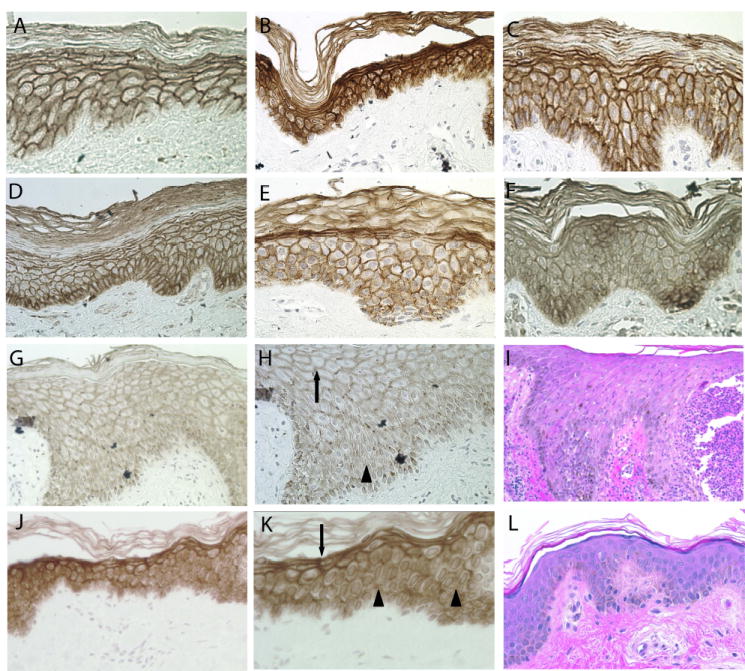
PERP expression is altered in epidermis of a subset of AEC patients. (A) PERP immunohistochemistry on epidermis from an unaffected individual shows robust PERP expression at the plasma membrane. (B, C, D, E) PERP shows clear membrane staining in the epidermis from a number of AEC patients (#18, 4, 21, and 17, respectively). (F) The levels of PERP membrane staining are potentially reduced in the epidermis of AEC patient #12. (G) PERP plasma membrane staining in the lower layers of the skin is lost in AEC patient sample #19. (H) Higher magnification of (G). Arrow indicates region of staining at cell-cell borders, and arrowhead indicates areas where cell-cell border staining is lost. (I) Hematoxylin and Eosin staining of sample #19, showing disorganization of the epidermal layer. (J) PERP plasma membrane staining in the lower layers of the epidermis is lost in AEC patient sample #13. (K) Higher magnification of (J). Arrow indicates regions of normal cell-cell border staining, and the arrowheads indicate loss of plasma membrane staining. (L) Hematoxylin and Eosin staining of sample #13 showing relatively normal epidermal architecture despite perturbances in PERP staining.
Table I. Summary of PERP Immunohistochemistry Staining on Human Tissue Samples.
| Sample | Nucleotide | Amino Acid | PERP Staining |
|---|---|---|---|
| 1 | 1630G>T | D544Y | Normal |
| 2 | 1793G>T | R598L | Normal |
| 3 | 1682G>A | G561D | Undetermined |
| 4 | 1682G>T | G561V | Normal |
| 5 | 1793G>T | R598L | Normal |
| 6 | 1682G>A | G561D | Normal |
| 7 | 1793G>T | R598L | Normal |
| 8 Unaffected individual |
Not tested | Not tested | Normal |
| 9 | 1802A>T | D601V | Normal |
| 10 | 1682G>A | G561D | Normal |
| 12 | 1793G>T | R598L | Potentially reduced levels |
| 13 | 1578C>A | F526L | Loss at membrane in basal and suprabasal layers |
| 14 | 1793G>T | R598L | Undetermined |
| 15 | 1631A>T | D544V | Normal |
| 16 | 1652C>T | P551L | Normal |
| 17 | 1578C>A | F526L | Normal |
| 18 | 1634T>C | L545P | Normal |
| 19 | 1682G>T | G561V | Loss at membrane in basal and suprabasal layers |
| 21 | 1610C>T | I537T | Normal |
Each patient sample is numbered, and specific nucleotide and amino acid changes are presented. The right-hand column summarizes the PERP staining pattern in each patient sample.
Analysis of individual TP63 target genes will likely have general utility for understanding the phenotypes exhibited in a specific TP63-associated syndrome as well as the variation in symptoms observed between different syndromes. For example, differential regulation of the DLX5/6 transcription factors, TP63 targets critical for limb development, might explain the severe limb phenotypes associated with EEC but not AEC, as the AEC-derived L514F TP63 mutant can transactivate DLX5/6 to near wild-type levels whereas EEC-derived TP63 mutants are completely deficient for this function [Lo Iacono et al. 2008]. In addition, as mentioned above, compromised expression of PERP may be associated with adhesion-related phenotypes in certain patients. PERP dysregulation in the context of certain AEC mutants may impair desmosome function, leading to an increased susceptibility to acantholysis and blistering in the epidermis as well as some of the other observed defects in ectodermal derivatives, as seen in patients bearing mutations in desmosomal proteins [Kottke et al. 2006; McGrath et al. 1997].
Interestingly, upon analysis of the TP63 mutations found in AEC patients with disrupted PERP staining, we discovered that different patients harboring the same TP63 mutation did not consistently show the same PERP expression pattern. (Fig. 2E and J, C and G, Table I, Data from Hans van Bokhoven, see article in this issue). This differential effect of a given TP63 mutation on PERP depending on the patient may be attributable to modifier genes influencing PERP expression. The idea of modifiers affecting phenotype expressivity has been documented, as the same mutation in TP63 has been associated with distinct syndromes [van Bokhoven and Brunner 2002].
These studies also provide insight into the mode of TP63 action in these diseases. The fact that PERP expression is maintained in the presence of some AEC-derived mutants but not others indicates that AEC is not caused by complete loss of function of TP63. In support of this notion, previous studies have shown that specific AEC mutants, including L530V, L514F, and Q536L, are compromised for transactivation of some but not all TP63 target genes [Ghioni et al. 2002; Huang et al. 2005; Lo Iacono et al. 2006]. Together, these observations suggest that AEC mutants are not total loss-of-function mutants completely defective for transactivation but rather are partial loss-of-function mutants retaining activity on at least some TP63 target genes. Loss of activity on select target genes would allow these mutants to serve as dominant negatives on those genes. It is also possible that there may be novel, gain-of-function effects of these mutants.
PERP is only one of many TP63 target genes whose expression may be altered in patients with AEC, and the continued identification of additional TP63 target genes affected in this disease is crucial to understanding its pathology. Elucidating how specific mutations in TP63 affect its ability to induce particular target genes may ultimately help predict the severity of phenotypes seen in different AEC patients and may lead to novel interventions to better manage the disease.
Acknowledgments
We would like to thank Hans van Bokhoven for the TP63 expression constructs and Rachel Dusek for critical reading of the manuscript. This work was supported by the NCI to VGB (1F1CA119944-01), the NIH to LDA (1113604-100-PABFR), and the National Foundation for Ectodermal Dysplasias (NFED) to LDA.
Abbreviations
- AEC
Ankyloblepharon Ectodermal Dysplasia and Cleft Lip/Palate Syndrome
- EEC
Ectrodactyly-Ectodermal Dysplasia and Cleft lip/Palate Syndrome
- ADULT
Acro-Dermato-Ungual-Lacrimal-Tooth Syndrome
- RHS
Rapp-Hodgkin’s Syndrome
- LMS
Limb-Mammary Syndrome
References
- Amiel J, Bougeard G, Francannet C, Raclin V, Munnich A, Lyonnet S, Frebourg T. TP63 gene mutation in ADULT syndrome. Eur J Hum Genet. 2001;9(8):642–5. doi: 10.1038/sj.ejhg.5200676. [DOI] [PubMed] [Google Scholar]
- Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, Jacks T. PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev. 2000;14(6):704–18. [PMC free article] [PubMed] [Google Scholar]
- Carroll DK, Brugge JS, Attardi LD. p63, cell adhesion and survival. Cell Cycle. 2007;6(3):255–61. doi: 10.4161/cc.6.3.3799. [DOI] [PubMed] [Google Scholar]
- Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, Newbury-Ecob R, Hennekam RC, Van Buggenhout G, van Haeringen A, Woods CG, van Essen AJ, de Waal R, Vriend G, Haber DA, Yang A, McKeon F, Brunner HG, van Bokhoven H. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999;99(2):143–53. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- Ghioni P, Bolognese F, Duijf PH, Van Bokhoven H, Mantovani R, Guerrini L. Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Mol Cell Biol. 2002;22(24):8659–68. doi: 10.1128/MCB.22.24.8659-8668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YP, Kim Y, Li Z, Fomenkov T, Fomenkov A, Ratovitski EA. AEC-associated p63 mutations lead to alternative splicing/protein stabilization of TP63 and modulation of Notch signaling. Cell Cycle. 2005;4(10):1440–7. doi: 10.4161/cc.4.10.2086. [DOI] [PubMed] [Google Scholar]
- Ihrie RA, Bronson RT, Attardi LD. Adult mice lacking the p53/p63 target gene PERP are not predisposed to spontaneous tumorigenesis but display features of ectodermal dysplasia syndromes. Cell Death Differ. 2006 doi: 10.1038/sj.cdd.4401871. [DOI] [PubMed] [Google Scholar]
- Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, Bronson RT, Mills AA, Attardi LD. PERP is a p63-regulated gene essential for epithelial integrity. Cell. 2005;120(6):843–56. doi: 10.1016/j.cell.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Kantaputra PN, Hamada T, Kumchai T, McGrath JA. Heterozygous mutation in the SAM domain of p63 underlies Rapp-Hodgkin ectodermal dysplasia. J Dent Res. 2003;82(6):433–7. doi: 10.1177/154405910308200606. [DOI] [PubMed] [Google Scholar]
- Kottke MD, Delva E, Kowalczyk AP. The desmosome: cell science lessons from human diseases. J Cell Sci. 2006;119(Pt 5):797–806. doi: 10.1242/jcs.02888. [DOI] [PubMed] [Google Scholar]
- Lo Iacono M, Di Costanzo A, Calogero RA, Mansueto G, Saviozzi S, Crispi S, Pollice A, La Mantia G, Calabro V. The Hay Wells syndrome-derived TAp63alphaQ540L mutant has impaired transcriptional and cell growth regulatory activity. Cell Cycle. 2006;5(1):78–87. doi: 10.4161/cc.5.1.2268. [DOI] [PubMed] [Google Scholar]
- Lo Iacono N, Mantero S, Chiarelli A, Garcia E, Mills AA, Morasso MI, Costanzo A, Levi G, Guerrini L, Merlo GR. Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development. 2008;135(7):1377–88. doi: 10.1242/dev.011759. [DOI] [PubMed] [Google Scholar]
- McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, Wessagowit V, Kelly A, Atherton DJ, Griffiths WA, Orlow SJ, van Haeringen A, Ausems MG, Yang A, McKeon F, Bamshad MA, Brunner HG, Hamel BC, van Bokhoven H. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001;10(3):221–9. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- McGrath JA, McMillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, Garrod DR, Eady RA. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet. 1997;17(2):240–4. doi: 10.1038/ng1097-240. [DOI] [PubMed] [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398(6729):708–13. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- Payne AS, Yan AC, Ilyas E, Li W, Seykora JT, Young TL, Pawel BR, Honig PJ, Camacho J, Imaizumi S, Heymann WR, Schnur RE. Two novel TP63 mutations associated with the ankyloblepharon, ectodermal defects, and cleft lip and palate syndrome: a skin fragility phenotype. Arch Dermatol. 2005;141(12):1567–73. doi: 10.1001/archderm.141.12.1567. [DOI] [PubMed] [Google Scholar]
- Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20(22):3185–97. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, Brunner HG. Splitting p63. Am J Hum Genet. 2002;71(1):1–13. doi: 10.1086/341450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, Jung M, Smits AP, van Beersum S, Ruschendorf F, van Steensel M, Veenstra M, Tuerlings JH, Mariman EC, Brunner HG, Wienker TF, Reis A, Ropers HH, Hamel BC. Limb mammary syndrome: a new genetic disorder with mammary hypoplasia, ectrodactyly, and other Hand/Foot anomalies maps to human chromosome 3q27. Am J Hum Genet. 1999;64(2):538–46. doi: 10.1086/302246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, McKeon F. Mutations in the p53 homolog p63: allele-specific developmental syndromes in humans. Trends Mol Med. 2002;8(3):133–9. doi: 10.1016/s1471-4914(01)02260-2. [DOI] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–8. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, Struhl K. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell. 2006;24(4):593–602. doi: 10.1016/j.molcel.2006.10.018. [DOI] [PubMed] [Google Scholar]