

Abstract
In the past few years, therapies targeted at the von Hippel-Lindau (VHL) and hypoxia-inducible factor (HIF) pathways, such as sunitinib and sorafenib, have been developed to treat clear cell renal cell carcinoma (ccRCC). However, the majority of patients will eventually show resistance to antiangiogenesis therapies. The purpose of our study was to identify novel pathways that could be potentially used as targets for new therapies. Whole transcriptome sequencing (RNA-Seq) was conducted on eight matched tumor and adjacent normal tissue samples. A novel RUNX1-RUNX1T1 pathway was identified which was upregulated in ccRCC through gene set enrichment analysis (GSEA). We also confirmed the findings based on previously published gene expression microarray data. Our data shows that upregulated of the RUNX1-RUNX1T1 gene set maybe an important factor contributing to the etiology of ccRCC.
1. Introduction
Renal cell carcinoma (RCC) is one of the most common malignancies with the highest mortality rate among genitourinary cancers. Approximately 65,000 people were diagnosed and 14,000 deaths were attributed to cancers of the kidney and renal pelvis in 2010 in the United States [1]. While kidney cancer can be divided into several histological subtypes, the majority of the cases (about 75%) are clear cell renal cell carcinoma (ccRCC) [2]. Surgery offers the best opportunity to cure localized ccRCC. In the past few years, therapies targeted at VHL/HIF pathways, such as sunitinib and sorafenib, have been developed to treat ccRCC. However, most patients who either experience recurrence after surgery or have metastatic disease at the time of diagnosis will ultimately succumb to the disease. Thus, there remains a great need for novel therapies that depend on the identification of novel pathways in individuals with ccRCC.
Gene expression profiling, based on microarray hybridization, has been successfully used for the identification of genes that are differentially expressed among RCC subtypes and in the search for new therapeutic targets [3–6]. This method has also been correlated with chromosomal abnormalities and deregulated oncogenic pathways. However, the complimentary deoxyribonucleic acid cDNA microarray technique suffers from its inherent high background signals and depends on predesigned probes against known target transcripts, which makes it unable to detect novel transcript regions and it can only cover a portion of annotated transcriptome. The global detection of whole transcriptome is now possible with the recent development of next generation high-throughput RNA sequencing techniques (RNA-Seq). RNA-Seq has high technical and biological reproducibility. In addition, researchers have found RNA-Seq to be a powerful tool for the detection of differentially expressed genes, rare transcripts, novel isoforms, and mutations in tissues [7–10].
In this study, we performed whole transcriptome sequencing on eight pairs of ccRCC tumor and adjacent normal tissues in a Chinese population. Our goal was to identify novel gene pathways that have altered expression by comparing the expression patterns between the tumor and adjacent normal samples.
2. Materials and Methods
2.1. Patients and Samples
A total of 16 patients were treated with radical nephrectomy (RN) for RCC at Huashan Hospital of Fudan University. The 11 men and 5 women had a median age of 55 years (range of 44 to 75 years). Histological characterization for tumor type, such as ccRCC, was determined according to the Heildelberg classification, and staging was based on the American Joint Committee on Cancer (AJCC) TNM 2009 system. Twelve patients in the study group had pT1N0M0 tumors, two had T2N0M0 tumors, and the remaining two had T3N0M0 tumors. Clear cell renal cancer tumor and adjacent normal tissues were obtained from all 16 patients and a total of 16 pairs of tumor and adjacent normal tissues were available, from which 8 pairs of specimens were randomly selected for RNA sequencing to perform the gene profiling. Tumor tissues were selected from sites with high density of cancer without necrosis and normal tissues were sampled where no cancer contamination was found. All 16 pairs of samples were used to validate the genes differentially expressed between tumor and normal samples by quantitative real-time reverse transcription polymerase chain reaction. Specimens were frozen in liquid nitrogen immediately after operation and stored at −80°C. Detailed information of the study population was described in Table 1 and Supplementary Table 1 (see Supplementary Material available online at http://dx.doi.org/10.1155/2014/450621). The study was approved by the Institutional Review Board at Huashan Hospital of Fudan University, and all patients signed an informed consent form for inclusion of their samples.
Table 1.
Summary of 8 ccRCC patients for RNA sequencing.
| Patients ID | Age | Sex | TNM | Tumor grade | Sample type | Sample ID |
|---|---|---|---|---|---|---|
| 1 | 47 | Male | T1bN0M0 | 2 | Normal | HS1N |
| Tumor | HS1C | |||||
|
| ||||||
| 2 | 58 | Female | T1aN0M0 | 3 | Normal | HS2N |
| Tumor | HS2C | |||||
|
| ||||||
| 3 | 64 | Male | T1bN0M0 | 2 | Normal | HS3N |
| Tumor | HS3C | |||||
|
| ||||||
| 4 | 44 | Female | T1bN0M0 | 3 | Normal | HS4N |
| Tumor | HS4C | |||||
|
| ||||||
| 5 | 62 | Male | T1aN0M0 | 1 | Normal | HS5N |
| Tumor | HS5C | |||||
|
| ||||||
| 6 | 61 | Female | T1bN0M0 | 1 | Normal | HS6N |
| Tumor | HS6C | |||||
|
| ||||||
| 7 | 61 | Male | T1aN0M0 | 1 | Normal | HS7N |
| Tumor | HS7C | |||||
|
| ||||||
| 8 | 48 | Male | T1bN0M0 | 2 | Normal | HS8N |
| Tumor | HS8C | |||||
2.2. cDNA Library Construction and Sequencing
Total RNA was isolated from frozen tumor and matched normal tissues using the reagent Trizol (Invitrogen). The sequencing library was constructed according to Illumina's TruSeq RNA Sample Preparation Protocol. Poly-A containing mRNA was purified from total RNA using magnetic beads with oligo-dT, followed by fragmentation. First-strand cDNA was synthesized using random hexamers and reverse transcriptase. Second-strand cDNA was synthesized with high quality deoxyribonucleotide triphosphates (dNTPs), ribonuclease H (RNase H), and DNA polymerase. Then the new double-strand cDNA was end-repaired and a single nucleotide “A” was added. Different in-house designed 6 bp adapters were ligated to the corresponding samples. DNA fragments with selected size and adapters were purified and amplified by PCR. After normalization, the DNA sample libraries were pooled into 4 libraries, and the pooled libraries were sequenced on an Illumina HiSeq 2000 sequencing machine.
2.3. Reads Mapping
Reads were processed and aligned to the University of California Santa Cruz (UCSC) H. sapiens reference genome and transcriptome (build hg19) using the RNA-Seq unified mapper (RUM) v1.0.9 [11]. RUM is an alignment program that maps reads in three phases. All reads were respectively aligned to the reference genome and transcriptome using Bowtie (v.12.7) [12]. The unmapped reads were then mapped to genome sequence with BLAT tool. Data collected from all the three mappings was then combined into a single mapping. This leverages the advantages of both genome and transcriptome mappings as well as combining the speed of Bowtie with the sensitivity and flexibility of Blat. The default parameters for RUM were used and more than 3 mismatches were allowed in the alignment. Finally, we used the Samtools software package (v0.1.17) to change the output of Bowtie to sorted bam files, which were used for further analysis [13].
2.4. Differential Expressed Gene (DEG) Analysis
The annotation for the UCSC known genes dataset was used for DEG data analysis for known genes. The exon union method was used to estimate the counts for each gene [14]. The genes differentially expressed between normal and tumor samples were then identified using negative binomial test as stated in the DESeq package [15]. Briefly, to compare differentially expressed genes between tumor and normal RCC samples, read counts of each of the identified genes were normalized to the total number of reads. The statistical significance (P value) was inferred based on the Bayesian method, a method specifically developed for the analysis of digital gene expression profiles and could account for the sampling variability of tags with low counts. A specific gene was deemed to be significantly differentially expressed if the P value given by this method was ≤0.05.
2.5. Pathway Analysis for RNA-Seq Data
The products of the negative log transformed P values, based on DEseq analysis plus the sign of the log2-fold change of each gene, were used as input to perform gene set enrichment analysis (GSEA) as implemented in the limma package. Specifically, the permutation test was used. The gene sets based on published papers were either generated in our lab or downloaded from the Molecular Signature Database (MsigDB, http://www.broadinstitute.org/gsea/msigdb/). These gene sets were curated from multiple sources including online pathway databases, biochemical literature, and mammalian microarray studies. Our main analysis was performed based on a class 2 database, which contains about 3,600 gene sets, which were curated from experimental data. The P values for each gene set were used to rank the functional representation of the significant genes in each gene set by their significance to the list of targets, thereby identifying biological processes likely to be affected.
2.6. Pathway Analysis for Microarray Data: PGSEA Analysis
Microarray databases (i.e., GSE17895 and GSE11024) were downloaded as described in the Section 3 and parameter gene set enrichment analysis (PGSEA) was used to generate enrichment scores for each pathway within each tumor sample using corresponding nondiseased kidney tissue as a reference [16]. A moderated t-statistic as implemented in the limma package was used to identify gene set enrichment scores that could discriminate between subtypes [5, 17].
2.7. Quantitative RT-PCR
Total RNA was isolated from tumor or adjacent normal tissues, followed by reverse transcription to cDNA using universal primers and a TaqMan Gene Expression Cells-to-CT Kit (Applied Biosystems). The qRT-PCR reaction was performed as previously described [18]. The primer sequences used for qRT-PCR were available upon request. β-Actin gene was used as internal quantitative control, and each assay was done in triplicate.
3. Results
3.1. RNA Sequencing
RNA sequencing was performed on eight matched pairs of ccRCC tumor and adjacent normal tissues. We generated an average of 14,306,899 (range: 3,186,698–27,215,607) single end reads with length of 100 bp, including 6 bp barcode sequence. The median total raw reads for the normal and tumor samples were approximately 11.8 million and 16.3 million separately, and the median alignment rates for the normal and tumor samples were 93.01% and 92.48% separately (Supplementary Table 2 and Supplementary Table 3).
We then quantified gene expression values in reads per kilobase of exon model per million mapped reads (RPKM) and observed that 56.30% of normal tissues and 55.48% of tumor tissues were less than 0.25 RPKM. In addition, approximately 1.11% and 1.12% of the genes separately for normal and tumor tissues were more than 5 RPKM (Supplementary Table 4). This suggests that there were only a very small number of genes expressed in relative high copies in those samples.
3.2. RNA-Seq Reveals Known DEG Changes in ccRCC
Using a cutoff of 0.05 for false discovery rate (FDR), we found that 3,514 (17.8%) out of 19,776 known genes were differentially expressed in tumor samples, including 2,054 (10.4%) upregulated genes and 1,460 (7.4%) downregulated genes. The top upregulated and downregulated genes are summarized in Table 2. Known HIF transcription targets, such as EGLN3 [19, 20], CA9 [21, 22], and VEGFA [23, 24], were in the top upregulated lists (Table 2). In addition, a set of known kidney differential markers such as KCNJ1 [25] and SLC22A8 were found in the top downregulated genes (Table 2). The top DEG list indicates that our whole transcriptome analysis recapitulates the known gene expression changes in ccRCC tumors.
Table 2.
Summary of selected genes of interest differentially expressed in ccRCC samples.
| Symbol | Entr. ID | log2-fold change | P value | Notes |
|---|---|---|---|---|
| SLC6A3 | 6531 | 8.41 | 1.59E − 139 | |
| CA9 | 768 | 7.78 | 6.24E − 82 | Hypoxia |
| FGG | 2266 | 7.42 | 8.98E − 62 | |
| NDUFA4L2 | 56901 | 6.95 | 7.34E − 122 | |
| EGLN3 | 112399 | 5.89 | 7.19E − 97 | Hypoxia |
| LOC100131551 | 100131551 | 4.38 | 1.18E − 42 | |
| C3 | 718 | 5.31 | 1.81E − 89 | |
| CYP2J2 | 1573 | 5.53 | 2.41E − 88 | |
| ANGPTL4 | 51129 | 5.63 | 3.39E − 87 | Hypoxia |
| FCGR3A | 2214 | 4.72 | 2.88E − 57 | |
| NETO2 | 81831 | 4.43 | 5.52E − 49 | RUNX1 |
| UBD | 10537 | 4.33 | 1.21E − 59 | |
| CP | 1356 | 4.47 | 1.80E − 67 | |
| PLIN2 | 123 | 4.12 | 3.01E − 53 | Myc |
| PVT1 | 5820 | 3.79 | 6.24E − 56 | Activator |
| ENO2 | 2026 | 3.19 | 2.19E − 21 | Hypoxia |
| VEGF-A | 7422 | 3.15 | 3.48E − 30 | Hypoxia |
| SLC1A3 | 6507 | 3.04 | 8.64E − 24 | RUNX1 |
| PLK2 | 10769 | 2.82 | 2.57E − 22 | RUNX1 |
| GBP2 | 2634 | 2.62 | 1.37E − 24 | RUNX1 |
| VCAN | 1462 | 2.44 | 4.65E − 24 | RUNX1 |
| COL1A1 | 1277 | 2.31 | 1.03E − 22 | SWI/SNF |
| SEL1L3 | 23231 | 1.84 | 5.38E − 13 | SWI/SNF |
| BCAT1 | 586 | 1.83 | 7.89E − 08 | SWI/SNF |
| SIM2 | 6493 | −5.70 | 2.02E − 53 | TF |
| SLC13A3 | 64849 | −5.97 | 4.53E − 52 | |
| SERPINA5 | 5104 | −5.81 | 2.08E − 42 | |
| SFRP1 | 6422 | −5.91 | 5.30E − 48 | Wnt |
| KCNJ1 | 3758 | −7.26 | 4.84E − 70 | Kidney |
| SCNN1G | 6340 | −8.95 | 7.28E − 64 | |
| TFAP2B | 7021 | −7.64 | 5.21E − 44 | TF |
| KNG1 | 3827 | −8.97 | 4.07E − 91 | |
| SLC12A1 | 6557 | −8.16 | 1.85E − 90 | Kidney |
| HSFY2 | 159119 | −7.41 | 4.54E − 42 | TF |
| SLC22A8 | 9376 | −8.10 | 1.94E − 64 | Kidney |
| GP2 | 2813 | −11.51 | 1.74E − 57 | |
| AQP2 | 359 | −11.64 | 9.68E − 118 | Kidney |
| UMOD | 7369 | −12.12 | 3.25E − 127 | Kidney |
3.3. RNA-Seq Reveals Novel RNX1/RNX1T1 Pathway/Signature Upregulated in ccRCC
A total of 206 pathway/signatures were upregulated in tumor samples and 23 pathway/signatures were downregulated in tumor samples compared with those in normal samples (Supplementary Table 5), using a P value cutoff of 0.001. The 35 upregulated and 2 downregulated pathways with most biological implications were selected and the relative deregulation of each gene sets for individual samples based on the enrichment P values were shown in Supplementary Figure 1.
Again, the known VHL/HIF pathway related gene sets were upregulated in tumor samples (Supplementary Figure 1). The gene sets related to kidney function were found downregulated in tumor samples, which indicates the loss of normal kidney function in these tumor samples. Both of these results showed the validity of our analytic approach.
The RUNX1-RUNX1T1 pathway was a novel gene set implicated in the pathway analysis. We then performed in silico confirmation study using a cohort with microarray data. The cohort included 90 ccRCC tumor tissue samples and 13 normal adjacent tissue samples [6]. Using PGSEA analysis, we found that RUNX1-RUNX1T1 was also upregulated in most of these 90 ccRCC cases and the upregulation of RUNX1-RUNX1T1 is not related to tumor grades (Figure 1(a)).
Figure 1.
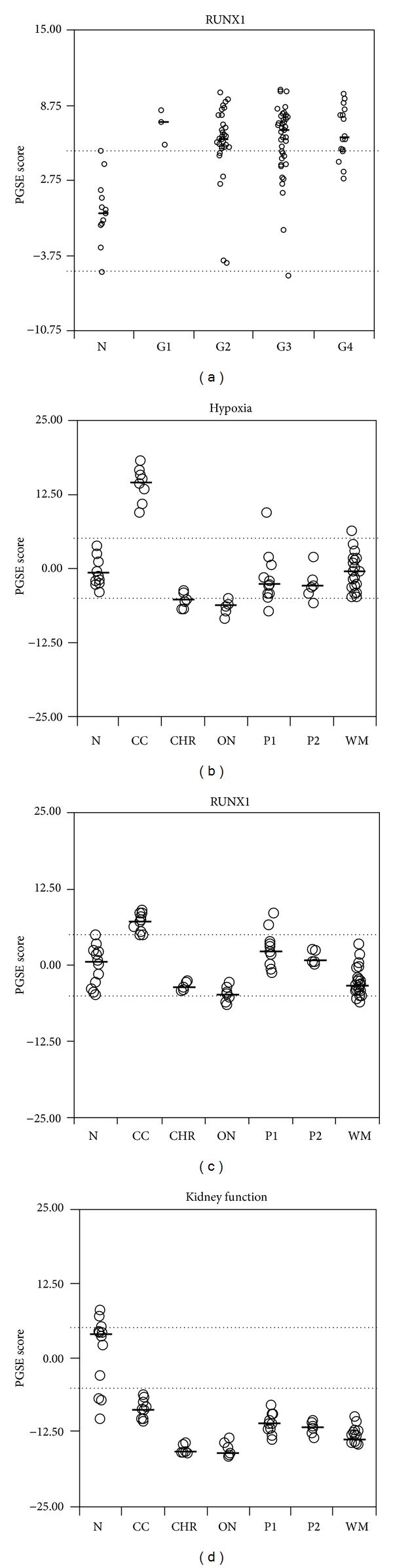
RUNX1-RUNX1T1 signature was upregulated in ccRCC. (a) PGSEA score from microarray data comprising 90 tumor and 13 normal tissues shows RUNX1-RUNX1T1 signature is up-regulated in ccRCC and shows no correlation to tumor grade. (b) Hypoxia related signature was specifically upregulated in ccRCC, and no significant upregulation was found in other kinds of RCC; (c) RUNX1-RUNXT1 related signature was specifically upregulated in ccRCC, and no significant upregulation was found in other kinds of RCC; (d) kidney function related signature was downregulated among all subtypes of RCC. N, normal samples; G1, tumor grade1; G2, tumor grade1; G3, tumor grade4; CC: clear cell; CHR: chromophobe; ON: oncocytoma; P: papillary; WM: Wilms' tumor.
A total of 66 genes of the RUNX1-RUNX1T1 pathway were differentially expressed. We plotted the relative gene expression levels of 66 genes which were differentially expressed between tumor and normal tissues in the eight paired ccRCC samples based on RNAseq (Supplementary Figure 2(a)). The top 5 upregulated genes are NETO2, GBP2, VCAN, SLC1A3, and PLK2. The expression levels of these 66 genes from 90 ccRCC microarray data showed the same pattern of expression of these genes in tumor tissues (Supplementary Figure 2(b)). Three genes, including NETO2, GBP2, and VCAN, showed consistent high expression across these 90 tumor samples.
To determine whether the RUNX1-RUNX1T1 gene sets were specifically upregulated in the clear cell subtype, we performed another in silico study based on a second cohort of microarray data which included the most common subtypes of kidney cancers, including 27 Wilms' tumor, 10 ccRCC, 6 chromophobe, 7 oncocytoma, 17 papillary renal cell carcinoma, and 12 normal kidney samples [26]. The upregulation of VHL/HIF and RUNX1-RUNX1T1 gene sets was only observed in the clear cell subtype (Figures 1(b) and 1(c)). As a control measure, kidney function gene sets were lost in all of the subtypes of kidney cancers (Figure 1(d)). These results suggest that RUNX1-RUNX1T1 gene sets were specifically upregulated in ccRCC.
3.4. Validation of RUNX1-RUNX1T1 Genes in ccRCC by qRT-PCR
To confirm the upregulation of RUNX1-RUNXT1 genes in ccRCC, we examined the expression level of three upregulated genes of NETO2, VCAN, and GBP2 in 16 pairs of ccRCC tumors and adjacent normal tissues using quantitative real-time PCR (qRT-PCR), including 8 pairs used for RNA sequencing and an additional 8 pairs of ccRCC tumor and normal tissues. The results of qRT-PCR were highly consistent with the RNA sequencing results (Figure 2). Overexpression of NETO2, VCAN, and GBP2 in tumor tissues compared to normal tissues were observed in 93.75% (15/16), 93.75 (15/16), and 87.5% (14/16), respectively.
Figure 2.
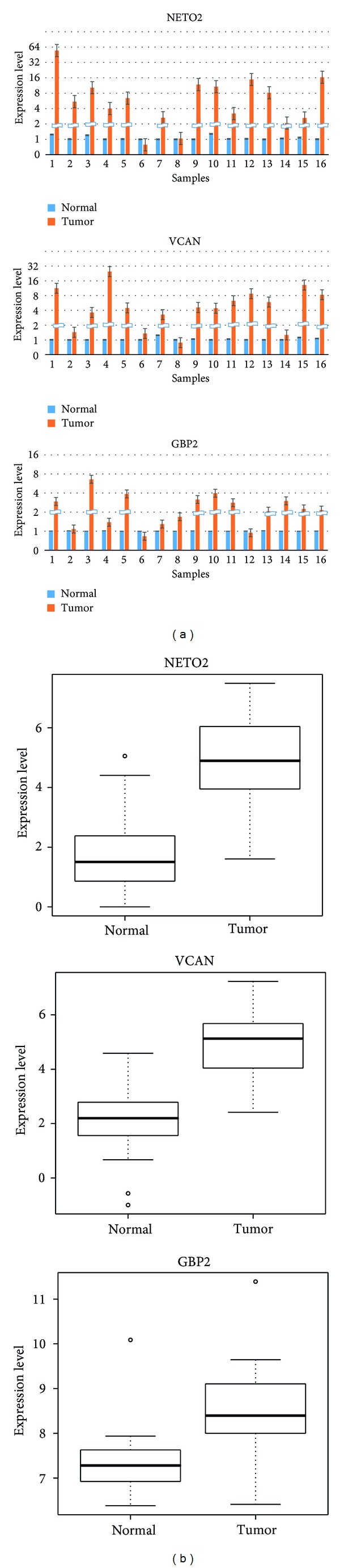
Validated upregulation of three RUNX1-RUNX1T1 genes in ccRCC by qRT-PCR. (a) Expression level of NETO2, VCAN, and GBP2 in 16 pairs of ccRCC tumor and normal tissues, including 8 pairs used for RNA-Seq and additional 8 pairs. β-Actin gene was used as reference gene. (b) Comparison of the expression level of NETO2, VCAN, and GPB2 between ccRCC tumor and adjacent normal tissues by qRT-PCR.
4. Discussion
In the current study, we have applied next generation sequencing technology along with RNA-Seq data in eight matched tumor plus normal tissues to identify novel pathways for ccRCC. We have successfully identified a novel gene set for the fusion transcription factor, RUNX1-RUNX1T1, which is upregulated in ccRCC. We have also replicated the findings based on previously published gene expression microarray data.
Our study has been the first to establish a role of RUNX1-RUNX1T1 gene set in the carcinogenesis of ccRCC. A previous functional study showed that this fusion transcription factor disrupted the natural function of transcription factor RUNX1 and induced altered expression of genes such as AP-1 [27]. RUNX1-RUNX1T1, also called AML1/ETO, is considered a leukemia-specific chimeric fusion transcription factor. It is one of the most common chromosomal translocations in acute myeloid leukemia (AML) subtype 2 and is found in about 12% of AML patients [28]. However, its association with ccRCC has yet been reported and, through our work and the database online, this fusion has not been found in ccRCC.
Although the direct clinical association between ccRCC and AML remains unclear, there are still some similarities in the response to chemotherapy in both diseases. Since the 1980s, ccRCC has been established as a prototype of a chemotherapy-resistant tumor [29]. Fojo et al. (1987) reported that the expression of the multiple drug resistant- (MDR-)associated glycoprotein on the surface of RCC cells might contribute to this feature [30]. It would be interesting if future studies could reveal an inherent relation between these two diseases.
As aforementioned, we have revealed the upregulation of NETO2, GBP2, and VCAN in ccRCCs (Supp. Figure 2). NETO2 encodes a predicted transmembrane protein containing two extracellular CUB domains followed by a low-density lipoprotein class A (LDLa) domain. Expression of this gene may be a biomarker for proliferating infantile hemangiomas. Thus far, its expression in kidney tissue remains unidentified, which makes it more elusive in the functional prediction. Likewise, expression of GBP2 in RCC has not been reported either. GBP2 belongs to the family of GTP-binding proteins with limited known function. Recently, it has been reported that GBP2 may be mediated by p53 and become a disease marker in esophageal cancer [31]. VCAN, however, has been previously reported to be upregulated in RCC. Moreover, VCAN is associated with proliferation, survival, apoptosis, and migration in a variety of malignancies. Investigation of these rarely studied genes and their interrelations in RCC appears attractive.
Recently, the Cancer Genome Atlas (TGCA) has released a comprehensive molecular profile of ccRCC with astronomical matrix of data [32]. Using the online analytic software developed by Memorial Sloan Kettering Cancer Center (http://www.cbioportal.org/public-portal/), we have externally validated our top 3 hits in the TCGA database, in which the expressions of NETO2, GBP2, and VCAN studied by RNA-seq were consistently upregulated, respectively, in ccRCC patients. Moreover, NETO2 and GBP were associated with worsened prognosis in the TCGA cohort, further signifying our findings as potential disease markers or targets in ccRCC.
Our study has limitations. First, selected candidate genes should have been validated by quantitative PCR (Q-PCR) and, if better, by western blots. In lieu of Q-PCR, we have incorporated 2 sets of independent array data and have validated our results in expanded cases in silico (see Section 3). These validations further support the extrapolation of our findings. Nonetheless, future PCR validation and subsequent functional analysis are warranted. Second, due to our limited sample size, the interpretation of our results could be skewed. We are at present collecting more tissue samples for further investigation.
In summary, our novel findings of upregulation of genes within RUNX1-RUNX1T1 signature in ccRCC indicate that this gene set is critical for the tumorigenesis of ccRCC. Additional functional studies are required to delineate their functions in ccRCC.
Supplementary Material
All the patients listed in the table were treated with radical nephrectomy (RN) for RCC at Huashan Hospital of Fudan University. The tissues experimented in RNA-seq test were from the first 8 cases. And we used the tissues for all 16 cases in qRT-PCR examination.
Acknowledgments
The authors would like to thank all participants in this study. They would also like to thank Dr. Kyle Furge, Karl Dykema, and Yongpan Yan from Van Andel Institute for their advice on the data analysis.
Disclosure
Zuquan Xiong, Hongjie Yu, Yan Ding, and Chenchen Feng contributed equally to this work.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
All authors participated in the design, interpretation of the studies, analysis of the data, and review of the paper; Siqun Lilly Zheng, Zuquan Xiong, and Hanming Wei conducted the experiments, Zujun Fang, Jianfeng Xu supplied critical reagents and funds, Zuquan Xiong, Hongjie Yu, Yan Ding, Jielin Sun, Chenchen Feng, and Jianfeng Xu wrote the paper, and Sha Tao and Dan Huang contributed to bioinformatics analysis.
Funding
The authors thank all the study subjects who participated in this study. The study is supported by a fund from National Science Foundation of China to Zujun Fang and an intramural fund from Fudan University to Jianfeng Xu.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer Journal for Clinicians. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Cohen HT, McGovern FJ. Renal-cell carcinoma. The New England Journal of Medicine. 2005;353(23):2477–2490. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi M, Yang XJ, Sugimura J, et al. Molecular subclassification of kidney tumors and the discovery of new diagnostic markers. Oncogene. 2003;22:6810–6818. doi: 10.1038/sj.onc.1206869. [DOI] [PubMed] [Google Scholar]
- 4.Furge KA, Lucas KA, Takahashi M, et al. Robust classification of renal cell carcinoma based on gene expression data and predicted cytogenetic profiles. Cancer Research. 2004;64(12):4117–4121. doi: 10.1158/0008-5472.CAN-04-0534. [DOI] [PubMed] [Google Scholar]
- 5.Furge KA, Chen J, Koeman J, et al. Detection of DNA copy number changes and oncogenic signaling abnormalities from gene expression data reveals MYC activation in high-grade papillary renal cell carcinoma. Cancer Research. 2007;67(7):3171–3176. doi: 10.1158/0008-5472.CAN-06-4571. [DOI] [PubMed] [Google Scholar]
- 6.Ding Y, Huang D, Zhang Z, et al. Combined gene expression profiling and RNAi screening in clear cell renal cell carcinoma identify PLK1 and other therapeutic kinase targets. Cancer Research. 2011;71:5225–5234. doi: 10.1158/0008-5472.CAN-11-0076. [DOI] [PubMed] [Google Scholar]
- 7.Sultan M, Schulz MH, Richard H, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 8.Chepelev I, Wei G, Tang Q, Zhao K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic Acids Research. 2009;37(16) doi: 10.1093/nar/gkp507.e106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 10.Greif PA, Eck SH, Konstandin NP, et al. Identification of recurring tumor-specific somatic mutations in acute myeloid leukemia by transcriptome sequencing. Leukemia. 2011;25(5):821–827. doi: 10.1038/leu.2011.19. [DOI] [PubMed] [Google Scholar]
- 11.Grant GR, Farkas MH, Pizarro AD, et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27(18):2518–2528. doi: 10.1093/bioinformatics/btr427.btr427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10(3, article R25) doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nature Methods. 2011;8(6):469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 15.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010;11(10, article R106) doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furge KA, Dykema KJ, Ho C, Chen X. Comparison of array-based comparative genomic hybridization with gene expression-based regional expression biases to identify genetic abnormalities in hepatocellular carcinoma. BMC Genomics. 2005;6, article 67 doi: 10.1186/1471-2164-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klomp JA, Petillo D, Niemi NM, et al. Birt-Hogg-Dube renal tumors are genetically distinct from other renal neoplasias and are associated with up-regulation of mitochondrial gene expression. BMC Med Genomics. 3, article 59 doi: 10.1186/1755-8794-3-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nature Cell Biology. 2005;7(6):591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Jimenez E, Gómez-López G, Leandro-García LJ, et al. Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Molecular Endocrinology. 2010;24(12):2382–2391. doi: 10.1210/me.2010-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aprelikova O, Chandramouli GVR, Wood M, et al. Regulation of HIF prolyl hydroxylases by hypoxia-inducible factors. Journal of Cellular Biochemistry. 2004;92(3):491–501. doi: 10.1002/jcb.20067. [DOI] [PubMed] [Google Scholar]
- 21.Grabmaier K, Vissers JL, de Weijert MC, et al. Molecular cloning and immunogenicity of renal cell carcinoma-associated antigen G250. International Journal of Cancer. 2000;85:865–870. doi: 10.1002/(sici)1097-0215(20000315)85:6<865::aid-ijc21>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 22.Oosterwijk E, Ruiter DJ, Hoedemaeker J. Monoclonal antibody G250 recognizes a determinant present in renal-cell carcinoma and absent from normal kidney. International Journal of Cancer. 1986;38(4):489–494. doi: 10.1002/ijc.2910380406. [DOI] [PubMed] [Google Scholar]
- 23.Siemeister G, Weindel K, Mohrs K, Barleon B, Martiny-Baron G, Marmé D. Reversion of deregulated expression of vascular endothelial growth factor in human renal carcinoma cells by von Hippel-Lindau tumor suppressor protein. Cancer Research. 1996;56(10):2299–2301. [PubMed] [Google Scholar]
- 24.Iliopoulos O, Levy AP, Jiang C, Kaelin WG, Jr., Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):10595–10599. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giebisch G. Renal potassium transport: Mechanisms and regulation. The American Journal of Physiology—Renal Physiology. 1998;274(5):F817–F833. doi: 10.1152/ajprenal.1998.274.5.F817. [DOI] [PubMed] [Google Scholar]
- 26.Kort EJ, Farber L, Tretiakova M, et al. The E2F3-oncomir-1 axis is activated in Wilms’ tumor. Cancer Research. 2008;68(11):4034–4038. doi: 10.1158/0008-5472.CAN-08-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tonks A, Pearn L, Musson M, et al. Transcriptional dysregulation mediated by RUNX1-RUNX1T1 in normal human progenitor cells and in acute myeloid leukaemia. Leukemia. 2007;21(12):2495–2505. doi: 10.1038/sj.leu.2404961. [DOI] [PubMed] [Google Scholar]
- 28.Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004;23(24):4255–4262. doi: 10.1038/sj.onc.1207727. [DOI] [PubMed] [Google Scholar]
- 29.Yagoda A, Abi-Rached B, Petrylak D. Chemotherapy for advanced renal-cell carcinoma: 1983–1993. Seminars in Oncology. 1995;22(1):42–60. [PubMed] [Google Scholar]
- 30.Fojo AT, Shen D-W, Mickley LA, Pastan I, Gottesman MM. Intrinsic drug resistance in human kidney cancer is associated with expression of a human multidrug-resistance gene. Journal of Clinical Oncology. 1987;5(12):1922–1927. doi: 10.1200/JCO.1987.5.12.1922. [DOI] [PubMed] [Google Scholar]
- 31.Guimaraes DP, Oliveira IM, de Moraes E, et al. Interferon-inducible guanylate binding protein (GBP)-2: a novel p53-regulated tumor marker in esophageal squamous cell carcinomas. International Journal of Cancer. 2009;124(2):272–279. doi: 10.1002/ijc.23944. [DOI] [PubMed] [Google Scholar]
- 32.Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All the patients listed in the table were treated with radical nephrectomy (RN) for RCC at Huashan Hospital of Fudan University. The tissues experimented in RNA-seq test were from the first 8 cases. And we used the tissues for all 16 cases in qRT-PCR examination.