

Abstract
The objective of this work was to develop an oral mucosal drug delivery system to facilitate the local and systemic delivery of acyclovir for the treatment of oral herpes infection caused by the herpes simplex virus (HSV). An in situ gelling system was used to increase the residence time and thus the bioavailability of acyclovir in oral mucosa. Temperature and pH trigged in situ gel formulations were prepared by cold method using polymers like poloxamer 407, carbopol 934, and HPMC. Glycerin and a mixture of tween 80 and ethanol (1 : 2 ratio) were used as the drug dissolving solvent. The pH of carbopol containing formulation was adjusted to pH 5.8 while the pH of poloxamer solution was adjusted to pH 7. These formulations were evaluated for sol-gel transition temperature, gelling capacity, pH, viscosity, spreadability, gel strength, drug content, ex-vitro permeation, and mucoadhesion. The gelation temperatures of all the formulations were within the range of 28–38°C. All the formulations exhibited fairly uniform drug content (98.15–99.75%). Drug release study of all the formulations showed sustained release properties. The release of drug through these in situ gel formulations followed the Higuchi model and Korsmeyer peppas model mechanism.
1. Introduction
Acyclovir (ACY), a widely used antiviral agent, is a synthetic purine nucleotide analog derived from guanine. It is effective in the treatment of herpes simplex virus (HSV), mainly HSV 1 and HSV 2, and varicella zoster [1, 2]. Acyclovir exerts its antiviral activity by competitive inhibition of viral DNA through selective binding of acyclovir to HSV-thymidine kinase [3, 4]. Currently available dosage forms of acyclovir are intended for intravenous, oral, and topical administration. Systemic delivery of the drug following administration by these routes is far from optimal. However, the oral absorption of acyclovir is dose dependent and highly variable with bioavailability ranging from 15 to 30% [5, 6].
The percutaneous penetration is poor; and because of its limited solubility in water (1.62 mg/mL at 22°C) it cannot be given as eye drops or by intramuscular injection. As parenteral administration acyclovir is presently available as infusion or as bolus intravenous injection in the form of strong alkaline (pH 10-11) solution of sodium salt. Consequently, administration of this dosage form may cause thrombophlebitis or perivascular inflammation [5].
There have been many attempts for improving the physicochemical properties of acyclovir by chemical modifications which are documented in the literature. These include using acyclovir as prodrug, employing novel redox-based chemical targeting systems to enhance ocular, parenteral, nasal, and intradermal delivery of drug for greater oral bioavailability. Because of its poor oral and transdermal absorption of acyclovir, oral mucosa is a logical choice for local and systemic delivery [6, 7]. There are several reasons due to which oral mucosa is an attractive site for the delivery of therapeutic agents that include its accessibility, excellent blood supply, by-pass of hepatic first-pass metabolism, rapid repair, and permeability profile [8].
The limitation of oral mucosal drug delivery is the dilution and rapid elimination of topically applied drugs due to the flushing action of saliva. The delivery system in which the drug is incorporated is therefore an important consideration and should be formulated to prolong the retention of the drug in the oral cavity. Bioadhesive polymers have been utilized in gel forms to prolong the residence time on oral mucosa and to reduce the frequency of application and the amount of drug administered. This might improve patient's compliance and acceptance of the drug product [9].
In situ is a Latin phrase which can be translated literally as “In process.” In situ gels are drug delivery systems that are in solution forms before administration in the body, once administered they undergo gelation in situ to form a gel. It is basically a polymeric drug delivery system.
Administration routes for in situ gels are oral, ocular, rectal, vaginal, injectable, and intraperitoneal. Advantages of in situ forming mucoadhesive polymeric delivery systems include ease of administration, improved local bioavailability, reduced dose concentration, reduced dosing frequency, and improved patient compliance and comfort. Also the formulation is less complex which lowers the investment and manufacturing cost [10]. There are several possible mechanisms that lead to in situ gel formation: solvent exchange, UV irradiation, ionic cross-linkage, pH change, and temperature modulation [11].
The aim of this study was to develop an in situ gel formulation containing acyclovir for local and systemic delivery from oral mucosal route. This was needed to increase absorption of the drug leading to an improvement in its bioavailability, to reduce its dosing frequency, and to achieve sustained release effect.
2. Materials and Methods
2.1. Materials
Acyclovir was purchased from Jackson Laboratories (P) Ltd., India. Poloxamer 188 and poloxamer 407 were received as gift samples from BASF chemical company, India. Carbopol 934, methyl paraben, and propyl paraben were purchased from Central Drug House (P). Ltd. India. HPMC K-100, glycerin, and ethanol were procured from LOBA Chemie pvt, Ltd., India. Tween 80 and triethanolamine were obtained from Molychaem, India.
2.2. Solubility Studies
The solubility of acyclovir was studied in water and in buffer solutions of different pH. These were HCl buffer (pH 1.2), acetate buffer (pH 4.5), phosphate buffer (pH 6.8 and 5.5), and borate buffer (pH 9.8). For evaluating the solubility in a particular solvent, an excessive amount of the drug was dissolved in 5 mL solvent and the solution was stirred using magnetic stirrer for 24 hrs at room temperature (25°C). After 24 hrs the sample was removed from stirrer and allowed to settle down. The supernatant solution was separated and filtered, and appropriate dilution was made with the respective solvents. Absorbance of diluted solution was measured at 255 nm and the concentration of soluble drug was calculated.
Similarly, the solubility of acyclovir was studied in surfactants like tween 80, tween 20, and SLS and cosurfactants like propylene glycol, PEG 400, and glycerin. Solubility was also studied in oleic acid, castor oil, and the mixture of tween 80 and ethanol (1 : 2 ratio). In this case solubility study was conducted for 48 hrs at 37 ± 5°C.
2.3. Preparation of In Situ Gel Formulation
In situ gel was prepared by the cold method. A weighed amount of poloxamer 407 and poloxamer 188 (15–20% w/v) was slowly added to 15 mL water (at 4 ± 2°C) in a beaker with continuous stirring using a magnetic stirrer at a speed of 500 rpm for 2 hrs. The temperature of water was maintained at 4 ± 2°C throughout the preparation. This solution was kept overnight in refrigerator. HPMC K-100 (0.5% w/v), carbopol 934 (0.1%, 0.3%, and 0.5% w/v), and the preservatives (methyl and propyl paraben 0.1% and 0.01%, w/v resp.) were added to poloxamer dispersion with continuous stirring. The preservative solution was prepared by solubilizing it in hot water. It was mixed with above dispersion after cooling. The weighed amount of drug (2% w/v) was dissolved in the mixture of tween 80 and ethanol (1 : 2) or glycerin. The drug solution was then mixed in the above described poloxamer dispersion. The final volume was made up and pH of the poloxamer dispersion was adjusted to 7 using triethanolamine, whereas the dispersion containing carbopol was adjusted to pH 5.8. The composition of the in situ gel formulations is shown in Tables 1 and 2 [12, 13].
Table 1.
Composition of the drug loaded formulations containing tween 80 and ethanol.
| FC | Drug (gm) | T + E (mL) | P407 (gm) | P188 (gm) | C934 (gm) | HPMC K-100 (gm) | MP (gm) | PP (gm) | D/W (mL) |
|---|---|---|---|---|---|---|---|---|---|
| Ft1 | 0.6 | 6 | 6 | — | — | 0.15 | 0.03 | 0.003 | 30 |
| Ft2 | 0.6 | 6 | 4 | 2 | — | 0.15 | 0.03 | 0.003 | 30 |
| Ft3 | 0.6 | 6 | 4.5 | — | 0.09 | 0.15 | 0.03 | 0.003 | 30 |
| Ft4 | 0.6 | 6 | 6 | — | 0.03 | 0.15 | 0.03 | 0.003 | 30 |
| Ft5 | 0.6 | 6 | 6 | — | 0.09 | 0.15 | 0.03 | 0.003 | 30 |
| Ft6 | 0.6 | 6 | 6 | — | 0.15 | 0.15 | 0.03 | 0.003 | 30 |
Note: FC: formulation code, T: tween 80, E: ethanol, P407: poloxamer407, P188: poloxamer188, C934: carbopol934, HPMC K-100: hydroxypropyl methyl cellulose, MP: methyl paraben, PP: propyl paraben, and D/W: distilled water.
Table 2.
Composition of the drug loaded formulations containing glycerin.
| FC | Drug (gm) | Glycerin (mL) | P407 (gm) | P188 (gm) | C934 (gm) | HPMC K-100 (gm) | MP (gm) | PP (gm) | D/W (mL) |
|---|---|---|---|---|---|---|---|---|---|
| Fg1 | 0.6 | 6 | 6 | — | — | 0.15 | 0.03 | 0.003 | 30 |
| Fg2 | 0.6 | 6 | 4 | 2 | — | 0.15 | 0.03 | 0.003 | 30 |
| Fg3 | 0.6 | 6 | 4.5 | — | 0.09 | 0.15 | 0.03 | 0.003 | 30 |
| Fg4 | 0.6 | 6 | 6 | — | 0.03 | 0.15 | 0.03 | 0.003 | 30 |
| Fg5 | 0.6 | 6 | 6 | — | 0.09 | 0.15 | 0.03 | 0.003 | 30 |
| Fg6 | 0.6 | 6 | 6 | — | 0.15 | 0.15 | 0.03 | 0.003 | 30 |
Note: FC: formulation code, Gly: glycerin, P407: poloxamer 407, P188: poloxamer188, C934: carbopol934, HPMC K-100: hydroxypropyl methyl cellulose, MP: methyl paraben, PP: propyl paraben, and D/W: distilled water.
2.4. Determination of Sol-Gel Temperature (T sol-gel)
The gelation temperature was determined by placing the solution in test tube: the test tube was dipped in water bath whose temperature was maintained at 37 ± 5°C for 2 min. The temperature at which solution was converted to gel was noted down by placing the thermometer in the test tube. In case of formulations containing carbopol, the formulation was taken in test tube containing phosphate buffer solution pH 6.8. This mixture was thoroughly mixed and dipped into the water bath. The maximum limit for gelation was checked up to 60°C. The gel was said to have formed when there was no flow of the formulation when the container was overturned.
2.5. Determination of Gelling Capacity
The gelling capacity was determined based on the formulation behaviors like gelling time and erosion time of formed gel due to the environmental changes.
2.6. Determination of pH
The pH of the gel was determined using calibrated pH meter. Determinations were carried out in triplicate and an average of these determinations was taken as the pH of the gel.
2.7. Viscosity of Formulation at Solution State and Gel State
The viscosity was measured at 25°C and 37°C using Brookfield viscometer and spindle number 62 at 100 rpm. First, the viscosity of gel solution was measured. This solution was allowed to convert to gel by increasing the temperature of the solution with the help of water bath whose temperature was maintained at 37 ± 1°C. In the formulation containing carbopol pH was increased along with temperature. Then the viscosity of this formed gel was measured. The average of two determinations was taken.
2.8. Spreadability Test
To determine the spreadability of the gel, approximately 1 g of gel was placed at the center of the glass plate (20 cm × 20 cm). This glass plate was covered with another glass plate of the same size. Next, the weight of 1000 g was carefully applied on the upper side of the plate; as a result the gel was spread out in between the plates. After one minute the weight was removed and the diameter of the spread area (cm) was measured. This determination was carried out in triplicate [14, 15].
2.9. Gel Strength
An accurate weighed quantity of 30 g of gel was placed in a 50 mL graduated measuring cylinder and was allowed to form gel in a water bath at 37°C. By applying 50 g weight to the gel with the help of a cylinder, the time taken by the cylinder to sink 5 cm down through the gel was measured [16].
2.10. Drug Content
In situ gel formulation, equivalent to 10 mg of acyclovir (0.5 mL), was pipetted out. It was suitably diluted with 0.1 N NaOH solution and the absorbance of this mixture was measured at 255 nm. The drug content was calculated against the absorbance of control ACY solution of the same concentration at 255 nm.
2.11. Ex Vivo Permeation Study
Ex vivo permeation study was assessed by using Franz diffusion cell. The porcine oral mucosa was used as biological membrane for the study. Porcine oral mucosa was obtained from local slaughter house and stored in phosphate buffer (pH 7) at 4°C from the time of collection. It was used within 3 hrs of procurement. The receptor compartment was filled with phosphate buffer (pH 7.4). The compartment also containing magnetic bead for stirring purpose. The suitable size of membrane was placed in between the donor and the receptor compartment. The cell was agitated by magnetic stirrer at 600 rpm and maintained at 37 ± 1°C. Approximately 500 mg of gel as a sample was transferred to the donor compartment. About 3 mL sample was withdrawn at the different time intervals of 15, 30, 45, 60, 75, 90, 120, 180, and 360 min. After each withdrawal, equal volume of fresh phosphate buffer of pH 7.4 previously heated to 37 ± 1°C was incorporated in the receptor compartment to maintain the sink conditions. The samples were filtered and diluted, and their absorbance was measured at 255 nm [17, 18].
2.12. Analysis of Release Mechanism
The release kinetics of acyclovir from in situ gel formulation was evaluated considering five different models including zero order, first order Higuchi model, Hixson Crowell, and Korsmeyer's peppas model [19].
2.13. Mucoadhesion Studies
Mucoadhesive property was determined using modified physical balance. Porcine oral mucosa was used as biological membrane, which was fixed under one pan of the balance with the help of cyanoacrylate glue and was hydrated with 100μL of phosphate buffer pH 6.8 maintained at 37 ± 1°C. Accurately weighed amount of 1 g of gel was stuck to the inverted beaker (250 mL) using glue and the height of the balance was adjusted to accommodate a glass container below the pan where membrane was glued. A preload of 20 g was applied in order to allow the formation of mucoadhesive joints. After a 3 min rest period, the preload was removed and gradually the weight was added to the other pan until the gel was detached from the mucosal surface. The total weight required for the complete detachment of the gel was recorded [13, 20, 21].
3. Results and Discussions
3.1. Solubility Studies
Solubility of drug in water was found to be 2.35 mg/mL at room temperature (RT). The pH dependent solubility profile in Table 3 indicates that acyclovir had pH dependent solubility. It had higher solubility in solution of basic pH as compared to the solution at acidic pH. It had less solubility towards neutral pH.
Table 3.
Solubility of acyclovir in different pH buffer solutions.
| S. no. | Buffer solution | Solubility (microg/mL) |
|---|---|---|
| 1 | HCl buffer of pH1.2 | 18.315 |
| 2 | Acetate buffer of pH4.5 | 10.064 |
| 3 | Phosphate buffer of pH5.5 | 2.515 |
| 4 | Phosphate buffer of pH6.8 | 2.25 |
| 5 | Phosphate buffer of pH7.4 | 2.558 |
| 6 | Borate buffer of pH9.8 | 61.842 |
The solubility studies were also performed in different surfactants/cosurfactants/oils/mixtures of surfactants and cosurfactants. Tween 80, glycerin, and mixture of tween 80 and ethanol (1 : 2) had higher solubility of ACY than other vehicles. So, glycerin and mixture of tween 80 and ethanol (1 : 2) were used as solvents in formulations. Although ACY had higher solubility in tween 80 in comparison to its mixture with ethanol, its higher viscosity restricted not using this compound for the formulation development.
3.2. Evaluation of Gel
A 20% w/v concentration was found to be the optimum concentration for the poloxamer solution to form an in situ gel. However, the gelling temperatures were found to be 30°C and above. But this gel was found to have weak mechanical strength and the erosion occurred rapidly. Hence, hydrophilic polymers such as HPMC, carbopol 934, and poloxamer 188 were incorporated in the formulation to overcome the drawback.
3.3. Solution to Gel Transition Temperature (T sol-gel)
The gelation temperature of all the formulations was in the range of 28°C to 38°C, while the transition temperature of the formulations containing tween 80-ethanol (Ft) and glycerin (Fg) showed slight difference. The transition temperature of Ft was slightly higher than Fg formulation because ethanol might have increased the transition temperature.
3.4. Gelling Capacity
The gelling capacity data of prepared formulations presented in Table 4 represent that the formulations Ft1 and Fg1 had immediate gelation and underwent rapid dissolution. On the other hand the formulations Ft6 and Fg6 had immediate gelation but exist for an hour. This implied that by increasing the concentration of polymer, transition time was decreased and the erosion time of formed gel was increased.
Table 4.
Evaluation data 1 of in situ gel formulation.
| FC | T sol-gel | Gelling capacity | Appearance | pH | ||
|---|---|---|---|---|---|---|
| Solution state | Gel state | Observed | Adjusted to | |||
| Ft1 | 31.333 ± 1.155 | ++ | √ | € | 8.15 | 7 |
| Ft2 | 35.333 ± 1.145 | ++ | √ | € | 8.21 | 7 |
| Ft3 | 35.333 ± 0.577 | ++ | √ | € | 5.6 | 5.8 |
| Ft4 | 38.666 ± 1.154 | ++ | √ | €€ | 5.78 | 5.8 |
| Ft5 | 35.666 ± 0.577 | ++ | √√ | €€ | 5.34 | 5.8 |
| Ft6 | 29.666 ± 1.154 | +++ | √√ √ | €€€ | 5.56 | 5.8 |
| Fg1 | 28.333 ± 0.577 | ++ | ∆ | ○ | 8.13 | 7 |
| Fg2 | 34.666 ± 1.527 | ++ | ∆ | ○ | 8.18 | 7 |
| Fg3 | 37.666 ± 0.577 | ++ | ∆ | ○ | 5.17 | 5.8 |
| Fg4 | 35.333 ± 0.577 | ++ | ∆ | ○○ | 5.65 | 5.8 |
| Fg5 | 31.333 ± 1.155 | ++ | ∆∆ | ○○ | 5.48 | 5.8 |
| Fg6 | 30.333 ± 0.577 | +++ | ∆∆∆ | ○○ | 5.63 | 5.8 |
+: gel after few minutes dissolves rapidly.
++: immediate gelation remains for few mins.
+++: immediate gelation remains for nearly an hr.
√: yellowish solution with less viscosity.
√√: yellowish solution with moderate viscosity.
√√ √: yellowish solution with high viscosity.
∆: white colored solution with less viscosity.
∆∆: white colored solution with moderate viscosity.
∆∆∆: white colored solution with high viscosity.
FC: formulation code.
€: yellowish semifluid gel.
€€: yellow colored semistiff gel.
€€€: yellow colored stiff gel.
○: white colored semistiff gel.
○○: white colored stiff gel.
3.5. pH of the Formulation
The pH of the formulations was neutral. This indicated the nonirritancy of the formulation in oral cavity. The formulation containing carbopol 934 was slightly acidic and was maintained within pH 5.8 while the pH of poloxamer solution was adjusted to pH 7.
3.6. Viscosity of Formulation at Solution State and Gel State
The viscosity was proportional to the concentration of the mucoadhesive polymer in the formulation. All the formulations exhibited quite low viscosity at low temperature. However, upon increasing the temperature, a gel was formed in well-defined temperature and viscosity of the formulation was increased.
3.7. Gel Strength
The results obtained for strength test of all the formulations are mentioned in Table 5 and its graphical representation is shown in Figure 1. It has been observed that gel strength increased with the increase in the concentration of mucoadhesive polymer in the formulation. If comparison is made between the Ft and Fg formulations, Fg formulation will show higher gel strength than Ft. The reason can be attributed to the ethanol present in these formulations, as it has a tendency to decrease the gel strength.
Table 5.
Evaluation data 2 of in situ gel formulation.
| FC | Viscosity (Cp) | |||
|---|---|---|---|---|
| Solution state | Gel state | Spreadability | Gel strength | |
| Ft1 | 98.93 ± 0.40 | 1481.33 ± 6.80 | 6.63 ± 0.15 | 4.67 ± 0.57 |
| Ft2 | 104.88 ± 2.29 | 1540.67 ± 15.65 | 6.43 ± 0.05 | 5.66 ± 0.57 |
| Ft3 | 149.10 ± 2.73 | 1505.00 ± 6.00 | 5.70 ± 0.17 | 21.33 ± 1.53 |
| Ft4 | 111.61 ± 10.67 | 1564.00 ± 7.21 | 5.66 ± 0.12 | 23.66 ± 0.57 |
| Ft5 | 158.27 ± 6.68 | 1498.67 ± 14.22 | 5.26 ± 0.11 | 80.33 ± 0.57 |
| Ft6 | 199.74 ± 0.63 | 1594.33 ± 12.89 | 4.53 ± 0.12 | 99.66 ± 0.57 |
| Fg1 | 214.41 ± 3.08 | 1493.67 ± 5.03 | 6.56 ± 0.05 | 10.33 ± 1.52 |
| Fg2 | 220.47 ± 2.16 | 1495.67 ± 6.65 | 6.30 ± 0.02 | 13.66 ± 1.52 |
| Fg3 | 242.05 ± 2.16 | 1533.67 ± 19.50 | 5.66 ± 0.20 | 26.33 ± 1.52 |
| Fg4 | 263.60 ± 10.58 | 1586.33 ± 5.50 | 5.03 ± 0.15 | 65.66 ± 1.15 |
| Fg5 | 242.50 ± 14.90 | 1535.33 ± 11.01 | 5.23 ± 0.05 | 89.33 ± 0.57 |
| Fg6 | 344.87 ± 5.56 | 1621.33 ± 3.21 | 4.46 ± 0.05 | 119.3 ± 1.52 |
The bold data refer to four selected formulations with the best results.
Figure 1.
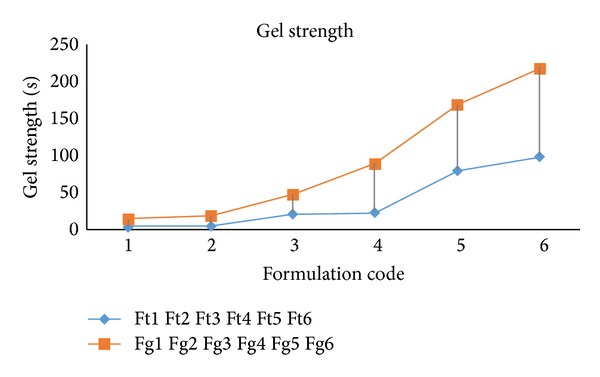
Graphical representation of gel strengths of formulations.
3.8. Spreadability Test
With increase in the concentration of the polymeric component, viscosity of the solution was increased. At the same time spreadability of the formulation was reduced. This can be observed from the evaluation tests data compiled in Table 5. Ft formulation showed a higher spreadability compared to Fg formulation because gel strength and viscosity of Fg formulation were higher. Consequently, its spreadability was less.
On the basis of gelling capacity, viscosity, gel strength, and spreadability results, formulations Ft1, Ft4, Fg1, and Fg4 showed optimum results within the desired range. So, these six formulations were subjected to further evaluation parameters.
3.9. Drug Content
All the formulations reflected fairly uniform drug content ensuring adequacy in the method of preparation of the in situ gel. Drug content was found to be within the range of 98.15–99.75%.
3.10. Ex Vivo Permeation Study
For more details see Table 6.
Table 6.
Ex vivo release data of different formulations.
| Time (min.) | %Release | ||||
|---|---|---|---|---|---|
| Ft1 | Fg1 | Ft4 | Fg4 | MC | |
| 15 | 25.209 | 22.203 | 17 | 16.584 | 8.234 |
| 30 | 35.645 | 32.549 | 24.625 | 27.125 | 20.963 |
| 45 | 49.48 | 45.489 | 39.641 | 39.648 | 31.839 |
| 60 | 67.457 | 62.626 | 53.982 | 52.637 | 46.71 |
| 75 | 78.393 | 75.279 | 68.892 | 64.135 | 52.45 |
| 90 | 89.742 | 87.993 | 83.891 | 80.453 | 69.45 |
| 120 | 97.638 | 97.729 | 90.678 | 87.825 | 75.85 |
| 180 | 97.892 | 97.773 | 95.365 | 90.135 | 83.95 |
| 360 | 97.845 | 97.715 | 95.632 | 93.899 | 95.87 |
Fg and Ft: formulation code and MC: marketed cream.
3.11. In Vitro Permeation Study
All the formulations including the marketed cream showed almost similar drug release rates. The drug release rate was high lasting for up to 3 hrs. Thereafter, it remained constant for up to 6 hrs. Initial burst release was higher in in situ gel formulations. Among the in situ gel formulations, a formulation containing carbopol 934 showed decreased burst release compared to a formulation without carpool 934. Further, kinetic models were applied on these formulations so as to analyze their release mechanism. Kinetic model was also applied in marketed cream (MC) in order to compare the release pattern with that of the prepared formulation (Table 7 and Figure 2).
Table 7.
Kinetic analysis of in vitro release data of different formulations.
| FC | Zero order | First order | Higuchi model | Hixson crowell model | Korsmeyer peppas model | Best fit model | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| K | R 2 | K | R 2 | K | R 2 | K | R 2 | K | R 2 | ||
| Ft4 | 16.88 | 0.91 | −0.25 | 0.904 | 48.69 | 0.98 | −0.52 | 0.96 | 0.58 | 0.98 | Higuchi model |
| Fg4 | 15.67 | 0.91 | −0.20 | 0.911 | 45.21 | 0.98 | −0.45 | 0.97 | 0.56 | 0.98 | Peppas model |
| Ft1 | 19.33 | 0.92 | −0.31 | 0.946 | 39.94 | 0.93 | −0.56 | 0.93 | 0.54 | 0.97 | Peppas model |
| Fg1 | 20.22 | 0.94 | −0.31 | 0.937 | 42.09 | 0.94 | −0.58 | 0.93 | 0.50 | 0.97 | Peppas model |
| MC | 17.66 | 0.94 | −0.20 | 0.928 | 43.60 | 0.99 | −0.45 | 0.98 | 0.72 | 0.97 | Higuchi model |
Figure 2.
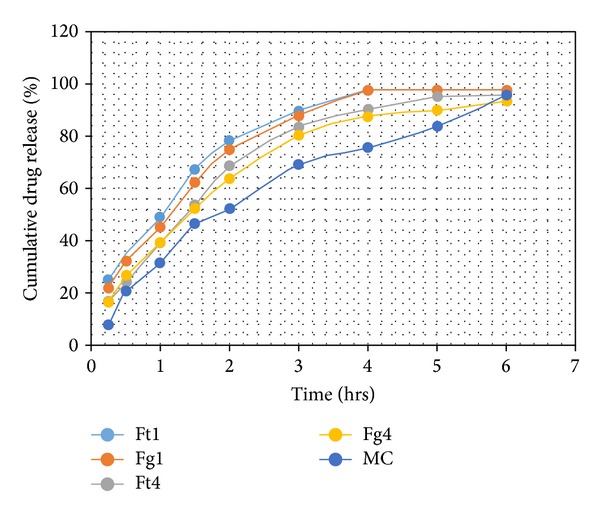
In vitro % drug release versus time plot of different formulations.
3.12. Mucoadhesion Studies
Mucoadhesion is an important feature of the formulations designed for the delivery of drug in oral cavity. Assessment of the mucoadhesive strength in terms of detachment stress showed that the adhesive properties of gel formulations increased with the increase in the concentration of carbopol 934. The high mucoadhesive strength of the delivery system may lead to prolonged retention and increased absorption across mucosal tissue.
4. Conclusion
Temperature and pH sensitive in situ gel of ACY (2% w/v) was successfully prepared by cold method using poloxamer 407, carbopol 934, HPMC, tween 80-ethanol (1 : 2 ratio), and glycerin as a drug dissolving solvent.
The gelation temperatures of all the formulations were within the range of 28–38°C. It was observed that the higher the concentration of polymer in the formulation the lower its transition temperature. The formulations containing carbopol 934 showed gelation only when their pH and temperature were raised simultaneously. With regards to the pH, the formulation containing carbopol was slightly acidic then the formulation containing poloxamer. Hence, acidic nature of the carbopol was used to prepare pH triggered in situ gel. The pH of carbopol containing formulation was adjusted to pH 5.8, while the pH of poloxamer solution was adjusted to pH 7. By addition or increase in the concentration of hydrophilic polymer, the gelling capacity, gel strength, viscosity, and mucoadhesion were increased whereas spreadability was decreased. All the formulations exhibited fairly uniform drug content. Drug release study of all the formulations showed sustained release properties. The release of drug through these in situ gel formulations followed a Higuchi model and Korsmeyer peppas model mechanisms. Delivery of drug through oral mucosa by in situ gel formulation avoids the first pass effect. So it is a logical choice for local and systemic delivery of drug, which eventually improves the bioavailability of drug. This approach can be used to treat oral herpes infection locally by improving the patient compliance. However, further studies are needed to be performed, in order to increase the dose of drug in the formulation.
Abbreviations
- ACY:
Acyclovir
- HSV:
Herpes simplex virus.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Charles PD. Oral Herpes (HSV-1) (Herpes of the Mouth) 2011, http://www.emedicinehealth.com/oral_herpes/article_em.htm#oral_herpes_hs v 1_overview.
- 2.Manish K, Nikhil M, Vilasrao K. Formulation development and evaluation of acyclovir orally disintegrating tablets. Journal of Applied Pharmaceutical Science. 2012;2(3):101–105. [Google Scholar]
- 3.Patel D, Sawant KK. Oral bioavailability enhancement of acyclovir by self-microemulsifying drug delivery systems (SMEDDS) Drug Development and Industrial Pharmacy. 2007;33(12):1318–1326. doi: 10.1080/03639040701385527. [DOI] [PubMed] [Google Scholar]
- 4.Tripathi KD. Essentials of Medical Pharmacology: Antiviral Drugs. 6th edition. New Delhi, India: Jaypee Brothers’s Medical Publishers (P) Ltd.; 2008. [Google Scholar]
- 5.Shojaei AH, Berner B, Li X. Transbuccal delivery of acyclovir: I. In vitro determination of routes of buccal transport. Pharmaceutical Research. 1998;15(8):1182–1188. doi: 10.1023/a:1011927521627. [DOI] [PubMed] [Google Scholar]
- 6.Drug Bank. 2012, http://www.drugbank.ca/drugs/DB00787.
- 7.Shojaei AH, Zhuo SL, Li X. Transbuccal delivery of acyclovir (II): feasibility, system design, and in vitro permeation studies. Journal of Pharmacy & Pharmaceutical Sciences. 1998;1(2):66–73. [PubMed] [Google Scholar]
- 8.Hearnden V, Sankar V, Hull K, et al. New developments and opportunities in oral mucosal drug delivery for local and systemic disease. Advanced Drug Delivery Reviews. 2012;64(1):16–28. doi: 10.1016/j.addr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Perchyonok VT, Shengmiao Z, Theunis O. Chitosan and gelation based prititype delivery systems for the treatment of oral mucositis: from materials to performance in vitro . Current Drug Delivery. 2013;10:144–150. doi: 10.2174/1567201811310010020. [DOI] [PubMed] [Google Scholar]
- 10.Tejas G, Vishal G, Vishal C, Umesh UA. Review on novel in-situ polymeric drug delivery system. International Journal of Pharmaceutical Research and Development. 2011;3(5):53–59. [Google Scholar]
- 11.Ruel-Gariépy E, Leroux J-C. In situ-forming hydrogels—review of temperature-sensitive systems. European Journal of Pharmaceutics and Biopharmaceutics. 2004;58(2):409–426. doi: 10.1016/j.ejpb.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 12.Gratieri T, Gelfuso GM, Rocha EM, Sarmento VH, de Freitas O, Lopez RFV. A poloxamer/chitosan in situ forming gel with prolonged retention time for ocular delivery. European Journal of Pharmaceutics and Biopharmaceutics. 2010;75(2):186–193. doi: 10.1016/j.ejpb.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 13.Alpana PK, Sarfaraz KAK, Mohammed HD. Evaluation of polaxomer-based in situ gelling system of articaine as a drug delivery system for anesthetizing periodontal pockets—an in vitro study. Indian Journal of Dentistry. 2012;3(4):201–208. [Google Scholar]
- 14.Lardy F, Vennat B, Pouget MP, Pourrat A. Functionalization of hydrocolloids: principal component analysis applied to the study of correlations between parameters describing the consistency of hydrogels. Drug Development and Industrial Pharmacy. 2000;26(7):715–721. doi: 10.1081/ddc-100101289. [DOI] [PubMed] [Google Scholar]
- 15.Garg A, Aggarwal D, Garg S, Singla AK. Spreading of semisolid formulations: an update. Pharmaceutical Technology. 2002;26(9):84–105. [Google Scholar]
- 16.Yong CS, Choi JS, Quan Q-Z, et al. Effect of sodium chloride on the gelation temperature, gel strength and bioadhesive force of poloxamer gels containing diclofenac sodium. International Journal of Pharmaceutics. 2001;226(1-2):195–205. doi: 10.1016/s0378-5173(01)00809-2. [DOI] [PubMed] [Google Scholar]
- 17.Ceschel GC, Maffei P, Sforzini A, Lombardi Borgia S, Yasin A, Ronchi C. In vitro permeation through porcine buccal mucosa of caffeic acid phenetyl ester (CAPE) from a topical mucoadhesive gel containing propolis. Fitoterapia. 2002;73(supplement 1):S44–S52. doi: 10.1016/s0367-326x(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 18.Xiang J, Fang X, Li X. Transbuccal delivery of 2′,3′-dideoxycytidine: in vitro permeation study and histological investigation. International Journal of Pharmaceutics. 2002;231(1):57–66. doi: 10.1016/s0378-5173(01)00865-1. [DOI] [PubMed] [Google Scholar]
- 19.Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Poloniae Pharmaceutica—Drug Research. 2010;67(3):217–223. [PubMed] [Google Scholar]
- 20.Aditya G, Gannesh KG, Manasa B, Subal D, Rajesham VV. Design and evaluation of controlled release Mucoadhesive buccal tablets of Lisinopril. International Journal of Current Pharmaceutical Research. 2010;2(4):24–27. [Google Scholar]
- 21.Dias RJ, Sakhare SS, Mali KK. Design and development of mucoadhesive acyclovir tablet. Iranian Journal of Pharmaceutical Research. 2009;8(4):231–239. [Google Scholar]