

Abstract
Pompe disease is an autosomal recessive disorder caused by mutations in the acid-α glucosidase (GAA) gene. Lingual dysfunction is prominent but does not respond to conventional enzyme replacement therapy (ERT). Using Pompe (Gaa−/−) mice, we tested the hypothesis that intralingual delivery of viral vectors encoding GAA results in GAA expression and glycogen clearance in both tongue myofibers and hypoglossal (XII) motoneurons. An intralingual injection of an adeno-associated virus (AAV) vector encoding GAA (serotypes 1 or 9; 1 × 1011 vector genomes, CMV promoter) was performed in 2-month-old Gaa−/− mice, and tissues were harvested 4 months later. Both serotypes robustly transduced tongue myofibers with histological confirmation of GAA expression (immunochemistry) and glycogen clearance (Period acid-Schiff stain). Both vectors also led to medullary transgene expression. GAA-positive motoneurons did not show the histopathologic features which are typical in Pompe disease and animal models. Intralingual injection with the AAV9 vector resulted in approximately threefold more GAA-positive XII motoneurons (P < 0.02 versus AAV1); the AAV9 group also gained more body weight over the course of the study (P < 0.05 versus AAV1 and sham). We conclude that intralingual injection of AAV1 or AAV9 drives persistent GAA expression in tongue myofibers and motoneurons, but AAV9 may more effectively target motoneurons.
Introduction
Pompe disease is an autosomal recessive disorder characterized by deficiency of acid-α glucosidase (GAA). This hydrolase degrades lysosomal glycogen, and reduced or absent GAA can cause profound glycogen accumulation, disruption of cellular architecture, and functional neuromuscular impairments. Respiratory-related motor units controlling the primary inspiratory muscle (e.g., diaphragm) and also the pharyngeal airway (e.g., tongue) are particularly affected in Pompe disease.1 Indeed, severe respiratory insufficiency and ventilator-dependence are common in both infantile and late-onset Pompe disease.2 Sleep-disordered breathing and tongue motor problems are also prevalent in the Pompe population.2,3,4,5 Since both respiratory skeletal muscle fibers6,7 and motoneurons8 show pathology in Pompe disease, therapeutic approaches should target the entire motor unit.1 In this regard, adeno-associated viral (AAV) vectors carrying the GAA gene provide an important therapeutic option since it is feasible to target both myofibers and motoneurons via intramuscular delivery.9,10,11
In the current study, we used the Gaa−/− mouse Pompe model12 to determine if a single intralingual injection of AAV encoding GAA could restore GAA enzyme activity in lingual myofibers and hypoglossal (XII) motoneurons. The tongue motor system was selected for these studies since XII motor dysfunction is common in Pompe disease. For example, tongue weakness is extremely prevalent and occurs even in Pompe patients who are otherwise asymptomatic.13 Patients also typically develop pharyngeal dysphagia and have difficulty with saliva management, secretions, and drooling.3 Difficulties with phonation are also common in advanced Pompe disease.14 Importantly, these abnormalities appear to be unaffected by enzyme replacement therapy (ERT), even if ERT is administered in the newborn period.15 Accordingly, “correction” of pathology in tongue motor units could have profound functional benefits in a clinical setting. The hypoglossal-tongue motor system also provides an ideal experimental model to evaluate retrograde gene delivery to motoneurons (i.e., muscle-to-motoneuron).11 Based on the above considerations, our primary aim was to test the hypothesis that intralingual delivery of AAV-GAA could restore GAA activity and reverse glycogen accumulation in both tongue myofibers and XII motoneurons in Gaa−/− mice. Our secondary aim was to compare and contrast the effectiveness of two different AAV serotypes at motoneuron transduction via retrograde movement after tongue injection. Thus, AAV serotypes 1 and 9 encoding human GAA were administered to separate groups of Gaa−/− mice via intralingual injection. The AAV1 serotype was selected since it is currently being tested in a phase 1/2 clinical trial in Pompe disease NCT00976352,16 and the AAV9 serotoype was evaluated since it appears to be particularly effective at retrograde transport.11,17
Results
Restoration of GAA activity and reduced glycogen accumulation in tongue myofibers
Periodic acid Schiff (PAS) staining indicated substantial glycogen accumulation within lingual myofibers of sham-treated Gaa−/− mice, but not wild-type mice, as expected (Figure 1). However, a considerable reduction in the extent of positive PAS staining was noted at or near the site of intralingual injection (i.e., the genioglossus muscle and inferior-posterior aspect of the tongue) in Gaa−/− mice receiving either AAV1-hGAA or AAV9-hGAA. Figure 2 shows consecutive histological sections from the posterior tongue that were incubated with GAA antibodies or PAS reagents, respectively. Note that those areas which are immunopositive for GAA (Panels a and g) show weak or entirely absent PAS staining (Panels b and h). Thus, intralingual delivery of either AAV1-hGAA or AAV9-hGAA reversed the pathological appearance that is typical of Gaa−/− myofibers. Note in particular that lingual myofibers expressing GAA have a homogenous appearing sarcoplasm and peripherally located nuclei (Figure 2). In addition, GAA-positive myofibers were no longer distended and did not show the phenotypical swelling of lysosomes (Figure 2).
Figure 1.

Representative examples of periodic acid Schiff (PAS) staining in genioglossus tissue sections from an adult (a) wild-type and (b) a sham-treated Gaa−/− mouse. The PAS reaction recognizes glycogen and is evident by the magenta coloring. Myofibers from the (b) sham-treated Gaa−/− mice are PAS-positive and have swollen vacuolar appearance with disruption of cellular architecture. Scale bars = 200 µm.
Figure 2.
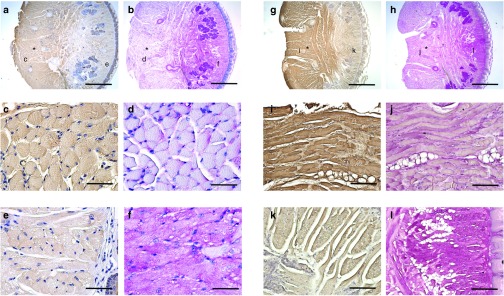
Immunohistochemical acid-α glucosidase (GAA) staining and periodic acid Schiff (PAS) staining of transverse lingual sections from the base of the tongue. Tissues were harvested at 4 months following intralingual injections with (a–f) AAV9-hGAA and (g–l) AAV1-hGAA. Alternating tissue sections were stained for GAA or PAS. The approximate site of the tongue AAV injection is indicated by the asterisk, and the sites marked c–f or i–l are shown in an expanded scale immediately below each top panel. Note that (c) GAA-positive myofibers are negative for (d) PAS. In contrast, the area negative for (e) GAA is also positive for (f) PAS. Similar results were seen in the AAV1-hGAA–injected lingual sections (panels g and h). Scale bars: 800 µm (a,b,g,h); 50 µm (c–f, i–l).
Biochemical assays confirmed robust GAA enzyme activity in the base of tongue (i.e., at the site of injection) as suggested by the histological results. Indeed, GAA activity was considerably increased after AAV-GAA treatment when compared to untreated Gaa−/− or wild-type control mice. Thus, at 4 months postinjection, GAA activity was 294 µmol/l/h*µg and 407 µmol/l/h*µg following AAV1-hGAA and AAV9-hGAA injection, respectively (P = 0.473). In comparison, control 129 mice averaged 59 µmol/l/h*µg (P < 0.05 versus AAV treated). Sham-treated Gaa−/− mice had an average GAA activity of 3 µmol/l/h*µg (P < 0.05 versus AAV treated).
GAA expression and glycogen clearance in XII motoneurons
The cytoarchitecture of XII motoneurons in wild-type mice was consistent with prior reports.18 Thus, the perikaryon around the nucleus was immunopositive for GAA, and an absence of glycogen accumulation was indicated by the lack of positive-PAS staining. In contrast, XII motoneurons in sham-treated Gaa−/− mice were negative for GAA immunostaining but robustly PAS-positive (Figure 3). XII motoneurons in the sham Gaa−/− group showed histopathologic features consistent with prior descriptions from Pompe animal models1,8,18 and human tissues.1,8 More specifically, as shown in Figure 3, XII motoneurons in the sham Gaa−/− group have a distended perikaryon filled with enlarged vacuoles.
Figure 3.

Immunohistochemical acid-α glucosidase (GAA) staining (panels a and c) and periodic acid Schiff (PAS) staining (panels b and d) of the XII motor nucleus in wild-type and sham-treated Gaa−/− tissues. The area highlighted by the box in the left panels is shown at a higher magnification in the right panels. (a) Positive GAA staining is evident in XII motoneurons with an absence of PAS staining (b) in wild-type tissues. In contrast, sham-treated Gaa−/− tissues show complete absence of (c) GAA immunostaining and (d) swollen PAS positive motoneurons. Scale bars = a–d left panels: 50 µm; right panels: 100 µm.
Quantification of medullary GAA immunostaining suggested differences in the retrograde transduction efficiency between AAV9-hGAA and AAV1-hGAA (Figure 4). More specifically, the AAV9 group had an almost threefold increase in the number of GAA-positive XII motoneurons when compared to the AAV1-treated mice (P = 0.02). When XII motoneurons were transduced following intralingual injection of AAV-GAA vectors (i.e., following retrograde transduction), there was a striking reversal of the histopathological features that are typical of Gaa−/− mice (Figure 5). Those XII motoneurons which were immunopositive for GAA in Gaa−/− mice were always negative for PAS staining. The GAA-positive cells had a “normal” appearing soma with a centrally located nucleus and an absence of vacuoles (i.e., compared to cells from wild-type and sham-injected Gaa−/− mice; Figure 3). In the AAV-treated tissues, GAA-positive motoneurons were often juxtaposed with GAA-negative cells, thus providing a contrast of “corrected” versus untreated cells (Figure 5). The histologic images shown in Figure 5 demonstrate that the perikaryon around the nucleus in the GAA-positive motoneuron lacks both the distended vacuoles and the positive PAS staining that are characteristics of the nearby GAA-negative cells (Figure 5, Panels b and d).
Figure 4.

Persistent acid-α glucosidase (GAA) expression in Gaa−/− XII motoneurons after intralingual injection of AAV9-hGAA or AAV1-hGAA. All tissues were harvested 4 months following AAV delivery to tongue. (a) AAV9-hGAA–treated Gaa−/− mice had more robust GAA expression in the XII motor nucleus as compared to (b) AAV1-hGAA. The average number of GAA-positive cells in the XII nucleus is shown in Panel c (*P < 0.05 compared to AAV9). Scale bars = a,b: 50 µm.
Figure 5.

Acid-α glucosidase (GAA)-positive XII motoneurons have no glycogen accumulation. Higher magnification views of XII motoneurons stained for GAA (brown) and periodic acid Schiff (PAS) (magenta) in alternate sections. The arrows indicate the same cell in consecutive histological sections. The juxtaposition of GAA-positive-GAA-negative cells allows direct comparison of the histological appearance. Note that GAA-positive cells lack PAS staining and do not have the vacuolar appearance typical of adjacent Gaa−/− motoneurons. Panels a and b: examples from AAV9-treated tissues; panels c and d: examples from AAV1-treated tissues. Scale bars = a–d: 200 µm.
Measurement of vector genomes and liver GAA activity
Real-time polymerase chain reaction (PCR) was used to quantify the distribution of AAV vector genomes after intralingual delivery (Figure 6). Consistent with the histological results (Figure 2), a similarly robust number of vector genomes was detected in the base of the tongue following both AAV1-hGAA and AAV9-hGAA injections (P = 0.7) (Figure 6a). However, the AAV9 vector had a greater neural distribution as compared to AAV1. Thus, increased AAV9 vector genomes were observed in the XII nerve (P = 0.007), the immediate area of the XII motor nucleus (P = 0.008), and the brain (P = 0.04) (all P values reflect comparison to AAV1; Figure 6b–d). No differences were noted, however, in vector genomes within the spinal cord (Figure 6e). Systemic distribution of the vectors was confirmed by liver expression (Figure 6f). Although not statistically significant, a tendency for increased liver vector genomes was seen following AAV9 versus AAV1 injections (P = 0.07). Importantly, liver GAA activity was not increased by intralingual injection of AAV9 or AAV1. Specifically, at 4 months postinjection, liver GAA activity (µmol/minute/mg protein) was 29.0 ± 7.6 in 129 mice, but only 4.8 ± 0.2, 3.7 ± 1.3, and 6.8 ± 0.9 in Gaa−/− mice treated with lactated ringers (LR), AAV1, and AAV9, respectively (all P < 0.05 versus 129 value). The values in LR-treated Gaa−/− mice were not statistically different than the AAV-treated Gaa−/− mice (P > 0.5).
Figure 6.

Mean number of vector genome copies 4 months after intralingual injection. Vector genome copies in the tongue were similar between (a) AAV1-hGAA– and AAV9-hGAA–treated Gaa−/− mice, but were greater in the (b) XII nerve, (c) medulla, (d) brain, (e) spinal cord, and (f) liver of AAV9-hGAA– versus AAV1-hGAA–treated Gaa−/− mice.
Body weight and ventilation
Following intralingual injection, a significant difference in weight gain was noted in the AAV9-treated Gaa−/− mice as compared to the AAV1 group. This was not due to a gender effect since there was an equal number of males and females in each group. Thus, over the course of the study, AAV9-hGAA–treated mice gained more weight when compared to either the sham Gaa−/− (LR group) (P = 0.028) or AAV1-hGAA groups (P = 0.006) (Figure 7). The body weight of both the LR- and AAV-treated Gaa−/− mice, however, remained below the age-matched wild-type control group (Figure 7).
Figure 7.

Mean increase in body weight following intralingual adeno-associated virus (AAV) injection. All experimental groups gained weight similarly over the first month of the study, but by the fourth month differences emerged between groups. The AAV9-hGAA–treated Gaa−/− mice had a significant increase in weight gain (*) compared to both LR- and AAV1-hGAA–injected Gaa−/− mice. The LR-treated mice were housed as cage mates with the AAV1-hGAA and AAV9-hGAA groups.
Although we had no a priori prediction that ventilation would differ between sham- and AAV-treated Gaa−/− mice, ventilation was assessed via whole-body plethysmography to confirm that the tongue injection did not impair breathing. During both baseline normoxic conditions (Table 1a) and a brief hypercapnic respiratory challenge (Table 2), the AAV1- and AAV9-treated Gaa−/− mice showed similar breathing frequency (br*minute−1), inspiratory tidal volume (ml*br−1), and minute ventilation (ml*minute−1), and both groups were comparable to sham-injected Gaa−/− mice. That is, there was no apparent impact of AAV1 or AAV9 on overall ventilation or breathing pattern. Compared to the wild-type mice, all Gaa−/− groups (i.e., AAV and sham) tended to have reduced minute ventilation during baseline (P = 0.17), and significantly lower minute ventilation during the respiratory challenge (P < 0.05). There was no indication for a negative impact on breathing after AAV delivery when compared to sham-treated Gaa−/− mice (Table 1).
Table 1. Ventilation measured in unanesthetized mice during baseline normoxic conditions.
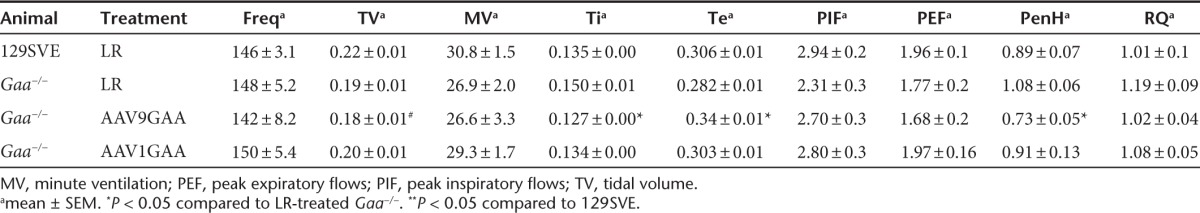
Table 2. Ventilation measured in unanesthetized mice during a brief hypercapnic challenge.
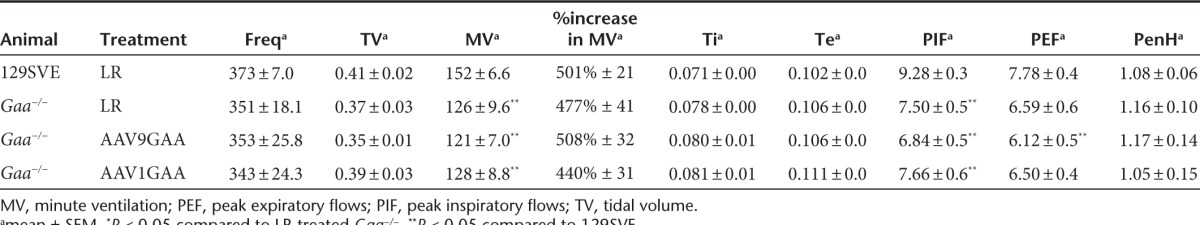
Some unexpected findings emerged from the whole body plethysmography measurements that suggest a positive impact of AAV treatment on pulmonary resistance. The PenH parameter is derived from analyses of the expiratory airflow signal and has been proposed to be an indirect measure of overall pulmonary airflow resistance.19 During the baseline condition, PenH tended to be reduced in wild-type mice as compared to sham-treated Gaa−/− mice (P = 0.07; Table 1). However, PenH was substantially reduced in Gaa−/− mice treated with the AAV9 vector when compared to sham animals (P = 0.001). In contrast, the AAV1-treated mice had similar PenH values to the sham Gaa−/− group (P = 0.19) (Table 1). The duration of the inspiratory effort (Ti, seconds) was also reduced in the AAV9 Gaa−/− mice when compared to sham Gaa−/− mice (Table 1). Although speculative, the decrease in PenH after AAV9 treatment may indicate an improvement19 in upper airway motor function resulting in decreased airflow resistance.
Immune response
There was histological evidence for a local immune response following injection of the viral vectors. Hematoxylin and eosin staining indicated a mild endomysial inflammatory reaction in tongue myofibers at or near the injection site, and this response was qualitatively similar in AAV1-hGAA– and AAV9-hGAA–treated animals (data not shown). In addition, CD3 immunochemistry indicated a higher proportion of T lymphocytes at the injection site as compared to the immediate surrounding tissues (Figure 8c,d), and again this was similar between AAV1- and AAV9-treated mice. Histological evidence of immune reactions, however, was absent in the medulla (Figure 8f,g). Finally, serum evaluations of anti-GAA immunoglobulins IgG1 (Figure 8e) and IgG2 (data not shown) showed that titers could be detected at 6 weeks post-AAV injection and persisted throughout the study. The antibody titers were significantly greater in AAV1- versus AAV9-treated mice at both 6 and 16 weeks following injection (P < 0.05).
Figure 8.

CD3 staining in the genioglossus and XII motor nucleus. No T-cell infiltrate was observed in sham-injected genioglossal myofibers of (a) wild-type or (b) Gaa−/− mice. In contrast, a mild T-cell infiltrate was noted in the genioglossus of both (c) AAV1-hGAA– and (d) AAV9-hGAA–injected mice. However, no T-cell infiltrate was observed in the medulla or XII motor nucleus of (e) AAV9-hGAA– or (f) AAV1-hGAA–treated animals. An IgG response against acid-α glucosidase (GAA) was noted in both (g) the AAV1-hGAA– and the AAV9-hGAA–treated mice. Scale bars = 200 µm.
Discussion
Our primary finding is that a single intralingual injection of single-stranded AAV-GAA (1 × 1011 vg) results in persistent GAA expression in both lingual myofibers and brainstem XII motoneurons. Moreover, GAA expression was associated with a marked reduction of glycogen accumulation (as evidenced through PAS staining), and reversal of the histopathologic features which characterize both Pompe tissues8,20 and Gaa−/− mouse skeletal muscle7 and motoneurons.8,18,21 The AAV9-hGAA vector resulted in more robust neural transduction as compared to AAV1-hGAA suggesting that it may be more appropriate for targeted gene delivery to motoneuron pools.
Retrograde transport of AAV-hGAA
In the present study, a direct injection in the base of the tongue with either AAV1-hGAA or AAV9-hGAA caused persistent GAA expression in XII motoneuron cell bodies. While the detection of vector genomes in the liver demonstrates that the vectors entered the systemic circulation as expected,22 GAA activity assays confirmed that there was no significant lysosomal GAA activity in the liver in AAV-treated Gaa−/− mice. Indeed, no differences in liver GAA activity levels were detected between the AAV- and sham-treated Gaa−/− mice. Therefore, we conclude that positive immunostaining for the transgene was not the result of liver secretion. Indeed, the focally restricted pattern of neuronal GAA expression within the XII motor nucleus (Figures 4 and 5) could only occur via retrograde movement of GAA protein or AAV along the XII nerve. There is no evidence that the GAA molecule can be transported retrogradely along motor axons,23 and we conclude that retrograde movement of the viral vector resulted in transgene expression in XII motoneurons. The specific transport mechanisms enabling AAV to move from myofibers to motoneurons have not been established, but retrograde transport has been repeatedly observed after direct skeletal muscle delivery.10,24 Accordingly, intramuscular injection with AAV is a viable means of delivering genetic material to motoneurons. This approach has potential clinical application beyond Pompe disease including other neurodegenerative motor disorders such as spinal cord injury,25 spinal muscular atrophy,17 and amyotrophic lateral sclerosis.10
A growing body of evidence indicates that AAV9 is an optimal serotype for gene delivery to the central nervous system.26,27 The AAV9 serotype effectively crosses the blood brain barrier,26 and results in extensive and persistent neuronal transduction in multiple species including primates.28,29,30 In terms of retrograde movement, the degree of motoneuron transduction with AAV9 demonstrated here and in prior studies11 exceeds the ~1–15% transduction rates noted in prior studies with other serotypes.9,10,25 A recent study from Gransee et al.25 reported that AAV7 was more effective at targeted delivery to respiratory motor neurons as compared to AAV9 following intrathoracic delivery in rats. In that study, however, there was an ~10% motoneuron transduction rate, and this is lower than our prior report using AAV9.11
Histopathology after AAV-hGAA
The reduction in Gaa−/− mice myofiber histopathology following intramuscular AAV delivery was entirely consistent with prior reports.6,7,31 Thus, the novel feature of the current data set was the reversal of the motoneuron histopathology following intralingual AAV dosing. Specifically, after the AAV injection, GAA-positive motoneurons did not show the “ballooned” and vacuolar appearance which is typical of humans with Pompe disease8,32 and multiple Pompe animal models.8,18,21,33 Accordingly, the intralingual AAV-hGAA injections were able to “correct” a characteristic histopathologic feature of Pompe motoneurons. Neuronal glycogen accumulation can be detected in Gaa−/− mice as early as 3 weeks of age.12,21 Therefore, it is difficult to say if the absence of XII motoneuron histopathology in GAA-positive cells in adult mice represented prevention of ongoing glycogen accumulation or reversal of a pathologic condition which existed prior to the gene delivery. However, since prior work indicates a progressive increase in neuronal glycogen as Gaa−/− mice age,8 we suggest that the current results indicate a combination of prevention and reversal of neuronal glycogen accumulation. The functional impact of motoneuron histopathology in Pompe disease is not definitively established, but both neurophysiological and behavioral evidence from rodent models,8,18,21 and neurophysiological evidence from humans34,35,36 indicate impairments in the neural regulation of skeletal muscle contraction. Moreover, neuronal lysosomal dysfunction, if left untreated, may lead to neuronal death.37 Accordingly, AAV-mediated neuronal GAA expression could have dramatic clinical consequences, particularly in light of the limited success of ERT (see Significance section). It must be emphasized, however, that motor dysfunction in Pompe disease is likely to reflect an interaction between myopathy and neuropathy. We recently comprehensively reviewed the literature in this area and concluded that long-term functional improvements in motor deficits will require treatments targeting the entire motor unit (e.g., myofibers and associated motoneurons).38 In the current study, this was achieved by intralingual AAV-hGAA treatment.
Based on quantitative assessment of tissues obtained 4 months following intralingual delivery of AAV-hGAA, we conclude that GAA expression in motoneurons is persistent. This conclusion was reinforced by additional experiments confirming GAA-positive motoneurons up to 12 months following intralingual delivery of AAV1-hGAA delivery (Figure 9). A similarly persistent GAA expression in XII motoneurons was obtained 12 months following intralingual delivery of AAV9-hGAA but using the chicken β actin promoter (data not shown). An interesting point of consideration that was not addressed in the current study is whether or not “cross correction” can occur in the vicinity of AAV-infected cells.39 In other words, can GAA protein be secreted from an AAV-transduced cell to “correct” a neighboring cell via receptor-mediated endocytosis? Prior work has established that a localized AAV transduction can indeed produce more widespread delivery of the transgene product.40 Inspection of brainstem histology at 12 months postintralingual AAV1-hGAA delivery suggests that XII motoneuron cross correction should be evaluated in future studies. Specifically, clusters of GAA-positive motoneurons are evident, with some cells showing robustly positive GAA staining and neighboring cells showing reduced staining intensity (Figure 9).
Figure 9.

Representative examples of medullary acid-α glucosidase (GAA) immunostaining 12 months following intralingual injection with AAV1-hGAA in a Gaa−/− mouse. Positive immunostaining for GAA is restricted to the XII motor nucleus in the rostral medulla at 12 months following intralingual injection of (a) AAV1-hGAA. A representative section caudal to the XII motor nucleus with no GAA labeling is shown in (b). Panels c and d show examples of “clustering” of GAA-positive motoneurons which was typical at 12 months postinjection. The contrast in the histological appearance of GAA-positive versus negative cells is illustrated in (e–f). The GAA-positive motoneurons have a central nucleus without evidence of vacuoles. In contrast, cells not expressing GAA (*) in panels e and f have an eccentric nucleus with an enlarged vacuole filled soma. Scale bars = a: 50 µm; b: 200 µm.
Immune response
Consistent with prior reports, we observed a mild inflammatory reaction following intramuscular delivery of AAV.9 There was no histological evidence of inflammation within the central nervous system, but mild persistent T-cell infiltration was apparent in lingual myofibers. As expected, a humoral response to human GAA was detected in the AAV-hGAA–treated mice. This is most likely because the GAA protein was not species-specific since this immune response has not been a problem in humans treated with AAV-hGAA.16 In any case, the humoral response validates that GAA protein was being produced by the transduced tissues. The significance of the greater immune response to AAV1 as compared to the AAV9 vector (Figure 8) is not clear, but may indicate an elevation in systemic levels of GAA in the AAV1 group. The lack of a cell-mediated immune response in the “immune privileged” central nervous system (CNS) following intramuscular delivery of AAV-hGAA is consistent with previous studies.9 In contrast to intraparenchymal injections of AAV which induce innate and adaptive immune responses,41 the intramuscular delivery method avoids temporary breaching of the blood–brain barrier and prevents mechanical stress and release of viral particles in extracellular spaces. Towne et al.9 have suggested that these factors result in humoral or cell-mediated immune responses in the CNS. When a CNS immune response occurs, it is likely to result in the elimination of transgene-expressing cells.9
Significance
The current standard of treatment for Pompe disease is ERT in which recombinant GAA is delivered intravenously every 2 weeks.2 This approach has improved ventilator-free survival rates in patients with infantile-onset disease, but substantial pulmonary and tongue motor problems remain.1 For example, there is a 35% mortality rate in ERT-treated children, and many patients show progressive losses in pulmonary function after ERT is initiated.42 In addition, children receiving ERT typically have impaired hypoglossal motor function as indicated by weak and ineffective swallowing43 and speech disorders.5 Rohrbach et al.44 reported that neurological symptoms including impaired language development persisted despite a 44 months period of ERT in a Pompe child. Adults with late-onset Pompe disease also continue to have lingual weakness with both dysphagia and dysarthria despite ongoing ERT.13
The persistent motor symptoms following ERT are likely to occur because intravenously delivered GAA protein does not cross the blood–brain barrier to treat the motoneuron pathology which is prominent in both Pompe patients8 and animal models including mice8,18,21 and quail.33 Another consideration is that ERT itself could exacerbate motoneuron disease as a result of preserved muscle function in the absence of neural correction. The limited effectiveness of ERT indicates that a better approach is needed. The persistent GAA expression and reversal of the histopathologic appearance of tongue myofibers and motoneurons shown in the present study suggests the potential to use AAV-hGAA to treat both peripheral (myopathy) and central (neuropathy) components of motor weakness.9,26,45
We did not directly assess the impact of the AAV-hGAA therapy on tongue motor function, but the plethysmography results were nevertheless intriguing. The “PenH” measurement has been proposed to noninvasively assess airway resistance in mice by comparing airflows in the early versus late phases of expiration.19 When the early expiratory airflow peak becomes more prominent, as happens with airway constriction, the PenH value increases.19 On the other hand, some groups have questioned the validity of this approach,46 and conclusions based on PenH need to be validated using more invasive methods. Accordingly, we had no a priori hypothesis regarding PenH, and we interpret the data cautiously. It is nevertheless intriguing that the sham-treated Gaa−/− mice showed an elevation in PenH, and this value was significantly reduced following AAV9-hGAA but not AAV1-hGAA tongue injection. One interpretation of these data is that airway resistance is elevated in Gaa−/− mice secondary to pharyngeal neuromuscular dysfunction, and the AAV9 tongue injections improved the neuromotor control of the oropharyngeal airway. Further study will be needed to validate this hypothesis.
A phase I/II study administering rAAV2/1-CMV-hGAA to the diaphragm in children with Pompe disease is ongoing at our institution (ClinicalTrials.gov Identifier: NCT00976352). This trial was initiated following extensive preclinical work documenting a positive impact of diaphragm application of this vector in animal models.6,7 Initial results are encouraging and confirm that diaphragm delivery of rAAV1-hGAA is safe. In addition, significant increases in inspiratory tidal volume as well as the duration of unassisted breathing (i.e., without ventilator support) have occurred.16 In the present work, we confirmed in the Gaa−/− Pompe mouse model that the clinically used vector does indeed move retrogradely to transduce motoneurons. Based on the retrograde transport described here (Figure 4), we suggest that the AAV9 vector may be a prime candidate for use in future clinical studies since this serotype may increase the overall retrograde transduction of motoneurons. Lastly, direct intralingual injection of AAV-GAA vectors could be considered as an adjunct therapy to conventional ERT, which has minimal impact on tongue motor dysfunction in Pompe disease.5,47
Materials and Methods
All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Florida. 129SVE (wild-type) mice were obtained from Taconic, Albany, NY. Gaa−/− mice, originally developed by Raben et al.12 were outbred to a 129SVE background (Taconic). An intralingual injection of vector or LR was administered at 8 weeks of age. Animals were weighed prior to injection, then 1 and 4 months after injection. Immunoglobulin G levels were performed 6 weeks and 4 months posttreatment. Plethysmography was performed at 4 months postdose and immediately prior to sacrifice. Animal tissues were processed for molecular, biochemical, and histological studies 4 months posttreatment. A subset of treated animals were maintained and sacrificed at 12 months of age to assess for persistence of GAA expression.
AAV vectors. Single-stranded AAV vectors encoding human GAA (hGAA), driven by the CMV promoter, were injected into the tongue at a vector dose of 1 × 1011 vg/ml. This concentration was determined by prior experience with tongue injections using AAV serotype 9 (AAV9).11 Two vector serotypes, were used for comparison: AAV serotype 1 (AAV1) which is the vector currently in use in a currently active clinical trial (NCT00976352),16 and AAV9, a vector considered to have greater neuronal transduction.11,30 All vectors were generated and titered at the University of Florida Powell Gene Therapy Center Vector Core Laboratory. Vectors were purified by iodixanol gradient centrifugation and anion-exchange chromatography as described previously.48 Final formulations of AAV1-CMV-hGAA and AAV9-CMV-hGAA were in LR solution.
In vivo vector administration. The Gaa−/− mice were created by disruption of exon 6 as previously described. 12 Animals were 8 weeks of age at the time of dosing.49 Twelve 129SVE mice were used as controls. Forty Gaa−/− mice were randomly assigned to one of the three groups by a blinded investigator: an AAV1-hGAA–injected group, an AAV9-hGAA–injected group, a group injected with LR. An equal number of male and female mice were randomized into each group. Mice were anesthetized with 3% isoflurane in oxygen administered via a nose cone. All mice were placed on a heating pad during the injections to maintain body temperature at 37 °C. A surgical microscope was used to visualize the injection site and to administer the sham or vector injection. The tongue was gently retracted from the mouth using blunt forceps. A single injection of 30 µl of LR (sham injection) or vector was directly injected into the left side of the base of the tongue lateral to the lingual frenulum. The needle was held at a 45 degree angle and inserted to a depth of 2 mm. PE50 tubing was placed over the needle to insure that the depth of injection was uniform in each mouse. Sham- and vector-injected mice shared the same cages to minimize variability. These sham-injected mice were used as a control group for studies of ventilation, biochemical, molecular, and histological studies.
Vector pharmacology
Genomic DNA extraction and real-time PCR. Four months following intralingual injections, AAV genome copies using PCR were measured in the tongue, liver, brain, medulla, and spinal cord of wild-type and Gaa−/− mice (N = 3 in each group). Tissues were harvested in a manner that prevented crosscontamination, snap frozen in liquid nitrogen, and stored at −80 °C until genomic DNA (gDNA) was extracted. All tissue was divided equally, with half used for PCR and half for GAA activity assay (see below). The tongue was harvested and divided into anterior and posterior portions. gDNA was isolated using a DNeasy blood and tissue kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. gDNA concentrations were determined using the NanoDrop system (Wilmington, DE). AAV genome copies present in gDNA were quantified by real-time PCR using an ABI 7900 HT sequence detection system (Applied Biosystems, Carlsbad, CA) according to the manufacturer's instructions, and results were analyzed using the Sequence Detection Systems (SDS) 2.3 software. Briefly, primers and probe were designed to the SV40 poly-A region of the AAV vector as previously described.7 A standard curve was performed using plasmid DNA containing the same SV40 poly-A target sequence. PCR reactions contained a total volume of 100 µl and were run at the following conditions: 50 °C for 2 minutes, 95 °C for 10 minutes, and 45 cycles of 95 °C for 15 seconds and 60 °C for 1 minute. DNA samples were assayed in triplicate. In order to assess PCR inhibition, the third replicate was spiked with plasmid DNA at a ratio of 100 copies/µg gDNA. If this replicate was greater than 40 copies/µg gDNA, then the results were considered acceptable. If a sample contained greater than or equal to 100 copies/µg gDNA, it was considered positive for vector genomes. If a sample contained less than 100 copies/µg gDNA, it was considered negative for vector genomes. If less than 1 µg of gDNA was analyzed, the vector copy number reported was normalized per µg gDNA and the plasmid spike-in was reduced to maintain the ratio of 100 copies/µg gDNA. Data were reported as AAV genome copies per µg total genomic DNA ± SD.
GAA activity assay. Tongue, liver, brain, medulla, and spinal cord were harvested from 129 mice and Gaa−/− mice intralingually injected with either LR or AAV-GAA. Tissues were immediately harvested, frozen in liquid N2 and maintained at −80 °C until biochemical analyses were performed. Briefly, tissues were homogenized in water containing a complete protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN) and subjected to three freeze-thaw cycles. Homogenates were centrifuged at 14,000 rpm for 10 minutes at 4 °C and the resulting supernatant was assayed for GAA activity by measuring cleavage of 4-methylumbelliferyl-α-D-glucopyranoside after incubation for 1 hour at 37 °C. Protein concentration was measured using the Bio-Rad DC protein assay kit per manufacturer's instructions. Data were expressed relative to values measured in untreated GAA tissue levels (% control).
Immunoglobulin G quantification. Enzyme-linked immunosorbent assay for IgG1 and IgG2 were developed and optimized.50 Immulon 4HBX 96-well plates (Thermo Fisher Scientific, Waltham, MA) were coated with rhGAA protein and incubated overnight at 4 °C. The following standards IgG1κ (4,000 to 62.5 ng/ml), IgG2a (1,000 to 15 ng/ml) were coated overnight at 4 °C at twofold dilutions. Experimental mouse plasma at a 1:50 dilution was used for IgG1 and IgG2a. Plasma samples were incubated for 2 hours at room temperature. The secondary detection antibodies rat anti-mouse IgG1 heavy chain-HRP (AbD Serotec, UK) or goat antibody to mouse IgG2a- HRP (Abcam, MA) were incubated for 2 hours at 37 °C. Plates were allowed to develop for 5–10 minutes in a solution containing Sigmafast OPD tablets (Sigma, MO) for color production. Plates were washed three times between procedures with Tris wash buffer. A BD-colorimetric plate reader was used to read the 96-well clear ELISA plates.
Brainstem histology. Mice were anesthetized with isoflurane and urethane (1.5 g/kg) and euthanized via systemic perfusion with 4% paraformaldehyde. Tissues were harvested 4 months after injection of sham or vector. The brainstem and spinal cord tissues were extracted and postfixed by immersion in 4% paraformaldehyde (N = 4 animals per group) for 24 hours then transferred to 70% ethanol until processing for paraffin embedding. Paraffin serial sections (5 μm) were stained with PAS, hematoxylin and eosin staining and by immunohistochemistry to GAA. GAA IHC coupled with a Vectastain ABC secondary detection kit and (3,3′-Diaminobenzidine) DAB was performed. The tissue was incubated overnight in primary antibody against GAA, 1:2,000 (rabbit polyclonal GAA antibody (Covance, Emeryville, CA). On the following day, the tissue was washed in PBS, incubated in a biotinylated anti-rabbit IgG secondary antibody, 1:200 (Vector Laboratories, Burlingame, CA) and coupled with a Vectastain ABC Kit and DAB for bright field microscopy. In addition, inflammatory cell immunophenotype was determined with monoclonal antibodies that recognize CD3. CD3 is a protein complex important in transducing the signal that initiates a T-cell activation and differentiation pathway. The presence of CD3 indicates activation of a T-cell inflammatory response.
Tongue histology. After perfusion, the tongue was removed, postfixed in 4% paraformaldehyde for 24 hours then transferred to 70% ethanol until processing. Paraffin serial sections (5 μm) were stained with PAS, hematoxylin and eosin staining and by immunohistochemistry to GAA as described above. Inflammatory cell immunophenotype was assessed using monoclonal antibodies that recognize CD3.
Microscopy and quantitative analyses. Brightfield photographs were taken with a Zeiss AxioPhot microscope and an AxioCam HRc digital camera linked to a PC. By using brightfield microscopy, GAA-positive XII motoneurons with visible nuclei were counted at ×10 magnification in every 10th 5µm transverse section of the medulla from each animal. Only cells with a visible nucleus were counted.
Ventilation. These studies were undertaken to determine the impact of vector injection to the tongue on ventilation. Thus, ventilation was quantified by a blinded investigator using whole-body plethysmography in unrestrained, unanesthetized mice as previously described.8 Mice were placed inside a 3.5” × 5.75” Plexiglas chamber which was calibrated with known airflow and pressure signals before data collection. Data were collected in 10-second intervals and respiratory volumes including tidal volume and minute ventilation were calculated as described previously.8 During both a 30 minutes acclimation period and subsequent 30–60 minutes baseline period, mice were exposed to normoxic air (21% O2, 79% N2). At the conclusion of the baseline period, the mice were exposed to a brief respiratory challenge which consisted of a 10-minute hypercapnic exposure (7% CO2, balance O2). Experiments were conducted using wild-type 129SVE mice and Gaa−/− mice that had received tongue injection of sham or AAV-GAA. Data were collected 4 months postinjection.
Statistical analysis. The number of transduced neuronal cells was compared between AAV9-hGAA– and AAV1-hGAA–injected Gaa−/− mice using an unpaired t-test. A one-way analysis of variance was used for the analysis of PCR and GAA activity assay quantification, with statistical significance considered if P < 0.05. A two-way ANOVA was used to compare body weight between groups. The ventilation data were compared between all groups using a one-way analysis of variance, with a post hoc Tukey HSD analysis used for multiple pairwise comparisons. Data were considered to be statistically different when P < 0.05.
Acknowledgments
The authors gratefully acknowledge the University of Florida Powell Gene Therapy Center Vector Core Laboratory for production and titering of recombinant adeno-associated virus vectors and the University of Florida Toxicology Core (Thomas Conlon and Kirsten Erger) for vector genome analysis. This work was supported by the Parker B. Francis Foundation (MKE) and the NIH: 201HD052682-06A1 (DDF, BJB), MDA 216676 (DJF), and PO1 HL59412 (BJB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. B.J.B., D.D.F., The Johns Hopkins University, and the University of Florida could be entitled to patent royalties for inventions described in this manuscript. The authors declared no conflict of interest.
References
- Fuller DD, ElMallah MK, Smith BK, Corti M, Falk DJ, Byrne BJ. Pompe disease and the respiratory system. Res Physiol Neurobiol. 2013;189:241–249. doi: 10.1016/j.resp.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne BJ, Falk DJ, Pacak CA, et al. Pompe disease gene therapy. Hum Mol Genet. 2011;20 R1:R61–8. doi: 10.1093/hmg/ddr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HN, Muller CW, Lin M, et al. Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia. 2010;25:277–83. doi: 10.1007/s00455-009-9252-x. [DOI] [PubMed] [Google Scholar]
- Margolis ML, Howlett P, Goldberg R, Eftychiadis A, Levine S. Obstructive sleep apnea syndrome in acid maltase deficiency. Chest. 1994;105:947–9. doi: 10.1378/chest.105.3.947. [DOI] [PubMed] [Google Scholar]
- Muller CW, Jones HN, OGrady G, Suarez AH, Heller JH, Kishnani PS. Language and speech function in children with infantile Pompe disease. J Ped Neurol. 2009;7:147–156. [Google Scholar]
- Mah C, Pacak CA, Cresawn KO, et al. Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol Ther. 2007;15:501–7. doi: 10.1038/sj.mt.6300100. [DOI] [PubMed] [Google Scholar]
- Mah CS, Falk DJ, Germain SA, et al. Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Mol Ther. 2010;18:502–10. doi: 10.1038/mt.2009.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRuisseau LR, Fuller DD, Qiu K, et al. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci U S A. 2009;106:9419–24. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towne C, Schneider BL, Kieran D, Redmond DE, Jr, Aebischer P. Efficient transduction of non-human primate motor neurons after intramuscular delivery of recombinant AAV serotype 6. Gene Ther. 2010;17:141–6. doi: 10.1038/gt.2009.119. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–42. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- ElMallah MK, Falk DJ, Lane MA, et al. Retrograde gene delivery to hypoglossal motoneurons using adeno-associated virus serotype 9. Hum Gene Ther Methods. 2012;23:148–56. doi: 10.1089/hgtb.2012.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Nagaraju K, Lee E, et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 1998;273:19086–92. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- Dubrovsky A, Corderi J, Lin M, Kishnani PS, Jones HN. Expanding the phenotype of late-onset Pompe disease: tongue weakness: a new clinical observation. Muscle Nerve. 2011;44:897–901. doi: 10.1002/mus.22202. [DOI] [PubMed] [Google Scholar]
- Hobson-Webb LD, Jones HN, Kishnani PS. Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement. Neuromuscul Disord. 2013;23:319–23. doi: 10.1016/j.nmd.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Chien YH, Lee NC, Thurberg BL, et al. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics. 2009;124:e1116–25. doi: 10.1542/peds.2008-3667. [DOI] [PubMed] [Google Scholar]
- Smith BK, Collins S, Conlon T, Mah C, Lawson LA, Martin D, et al. Phase I/II trial of AAV1-GAA gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther. 2013. [DOI] [PMC free article] [PubMed]
- Benkhelifa-Ziyyat S, Besse A, Roda M, et al. Intramuscular scAAV9-SMN injection mediates widespread gene delivery to the spinal cord and decreases disease severity in SMA mice. Mol Ther. 2013;21:282–90. doi: 10.1038/mt.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KZ, Qiu K, Sandhu MS, et al. Hypoglossal neuropathology and respiratory activity in pompe mice. Front Physiol. 2011;2:31. doi: 10.3389/fphys.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomask M. Further exploration of the Penh parameter. Exp Toxicol Pathol. 2006;57 suppl 2:13–20. doi: 10.1016/j.etp.2006.02.014. [DOI] [PubMed] [Google Scholar]
- van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–40. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Taksir T, Fidler J, et al. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J Neuropathol Exp Neurol. 2008;67:803–18. doi: 10.1097/NEN.0b013e3181815994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick B, Ghosh A, Yue Y, Long C, Duan D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther. 2007;14:1605–9. doi: 10.1038/sj.gt.3303029. [DOI] [PubMed] [Google Scholar]
- Raben N, Danon M, Gilbert AL, et al. Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genet Metab. 2003;80:159–69. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Towne C, Raoul C, Schneider BL, Aebischer P. Systemic AAV6 delivery mediating RNA interference against SOD1: neuromuscular transduction does not alter disease progression in fALS mice. Mol Ther. 2008;16:1018–25. doi: 10.1038/mt.2008.73. [DOI] [PubMed] [Google Scholar]
- Gransee HM, Zhan WZ, Sieck GC, Mantilla CB. Targeted delivery of TrkB receptor to phrenic motoneurons enhances functional recovery of rhythmic phrenic activity after cervical spinal hemisection. PLoS One. 2013;8:e64755. doi: 10.1371/journal.pone.0064755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Salegio EA, San Sebastian W, Kells AP, Foust K, Bringas J, et al. AAV9 transduction in the central nervous system of non-human primates. Hum Gene Ther. 2012;23:382–389. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan AK, Duque S, Foust KD, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther. 2011;19:1971–80. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Salegio EA, San Sebastian W, et al. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum Gene Ther. 2012;23:382–9. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredsson FP, Rising AC, Mandel RJ. AAV9: a potential blood-brain barrier buster. Mol Ther. 2009;17:403–5. doi: 10.1038/mt.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah C, Cresawn KO, Fraites TJ, Jr, et al. Sustained correction of glycogen storage disease type II using adeno-associated virus serotype 1 vectors. Gene Ther. 2005;12:1405–9. doi: 10.1038/sj.gt.3302550. [DOI] [PubMed] [Google Scholar]
- Thurberg BL, Lynch Maloney C, Vaccaro C, et al. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest. 2006;86:1208–20. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Yang HW, Pennybacker M, et al. Clinical and metabolic correction of pompe disease by enzyme therapy in acid maltase-deficient quail. J Clin Invest. 1998;101:827–33. doi: 10.1172/JCI1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng YT, Su WJ, Hou JW, Huang SF. Infantile-onset glycogen storage disease type II (Pompe disease): report of a case with genetic diagnosis and pathological findings. Chang Gung Med J. 2004;27:379–84. [PubMed] [Google Scholar]
- Willemsen MA, Jira PE, Gabreëls FJ, van der Ploeg AT, Smeitink JA. Three hypotonic neonates with hypertrophic cardiomyopathy: Pompe's disease. Ned Tijdschr Geneeskd. 1998;142:1388–92. [PubMed] [Google Scholar]
- Gambetti P, DiMauro S, Baker L. Nervous system in Pompe's disease. Ultrastructure and biochemistry. J Neuropathol Exp Neurol. 1971;30:412–30. doi: 10.1097/00005072-197107000-00008. [DOI] [PubMed] [Google Scholar]
- Bellettato CM, Scarpa M. Pathophysiology of neuropathic lysosomal storage disorders. J Inherit Metab Dis. 2010;33:347–62. doi: 10.1007/s10545-010-9075-9. [DOI] [PubMed] [Google Scholar]
- Fuller DD, ElMallah MK, Smith BK, et al. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol. 2013;189:241–9. doi: 10.1016/j.resp.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung SD, Reddy RG, Whitley CB, McIvor RS. Enzymatic correction and cross-correction of mucopolysaccharidosis type I fibroblasts by adeno-associated virus-mediated transduction of the alpha-L-iduronidase gene. Hum Gene Ther. 1999;10:2163–72. doi: 10.1089/10430349950017158. [DOI] [PubMed] [Google Scholar]
- Skorupa AF, Fisher KJ, Wilson JM, Parente MK, Wolfe JH. Sustained production of beta-glucuronidase from localized sites after AAV vector gene transfer results in widespread distribution of enzyme and reversal of lysosomal storage lesions in a large volume of brain in mucopolysaccharidosis VII mice. Exp Neurol. 1999;160:17–27. doi: 10.1006/exnr.1999.7176. [DOI] [PubMed] [Google Scholar]
- Reimsnider S, Manfredsson FP, Muzyczka N, Mandel RJ. Time course of transgene expression after intrastriatal pseudotyped rAAV2/1, rAAV2/2, rAAV2/5, and rAAV2/8 transduction in the rat. Mol Ther. 2007;15:1504–11. doi: 10.1038/sj.mt.6300227. [DOI] [PubMed] [Google Scholar]
- Chakrapani A, Vellodi A, Robinson P, Jones S, Wraith JE. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J Inherit Metab Dis. 2010;33:747–50. doi: 10.1007/s10545-010-9206-3. [DOI] [PubMed] [Google Scholar]
- van Gelder CM, van Capelle CI, Ebbink BJ, et al. Facial-muscle weakness, speech disorders and dysphagia are common in patients with classic infantile Pompe disease treated with enzyme therapy. J Inherit Metab Dis. 2012;35:505–11. doi: 10.1007/s10545-011-9404-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach M, Klein A, Köhli-Wiesner A, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis. 2010;33:751–7. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]
- Lu YY, Wang LJ, Muramatsu S, et al. Intramuscular injection of AAV-GDNF results in sustained expression of transgenic GDNF, and its delivery to spinal motoneurons by retrograde transport. Neurosci Res. 2003;45:33–40. doi: 10.1016/s0168-0102(02)00195-5. [DOI] [PubMed] [Google Scholar]
- Mitzner W, Tankersley C. Interpreting Penh in mice. J Appl Physiol (1985) 2003;94:828–31; author reply 831. doi: 10.1152/japplphysiol.00815.2002. [DOI] [PubMed] [Google Scholar]
- Jones HN, Muller CW, Lin M, et al. Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia. 2010;25:277–83. doi: 10.1007/s00455-009-9252-x. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Zolotukhin I, et al. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–67. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- Obernier JA, Baldwin RL. Establishing an appropriate period of acclimatization following transportation of laboratory animals. ILAR J. 2006;47:364–9. doi: 10.1093/ilar.47.4.364. [DOI] [PubMed] [Google Scholar]
- Elder ME, Nayak S, Collins SW, et al. B-Cell depletion and immunomodulation before initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2013;163:847–54.e1. doi: 10.1016/j.jpeds.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]