

Abstract
Huntington's disease (HD) is a fatal neurological disorder caused by a CAG repeat expansion in the HTT gene, which encodes a mutant huntingtin protein (mHTT). The mutation confers a toxic gain of function on huntingtin, leading to widespread neurodegeneration and inclusion formation in many brain regions. Although the hallmark symptom of HD is hyperkinesia stemming from striatal degeneration, several other brain regions are affected which cause psychiatric, cognitive, and metabolic symptoms. Additionally, mHTT expression in peripheral tissue is associated with skeletal muscle atrophy, cardiac failure, weight loss, and diabetes. We, and others, have demonstrated a prevention of motor symptoms in HD mice following direct striatal injection of adeno-associated viral vector (AAV) serotype 1 encoding an RNA interference (RNAi) construct targeting mutant HTT mRNA (mHTT). Here, we expand these efforts and demonstrate that an intrajugular vein injection of AAV serotype 9 (AAV9) expressing a mutant HTT-specific RNAi construct significantly reduced mHTT expression in multiple brain regions and peripheral tissues affected in HD. Correspondingly, this approach prevented atrophy and inclusion formation in key brain regions as well as the severe weight loss germane to HD transgenic mice. These results demonstrate that systemic delivery of AAV9-RNAi may provide more widespread clinical benefit for patients suffering from HD.
Introduction
Huntington's disease (HD) is a fatal, genetic neurodegenerative disorder that results from a CAG repeat expansion in the HTT gene on chromosome 4.1 The encoded protein, mutant huntingtin (mHTT), contains a polyglutamine expansion on its N-terminus and takes on a toxic gain of function. The characteristic hallmarks of HD neuropathology include mHTT-containing cellular inclusion bodies, pronounced atrophy, and cellular degeneration in the brain.1,2,3 Neuropathology is most pronounced in the striatum, where up to 80% cell loss can be seen at the end stages of disease.4 This area of the brain is involved in the initiation, execution, and refinement of motor behaviors. Accordingly, a hallmark of HD disease progression is chorea, characterized by hyperkinetic movements of a patient's head, trunk, and limbs.5
Huntingtin is ubiquitously expressed, and it has become apparent that many other regions of the brain are also affected in HD, including the regions that comprise the rest of the basal ganglia (globus pallidus, subthalamic nucleus, substantia nigra, and thalamus) and also the frontal and motor cortex, hippocampus, hypothalamus, and cerebellum.3,4,6,7,8 Accordingly, it is not surprising that HD patients experience a wide array of symptomology potentially associated with these brain regions including cognitive deficits, psychiatric changes, and a severe metabolic phenotype that includes dramatic weight loss and cachexia at end stages of disease.9,10,11 Moreover, patients suffer from a variety of peripheral abnormalities including skeletal muscle wasting, reduced cardiac output and heart failure, liver dysfunction, insulin dysregulation, and decreased testosterone levels.12 Although HD is caused by a single gene mutation, a complex interaction of diseased tissues/systems leads to complex phenotypes for which the pathophysiological processes are not yet fully elucidated. It is not completely understood whether peripheral pathologies result secondarily to brain dysfunction, whether they are driven by mHTT expression specifically in those tissues or whether peripheral pathologies drive central dysfunction. However, these cumulative data suggest that strategies to treat HD may have a more profound therapeutic outcome if a broad range of tissues are targeted.
This year marks the 20th anniversary of the discovery of the HD-causing gene. Because the underlying genetic mutation in HD is known and the course of disease is well characterized, this provides the unique opportunity to intercede therapeutically prior to the onset of symptom manifestation. Therapies that reduce expression of the diseased gene will likely have the greatest success; the rationale being that preventing expression of the toxic protein will prevent the development of its numerous downstream cytotoxic effects. Prior studies have shown that adeno-associated viral vectors (AAVs) can successfully deliver mutant HTT mRNA (mHTT)-specific RNA interference (RNAi) constructs following direct injection into the striatum.13,14,15,16,17 Reducing mHTT expression by ~75% in the striatum of HD transgenic mice significantly prevents motor deficits and extends life span but does not affect weight loss.16 Similar therapeutic benefits have been reported using antisense oligonucleotides to reduce HTT expression by ~75% in the HD transgenic mouse brain following an injection into the lateral ventricle.18 Ongoing studies in our laboratory are assessing the safety and efficacy of a viral-mediated RNAi approach in non-human primates as a critical step toward advancing this approach into a Phase 1 clinical trial.19 Many of the studies to date using AAVs to deliver therapeutic agents, including RNAi, in animal models of HD have relied on stereotaxic-guided injections of serotypes 1, 2, or 5 directly into the striatum.19,20 While these studies highlight the utility of these AAV serotypes to confer long-term suppression of mHTT in the striatum, the widespread pathology observed in HD patients and mouse models of the disease makes a global delivery strategy an appealing approach.
AAV serotype 9 (AAV9) crosses the blood–brain barrier and transduces neurons and glia in the brain, as well as a variety of peripheral tissues, following a systemic, intravascular injection.21 AAV9 is currently being investigated as a tool for a variety of diseases that would benefit from a widespread gene therapy delivery system. In the current study, we performed a series of experiments using wild-type (WT) and HD transgenic mice to assess whether jugular vein administration of AAV9 expressing a mHTT-specific RNAi construct: (i) transduces the specific brain and peripheral tissues affected in HD, (ii) significantly reduces mHTT expression in brain and peripheral tissues, and (iii) has the capacity to prevent central and peripheral pathologies characteristic of HD.
Results
Jugular vein delivery of AAV9 results in the transduction of numerous tissues affected in HD
Although systemically administered AAV9 is known to have a broad tropism for peripheral tissues and crosses the blood–brain barrier to transduce cells in the central nervous system (CNS),21 it is unclear whether there is a natural tropism for the peripheral organs and brain regions that are particularly affected in HD. In our first experiment, we addressed this question by injecting C57BL/6J WT mice with a recombinant AAV9 encoding enhanced green fluorescent protein (AAV9-eGFP) into the right jugular vein (Figure 1a,b). AAV9-eGFP was diluted to a working titer of 3 × 1012 vg/ml, and the overall dose of AAV9 injected was 3.3 × 1010 viral genomes per gram body weight (vg/gram bw, Table 1). Transduction patterns in several peripheral organs and brain regions were observed at 4 weeks after injection. We found that AAV9 transduces many of the key brain regions most heavily affected in HD, evidenced by robust eGFP expression throughout the cortex, striatum, and hypothalamus (Figure 2a–d). Medium spiny neurons of the striatum, which are particularly vulnerable to mHTT toxicity, showed a high level of transduction (Figure 2c). Other brain areas also known to exhibit pathology in HD, including the globus pallidus, thalamus, hippocampus (Figure 2e–g), and cerebellum (not shown) also showed high levels of eGFP expression. In the HD brain, both neurons and glia are affected by mHTT expression. Therefore, we used double-label immunofluorescence staining to identify the specific cell types transduced. In all brain regions examined, AAV9 transduced NeuN-positive neurons (Figure 3a–c), glial fibrillary acidic protein (GFAP)–positive astrocytes (Figure 3d–f), and CD31-positive vascular endothelial cells (Figure 3j–l) but not Iba-positive microglia (Figure 3g–i) or myelin basic protein–positive oligodendrocytes (not shown).
Figure 1.

Adeno-associated viral vector serotype 9 (AAV9) design and delivery. (a) Wild-type (WT) and Huntington's disease transgenic mice were injected with AAV9 containing either a bicistronic construct including a U6-driven micro RNA (miRNA) (either mi2.1 or miSCM) and a cytomegalovirus (CMV)-driven enhanced green fluorescent protein (eGFP) reporter gene (top) or a monocistronic construct consisting of a CMV-driven eGFP (bottom). miRNA constructs consisted of mi2.1, a human mutant HTT-specific miRNA, and miSCM, a control miRNA. (b) Mice were anesthetized and placed in a recumbent position, and a small incision was made lateral to the ventral midline, from the pectoral muscle to the lower neck. The jugular vein was exposed with blunt dissection. Viral vectors, saline, and/or 25% mannitol were delivered into systemic circulation through a direct injection into the jugular vein. Intrajugular injections were made using a 30-gauge needle after passing through the pectoral muscle. ITR, inverted terminal repeat. pA, poly-A tail.
Table 1. Experimental details.
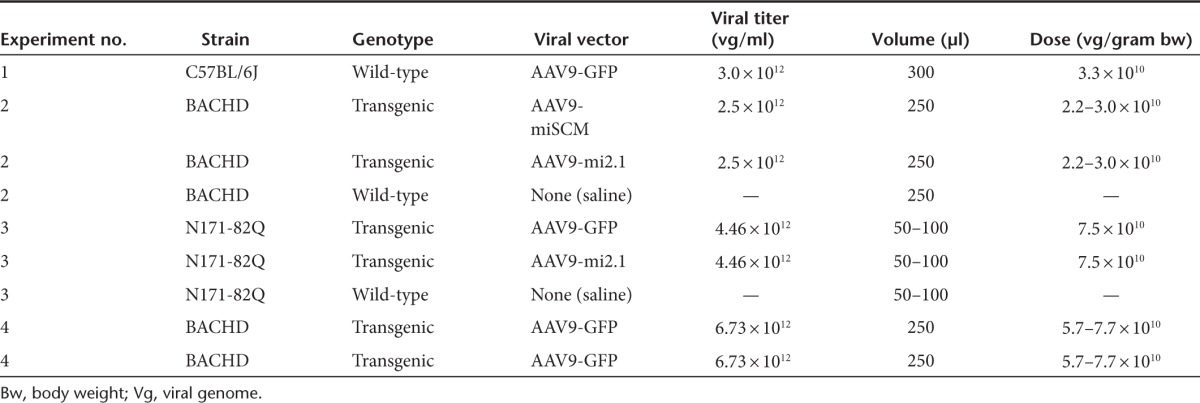
Figure 2.

Intravenous adeno-associated viral vector serotype 9 (AAV9) transduces numerous tissues affected in Huntington's disease (HD). Enhanced green fluorescent protein (eGFP) expression is shown for central and peripheral tissues following a single intrajugular vein injection of AAV9-eGFP in wild-type mice. Mice were injected with vector at 9 weeks of age and sacrificed at 26 weeks. Forty-micrometer brain sections were immunolabeled using an antibody against eGFP and a fluorescent secondary antibody. eGFP expression was found in widespread tissues affected in HD including (a) cortex, (b,c) striatum, (d) hypothalamus (arcuate nucleus and median eminence shown here), (e) globus pallidus, (f) thalamus, and (g) hippocampus. (c) Striatal medium spiny neurons, which are particularly affected in HD, showed robust eGFP expression. Both neurons and astrocytes were transduced throughout all brain regions. Additionally, AAV9 demonstrated a broad tropism for peripheral tissues that are affected in HD, including (h) skeletal muscle (quadriceps), (i) liver, (j) heart, (k) pancreas, (l) stomach, and (m) testis. Images were obtained using a confocal microscope, with a,b,e,f at a magnification of ×40; d,g,h at a magnification of ×20, and i–m at a magnification of 10×. Bar = 100 µm. AAV9-eGFP, AAV9 encoding enhanced green fluorescent protein.
Figure 3.

Intravenous adeno-associated viral vector serotype 9 (AAV9) transduces neurons, astrocytes, and vessels but not microglia in the brain. Following intrajugular vein administration of viral vectors at the titers/doses used in this study, double-label immunofluorescence demonstrates colocalization of (a,d,g,j) enhanced green fluorescent protein (eGFP)–positive cells: (b,c) NeuN-labeled neurons, (e,f) glial fibrillary acidic protein (GFAP)–labeled astrocytes, (k,l) CD31-labeled vascular endothelial cells but not (h,i) Iba1-labeled microglia in the mouse brain. Bar = 25 µm.
Although not as well characterized as the CNS pathology in HD, peripheral tissues are also affected by toxic mutant huntingtin expression. HD patients show dramatic muscle wasting, cardiac tissue atrophy, and altered hepatic function.12 We found particularly high eGFP expression in some of these affected peripheral organs, including skeletal and cardiac muscle, liver, pancreas, stomach, and testis (Figure 2h–m). Following the brain, testes show the second highest levels of mHTT expression in the body and male patients often show reduced levels circulating testosterone levels in late stages.22 As shown by immunofluorescence staining, eGFP-positive cells in the testes colocalized exclusively with the testosterone-producing (17-α hydroxylase positive) Leydig cells (Supplementary Figure S1a–d). Pancreatic exocrine and endocrine cells (islets of Langerhans) both express mutant huntingtin,23 and both were transduced by AAV9 (Supplementary Figure S1e–l). However, within the islets themselves, eGFP colocalized with insulin-producing β cells (Supplementary Figure S1e–h) but not with the glucagon-producing α cells (Supplementary Figure S1i–l). This proof-of-concept biodistribution study established a rationale for using AAV9 as a potent vehicle to deliver therapeutic agents in preclinical studies using mouse models of HD.
Intrajugular administration of AAV9-RNAi reduces mHTT mRNA in peripheral tissues and reduces serum LDH levels in BACHD transgenic mice
Prior work has demonstrated therapeutic efficacy following direct striatal injection of AAV1 vectors expressing mutant and/or WT HTT-specific miRNAs.13,16,24 In the current study, a sequence that specifically silences a region in exon 2 of human mHTT (first described by Harper et al.) and a control sequence (SCM) designed to have low off-targeting potential25 were cloned into an artificial miRNA backbone based on miR-30 and subsequently cloned into AAV9 to generate AAV9-mi2.1 and AAV9-miSCM, respectively. Expression of the mHTT-specific miRNA (mi2.1) and the control, scrambled miRNA (miSCM) was driven by a mouse U6 promoter, and eGFP was driven from a cytomegalovirus promoter (Figure 1a) to allow for assessment of vector distribution following injection into the jugular vein (Figure 1b).
We utilized the BACHD model in this first therapeutic study, a transgenic mouse that expresses full-length human mutant huntingtin, which is driven by the human huntingtin promoter.26 At 9 weeks of age, male and female transgenic mice were injected with 250 µl of either AAV9-mi2.1 (n = 11) or AAV9-miSCM (n = 10) into the jugular vein. Six minutes prior to vector injection, all mice received an injection of 25% mannitol (2 mg/g body weight) as this pretreatment strategy has previously been shown to temporarily open the blood–brain barrier and increase transduction in brain following a systemic injection of AAV9 and other AAV serotypes.27,28,29 Both viral preps were diluted to a working titer of 2.5 × 1012 vg/ml, and the overall dose of AAV9 injected ranged from 2.0–3.0 × 1010 vg/gram bw (Table 1). For comparison, a third group of WT littermates (n = 12) were injected with 25% mannitol followed by 250 µl of 0.9% sterile saline. Behavioral and physiological measures were taken until the mice reached 29 weeks of age, at which time they were sacrificed and their tissues analyzed.
Analysis of eGFP expression in transgenic BACHD mice showed a similar pattern of transduction compared with the WT mice assessed in experiment 1. Peripheral organs showed particularly high transduction levels in liver (Figure 4b,c), heart (Figure 4f,g), and skeletal muscle (Figure 4j,k). No eGFP expression was detected in tissues from saline-injected controls (Figure 4d,h,l). Using quantitative real-time polymerase chain reaction (qPCR), we assessed relative expression levels of mutant huntingtin, between mi2.1, miSCM and saline-treated animals. qPCR showed that relative mHTT mRNA expression was significantly reduced in key peripheral tissues affected in HD—15% in liver (Figure 4a; P < 0.01), 14% in heart (Figure 4e; P < 0.05), and 23% in quadriceps (Figure 4i; P < 0.05), indicating that this therapeutic strategy may be capable of preventing mHTT-related peripheral tissue pathologies. mHTT expression in testis was similar between mi2.1- and miSCM-injected mice (P > 0.05), likely owing to the fact that whole biopsies of testis were collected for qPCR analysis, and GFP expression is specifically restricted to the testosterone-producing Leydig cells. Future studies will use laser-capture microdissection to look at mHTT expression specifically in these cells.
Figure 4.

Adeno-associated viral vector serotype 9 (AAV9)–delivered microRNAs reduce mutant huntingtin protein (mHTT) expression in BACHD mouse peripheral tissues. Quantitative polymerase chain reaction demonstrated a significant reduction in relative mHTT messenger RNA (mRNA) levels in the (a) liver (15% knockdown; P < 0.01), (e) heart (14% knockdown; P < 0.05), and (i) quadriceps (23% knockdown; P < 0.05) of transgenic BACHD mice following AAV9-mi2.1 treatment compared with AAV9-miSCM treatment. Representative images of enhanced green fluorescent protein (eGFP) expression in the liver, heart, and quadriceps are shown for (b,f,j) miSCM and (c,g,k) mi2.1 and (d,h,l) saline-injected, wild-type controls. (m) The mean values for mHTT mRNA were consistently lower in mi2.1-treated mice relative to controls although the difference was not statistically significant (P > 0.05 for each brain region). However, there was a trend for mHTT reduction in the hypothalamus (P = 0.058), hippocampus (P = 0.063), and thalamus (P = 0.090). All images were taken at a magnification of ×20 (Bar = 100 µm). Values indicated in a,e,i,m are the mean ± SEM. *P < 0.05, **P < 0.01.
BACHD mice showed eGFP-positive neurons and glia in the striatum, cortex, thalamus, hippocampus, and hypothalamus, among other regions (not shown). While qPCR analysis demonstrated reduced mean levels of mHTT mRNA in mi2.1-treated animals in the striatum (12.8% reduction), frontal cortex (13.2%), thalamus (12.7%), hypothalamus (21.8%), and hippocampus (19.7%) compared with controls, values for each region did not reach statistical significance (P > 0.05; Figure 4m). However, there was a statistical trend for a reduction in mHTT mRNA in thalamus (P = 0.090), hypothalamus (P = 0.058), and hippocampus (P = 0.063) of mi2.1-treated mice. Coronal brain sections from animals in each group showed no signs of Iba1+ microgliosis or evidence of neuronal loss or glial activation in Nissl-stained sections (not shown), demonstrating the safety of delivering mi2.1 using a vascular delivery approach. This data suggest that, while well tolerated, delivery of a higher viral dose is necessary to achieve enough transduction in BACHD mouse brain to elicit robust reduction of mHTT in the brain regions analyzed.
Lactate dehydrogenase (LDH), a metabolic enzyme responsible for the bidirectional interconversion of pyruvate and lactate, is typically found in high levels in tissues and low levels in blood. Blood levels of LDH increase dramatically following muscle degradation, are often used clinically as a biomarker of tissue breakdown, and are increased in the plasma of human HD patients.30 We assayed plasma for LDH levels in female BACHD mouse plasma before surgery (8 weeks of age) and at 26 weeks of age. Similar to what is seen in HD patients, we found that plasma levels of LDH were significantly elevated in miSCM control-treated, female BACHD mice at 26 weeks of age, compared with mi2.1-treated (P < 0.01) and WT mice (P < 0.05) at the same timepoint (Figure 5a). LDH levels from male mice were not analyzed due to a low plasma sample number in this sex. LDH values did not differ between mi2.1 and WT mice (P > 0.05), demonstrating a therapeutic benefit from reducing mHTT in BACHD mouse muscle and liver. Presurgical levels did not differ between the three groups (P > 0.05). Elevated LDH has not been previously assessed as a biomarker in BACHD mice, and further studies will be needed to clarify whether this is a valid biomarker of HD progression in mouse models of HD.
Figure 5.
Peripheral administration of AAV9-mi2.1 prevents increases in plasma lactate dehydrogenase (LDH) in BACHD mice but does not prevent the weight-gain phenotype characteristic of this model. (a) Plasma levels of enzyme LDH, a biomarker for peripheral tissue breakdown, are significantly elevated in 26-week-old, control-treated, female BACHD mice, relative to mi2.1-treated, female BACHD mice (P < 0.01) and saline-injected, female wild-type (WT) mice (P < 0.05) at the same age (males not evaluated). LDH levels in adult female mi2.1 and WT mice were not significantly different (P > 0.05). (b,c) Reducing mHTT levels in peripheral tissues did not prevent weight gain in BACHD mice. AAV9-mi2.1–treated animals' weights were not significantly different compared with AAV9-miSCM control-treated mice (P > 0.05). Both groups of transgenic BACHD mice gained significantly more weight than WT littermates (P < 0.01 for males, P < 0.001 for females). Values indicated are the mean ± SEM. *P < 0.05, **P < 0.01. AAV9, adeno-associated viral vector serotype 9.
A severe symptom of human HD, particularly in later stages, is a metabolic phenotype in which patients lose tremendous amounts of weight, even in spite of nutritional intervention.9,10 Full-length human mutant huntingtin expression in transgenic mice, as shown by the BACHD and YACHD models, and direct gene transfer of mutant huntingtin fragments into the hypothalamus31 are all associated with a severe paradoxical weight gain phenotype. In spite of robust transduction of peripheral tissues in this study, accompanied by a significant reduction of mHTT expression, systemic AAV9-mi2.1 treatment did not lower BACHD body weight. mi2.1 and miSCM treated mice did not differ in body weight (P > 0.05 for males and females). WT mice did however show substantially lower body weights than mi2.1 and miSCM (P < 0.01 for males and P < 0.001 for females; Figure 5b,c).
Intrajugular AAV9-RNAi reduces mHTT mRNA expression in brain and prevents multiple neuropathological measures in N171-82Q mice
With significant reduction of mHTT achieved in peripheral tissues, but not in the CNS, of BACHD mice at a dose of 2.0–3.0 × 1010 vg/gram bw, we posited that increasing the total number of viral genomes delivered into the jugular vein would increase transduction in brain. To test this hypothesis, we performed a third experiment in which AAV9-mi2.1 was administered at a higher dose. We selected the N171-82Q mouse model of HD for the third experiment, which expresses a 171 amino acid N-terminal fragment of the mutant HTT allele containing an expanded 82 glutamine repeats.32 The rationale being that N171-82Q mice have a more aggressive phenotype, exhibit well-defined neurodegenerative biomarkers and also experience significant weight loss, similar to what is seen in HD patients. We rationalized that if a larger viral dose of AAV9-mi2.1 caused significant reduction of mHTT in the brain, this mouse model would provide us with the unique ability to assess potential prevention of atrophy, cell loss and mHTT inclusion body formation in various brain regions compared with BACHD mice.
Three-week-old male and female N171-82Q mice were injected with 7.5 × 1010 vg/g/bw (50–100µl from a titer of 4.46 × 1012 vg/ml, Table 1) of either AAV9-mi2.1 or AAV9-eGFP into the jugular vein. This dose is more than double of what was delivered to the WT mice in experiment 1 and the BACHD mice in experiment 2. Six minutes prior to vector injection, all mice received an injection of 25% mannitol (2 mg/g bw). For comparison, a third group of WT littermates were injected with 25% mannitol followed by 50–100 µl of 0.9% sterile saline. Behavioral and physiological measures were taken until 17 weeks of age, at which time mice were sacrificed and their tissues analyzed.
AAV9 transduction patterns (eGFP expression) in N171-82Q mice were similar to what was observed previously in both WT and BACHD mice, with respect to the neural regions and cell types transduced. Both AAV9-mi2.1– and AAV9-eGFP–treated N171-82Q mice showed robust transduction in cortex, striatum, thalamus, hippocampus, hypothalamus, and cerebellum (Figure 6a–x), among others. Unbiased stereological counts demonstrated that the mean number of eGFP-positive neurons in the cortex was 48,819 ± 4,732 for mi2.1-treated mice and 43,379 ± 4,485 for eGFP-treated mice (Figure 7a). Cortical astrocytes were transduced in much higher numbers, with a mean value of 134,641 ± 9,014 for mi2.1-treated and 138,055 ± 23,813 for eGFP-treated animals (Figure 7a). The estimated numbers of transduced striatal neurons were 28,090 ± 3,168 for mi2.1-treated mice and 30,606 ± 2,805 for eGFP-treated mice (Figure 7b). The mean estimated number of striatal astrocytes was lower at 18,354 ± 1,787 (mi2.1) and 21,921 ± 3,256 (eGFP) (Figure 7b). Interestingly, we observed region-specific differences in the type of cells transduced. In both treatment groups, significantly more astrocytes than neurons were transduced in cortical regions, roughly at a ratio of 3:1 (P < 0.001; Figure 7a). Conversely, AAV9 preferentially transduced significantly more neurons than astrocytes in the striatum at a ratio of 2:1 (P < 0.01; Figure 7b). Although not quantified here, AAV9 showed a similar preferential transduction of neurons to astrocytes in the hippocampus, hypothalamus, and cerebellum (Figure 6m–x). In other subcortical regions such as the thalamus and globus pallidus, neuronal and astrocytic transduction levels were particularly high and appeared to be transduced in equal numbers (Figure 6i–l). Sex did not have an effect on transduction patterns, in either the total number of cells transduced or the ratio of neurons to astrocytes in any of the regions measured. Coronal sections from mice in each group showed no evidence of glial fibrillary acidic protein–positive astrocytosis nor Iba1+ activated microglia throughout the brain, highlighting the safety of this therapeutic strategy (not shown).
Figure 6.

Transduction patterns in N171-82Q mouse brain following intrajugular injection of AAV9-mi2.1. Consistent with the transduction patterns observed in wild-type (WT) mice, there was robust enhanced green fluorescent protein (eGFP) expression in numerous regions of the central nervous system (CNS) affected in Huntington's disease (HD) following systemic AAV9-mi2.1-GFP and AAV9-GFP in N171-82Q mice. These regions include, but are not limited to, the following: (a–d) cortex; (e–h) striatum; (i–l) thalamus; (m–p) hippocampus; (q–t) hypothalamus, including the paraventricular nucleus; and (u–x) cerebellum. Forty-micrometer-thick brains were immunolabeled using an antibody against eGFP. All nuclei were labeled with Hoechst (blue). Images were taken using a confocal microscope either at ×20 magnification (Bar = 100 µm) or at ×40 magnification with a 2× digital zoom (Bar = 25 µm). AAV9, adeno-associated viral vector serotype 9; AAV9-GFP, AAV9 encoding green fluorescent protein.
Figure 7.

AAV9-mi2.1–mediated mHTT reduction in the central nervous system (CNS) of N171-82Q mice. Counts of enhanced green fluorescent protein (eGFP)–positive neurons and astrocytes in the striatum and cortex of AAV9-mi2.1– and AAV9-GFP–treated mice showed equal numbers of transduced cells between treatment groups (P > 0.05). Regardless of treatment, region-specific differences in the numbers of neurons and astrocytes were detected; significantly more astrocytes than neurons were transduced in the cortex (a, P < 0.001), and significantly more neurons than astrocytes were transduced in the striatum (b, P < 0.01). (c) Significantly reduced levels of mHTT messenger RNA (mRNA) levels in mi2.1-injected animals were detected in striatum (33%, P < 0.01), hypothalamus (11.5%, P < 0.05), and hippocampus (25.5%, P < 0.05), relative to AAV9-GFP–treated mice. Cortical, thalamic, and hippocampal relative mHTT mRNA levels are not different between groups (P > 0.05 for each region). (d) Optical density measures taken from mHTT-stained tissue were used to evaluate relative differences in mHTT protein expression levels. Staining intensity was significantly lower (P < 0.05 for each analysis) in AAV9-2.1–treated mouse striatum (65%), hippocampus (34%), frontal cortex (32%), and thalamus (32%). (e–g) Double-label immunofluorescence demonstrates that cortical cells transduced by AAV9-mi2.1 (eGFP positive) show a lack of colocalization with EM48+, mutant huntingtin-positive inclusion bodies. Frequent colocalization of eGFP and EM48 was seen in AAV9-eGFP–treated control transgenic mice (h–j). (k) Peripheral tissues from AAV9-mi2.1–treated mice showed a significant (P < 0.001 for each analysis) reduction in mHTT mRNA in the liver (54%), heart (85%), and quadriceps muscle (50%). Bar = 25 µm. Values indicated in (a–d, k) are the mean ± SEM. For all graphs, AAV9-mi2.1 treated animals are represented in blue and AAV9-eGFP in gray. *P < 0.05, **P < 0.01, ***P < 0.001. AAV9, adeno-associated viral vector serotype 9; AAV9-eGFP, AAV9 encoding enhanced green fluorescent protein.
Sixteen weeks after injection, we detected reductions in mHTT mRNA levels in AAV9-mi2.1–treated N171-82Q mouse brains compared with AAV9-eGFP–injected controls. There was a significant 33% reduction in striatum (P < 0.01), 12% in hypothalamus (P < 0.05), and 26% in hippocampus (P < 0.05) (Figure 7c). However, mHTT reduction in the frontal cortex and thalamus was not significant (P > 0.05 for both; Figure 7c). Integrated densitometry analysis (Image J) on human mHTT-stained brain sections demonstrated a significant reduction in human mHTT protein in the striatum (65%), hippocampus (34%), frontal cortex (32%), and thalamus (33%) (P < 0.05 for each; Figure 7d and Supplementary Figure S2). Although the mean level of mHTT protein was reduced in the hypothalamus by 28%, this did not reach statistical significance (P = 0.17). These findings demonstrate that, at a higher viral dose, AAV9-mi2.1 is able to cross the blood–brain barrier and transduce enough cells to elicit a significant reduction of pathogenic mHTT mRNA and protein in key brain regions affected in N171-82Q mice.
In the N171-82Q mouse model of HD, human mHTT is driven from a prion promoter and expressed most highly in brain. However, the prion promoter also directs transgene expression in a variety of peripheral tissues. Therefore, in addition to analysis in the CNS, we used qPCR to assess mHTT levels in N171-82Q mouse heart, liver, and skeletal muscle (Figure 7k). We found a robust and significant reduction in mHTT expression in AAV9-mi2.1–treated mice compared with AAV-eGFP controls in the heart (84%; P < 0.001), liver (54%; P < 0.001), and quadriceps muscle (50%; P < 0.001).
Transgenic N171-82Q mice exhibit a variety of neuropathological biomarkers reminiscent of human HD, including mHTT-positive inclusion formation, brain atrophy, striatal neuron loss, severe weight loss, and motor deficits.32,33 Double-label immunohistochemistry of N171-82Q brain sections indicated that AAV2/1-mi2.1–transduced cells (eGFP+) did not colocalize with EM48, an antibody specific to mHTT-positive fragments and inclusions (Figure 7e–g). In comparison, AAV9-eGFP transduced cells (eGFP+) showed robust colocalization with EM48 (Figure 7h–j). Examples of colocalized cells shown in Figure 7 are from the piriform cortex; however, we observed similar findings in other regions expressing EM48+ cells, such as the striatum and hippocampus. These results demonstrate that reducing mHTT expression with a systemic injection strategy prevents the formation of disease-associated inclusion bodies in transduced cells throughout the brain.
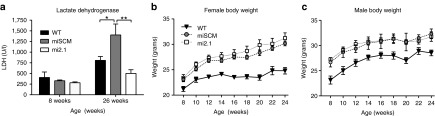
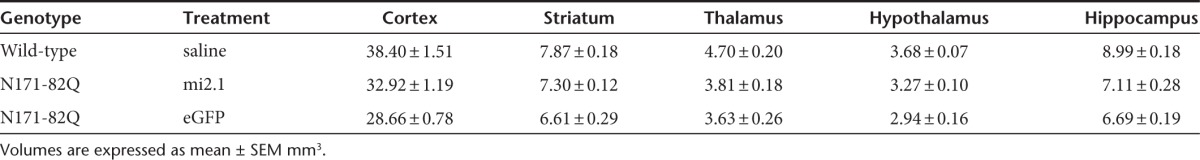
Previous studies have shown that human HD brains, as well as N171-82Q transgenic mouse brains, undergo significant atrophy and neuron loss as the disease progresses.34,35,36 To assess whether systemic AAV9-mi2.1 treatment prevented brain atrophy, regional brain volume was assessed utilizing the Cavalieri method. AAV9-mi2.1 treatment significantly reduced striatal and cortical atrophy relative to eGFP-treated mice (P < 0.05 for both regions; Figure 8a). Striatal volume was not different between mi2.1 mice and WT controls (P > 0.05). The mean estimated striatal volumes were 7.87 ± 0.18 mm3 for WT mice, 7.30 ± 0.12 mm3 for mi2.1-treated mice, and 6.61 ± 0.29 mm3 for eGFP-treated mice. Mean cortical volumes were 38.40 ± 1.51 mm3 for WT mice, 32.92 ± 1.19 mm3 for mi2.1-treated mice, and 28.66 ± 0.78 mm3 for eGFP-treated mice. There was no detectible reduction of thalamic, hypothalamic, or hippocampal brain atrophy in mi2.1-treated mice (P > 0.05) although regional brain atrophy was detected for both mi2.1- and eGFP-treated transgenic mice compared with WT mice (P < 0.05 for the thalamus, hypothalamus, and hippocampus, see Table 2 for regional volumetric data).
Figure 8.
AAV9-mi2.1 treatment reduced brain atrophy and prevented weight loss in N171-82Q mice. (a) In transgenic N171-82Q mice, regional brain volume is reduced and mi2.1 treatment significantly prevented atrophy in striatum (P < 0.05) and cortex (P < 0.05), but not in thalamus, hypothalamus, or hippocampus (P > 0.05 for the latter three regions), compared with controls. (b) There was detectable neuron loss in the striatum of AAV9-eGFP–injected controls relative to wild-type (WT) mice (P < 0.05) although AAV9-mi2.1 were not significantly different from either group (P > 0.05). (c) Weight loss characteristic of the N171-82Q mouse model was completely prevented in AAV9-mi2.1–treated mice compared with AAV9-eGFP controls (significantly different from AAV9-GFP–treated mice from week 9 to week 17, P < 0.01, data presented as percentage change from week 5 of life). (d) AAV9-mi2.1 treatment did not prevent motor deficits, analyzed using the rotorod assay, compared with AAV-eGFP–treated control transgenics (P > 0.05). Values indicated are the mean ± SEM. *P < 0.05 and #P < 0.05. AAV9, adeno-associated viral vector serotype 9; AAV-eGFP, AAV encoding enhanced green fluorescent protein.
Table 2. Regional volumetric analysis of N171-82Q brains.
Additionally, stereological counts of the number of NeuN+ neurons in the striatum revealed a significant 13% loss of neurons in AAV9-eGFP–treated control mice compared with WT mice (mean number of neurons was 1,294,100 ± 19,828 for WT mice and 1,122,348 ± 42,590 for eGFP-treated mice (P < 0.05). Neuronal counts for WT mice and m2.1-treated mice were not significantly different (P > 0.05) nor they were different between mi2.1- and eGFP-treated transgenic mice (mean number of neurons for mi2.1-treated mice was 1,259,084 ± 63,652; P = 0.135; Figure 8b).
Intrajugular AAV9-RNAi administration prevents severe weight loss in N171-82Q mice
Weight loss is a prevalent symptom of HD and by end stage of disease, patients are severely cachexic, despite painstaking efforts to maintain daily caloric intake.9,10 Similarly, N171-82Q mice fail to gain weight during development compared with WT littermates and experience a precipitous weight-loss phenotype at end stages.32 There was a robust effect of systemic AAV9-mi2.1 treatment on body weight; mi2.1-treated mice had significantly higher body weights compared with eGFP-treated mice from weeks 11 to 17 (P < 0.05) and gained weight similarly to WT mice (Figure 8c, data expressed as percentage change in weight relative to week 5).
To address whether AAV9-mi2.1 treatment had a similar protective effect on the motor deficits characteristic in HD patients and transgenic mice, we assessed behavior on the rotorod apparatus, a measure of motor coordination. eGFP-treated N171-82Q mice showed the typical phenotypic decline in rotorod performance (shorter latency to fall), compared with WTs (Figure 8d). Surprisingly, reducing mHTT in the striatum, among other regions, did not significantly prevent rotorod deficits as evidenced by similar rotorod performance between mi2.1- and eGFP-treated transgenic mice (P > 0.05 for each timepoint).
Mannitol pretreatment does not increase AAV9 transduction in the CNS
Twenty-five percent mannitol, an osmotic agent with the ability to temporarily disrupt the blood–brain barrier, has been shown to improve CNS transduction of AAV9, in addition to enabling other AAV serotypes to cross and transduce brain parenchyma that ordinarily cannot.27,28,29 We assessed whether a pretreatment with 25% mannitol, at 2 mg/gram body weight and administered 6 minutes before vector improved AAV9-eGFP (5.7–7.7 × 1010 vg/gram bw) transduction levels in BACHD mouse brain relative to pretreatment with equivalent volumes of 0.9% saline. Interestingly, we found no differences in mannitol pretreated mice compared with saline pretreated mice (P > 0.05; Supplementary Figure S3) in either the numbers of transduced cells in the motor cortex (neurons: 7,116 ± 1,418 in saline and 6,332 ± 680 in mannitol pretreated animals; astrocytes: 14,314 ± 629 in saline and 16,020 ± 1,305 in mannitol pretreated animals) or the striatum (neurons: 22,477 ± 3,131 in saline and 25,253 ± 1,639 in mannitol pretreated animals; astrocytes: 10,425 ± 735 in saline and 10,761 ± 1,253 in mannitol pretreated mice). However, we replicated the region-specific transduction pattern differences that we observed in N181-82Q mice. There were significantly more transduced neurons relative to astrocytes in the striatum in a ratio of 2:1 (P < 0.001). Contrarily, there were significantly more transduced astrocytes relative to neurons in the cortex in a ratio of 3:1 (P < 0.001). Sex did not influence the total number of transduced cells (P > 0.05) or the ratio of cell types transduced (P > 0.05) in either pretreatment group.
Discussion
Prior work has demonstrated mHTT suppression and associated therapeutic efficacy following injection of viral vectors encoding mHTT-specific hairpin RNAs into the striatum of rodent models of HD.13,14,15,16,17,24 However, given the widespread expression and consequences of mHTT expression outside of the striatum, developing a more comprehensive delivery strategy for RNAi therapeutics would likely be of tremendous benefit to HD patients. Here, we present novel data demonstrating the reduction of mHTT in key peripheral tissues and brain regions and the prevention of several hallmark pathological features of HD, resulting from a single intrajugular administration of AAV9 expressing a mHTT-specific miRNA. This study is a first step in demonstrating the utility of AAV9 as a delivery tool for global RNAi therapy for patients suffering from HD.
One of the most unexpected findings of the current study was the prevention of weight loss in N171-82Q mice. This is an important finding, in that severe weight loss is an extremely common and debilitating feature of the disease that has a severe impact on the quality of life of HD patients.9,10 AAV1-miRNA–mediated mHTT suppression in the striatum alone does not prevent weight loss in this transgenic mouse model of HD.16 In future studies, it will be of utility to investigate if the significant prevention of weight loss seen in AAV9-mi2.1–treated N171-82Q mice was mediated by a decrease of mHTT in multiple brain regions (cortex, striatum, thalamus, and hippocampus), mHTT reduction in peripheral tissues (skeletal muscle, cardiac muscle and liver) or the combination of reduction in both the CNS and periphery.
At the viral dose evaluated here, we did not prevent motor deficits, as measured by the rotorod assay, in N171-82Q mice. This finding is in contrast to the prevention of rotorod deficits observed following either a direct striatal injection of AAV2/1-miRNA targeting mHTT in N171-82Q mice or an intraventricular injection of antisense oligonucleotides into YAC128 mice, both of which resulted in ~75% reduction of mHTT expression.16,18 Further studies are required to address whether increasing the total number of viral genomes of AAV9-mi2.1 will prevent motor deficits, as well as other HD-associated phenotypes such as cognitive deficits, anxiety- and depressive-like behaviors. Moreover, significant reduction of mHTT in peripheral tissues of BACHD mice prevented build-up of plasma LDH, suggesting that this approach may prevent mHTT-related muscle pathology. Ongoing studies are currently evaluating whether an intravascular AAV9-mi2.1 injection prevents disease-associated pathology in N171-82Q muscle, such as altered gene expression, reduced muscle fiber diameter, and the conversion from fast-twitch to slow-twitch muscle.
An important consideration of a vascular AAV injection strategy is the potential for a mounted immune response to either the AAV capsid or the expressed transgene. Humans have appreciable levels of circulating neutralizing antibodies (Nabs) to many AAV serotypes although the prevalence of anti-AAV9 Nabs is appreciably lower compared with other serotypes.37 Previous studies, investigating a variety of animals typically used in preclinical research including non-human primates, have reported that preexisting circulating Nabs to AAV9 can prevent transduction in brain following intravascular or intracisterna magna injections of AAV9 expressing GFP.38,39 In contrast, we have previously demonstrated that preexisting Nabs do not abrogate transduction following a direct injection of AAV1 expressing a HTT-specific miRNA into the non-human primate putamen.19 Moving forward with a systemic approach, it will be important to (i) prescreen laboratory animals for circulating anti-AAV9 Nabs, (ii) address whether an increased viral dose can overcome neutralization, (iii) explore novel engineered capsids, or (iv) investigate mutation of the Nab binding site on the AAV9 capsid as a means to prevent neutralization. Furthermore, studies have shown that viral-mediated expression of a non-self protein in peripheral tissues induces a robust T-cell response.40 It was recently shown that a direct striatal injection of AAV9 transduces microglia and elicits a cell-mediated immune response as the expressed protein is recognized as non-self.41 In contrast, we found that a systemic AAV9 injection transduced neurons, astrocytes, and vascular endothelial cells in the striatum but not in microglia. We posit that this discrepancy may be a matter of dose and hypothesize that an increase in systemically delivered AAV9 viral genomes could potentially result in microglial transduction in brain. An appreciable advantage to both direct and systemic AAV9-mediated RNAi therapy for HD, and other disorders, is the lack of an expressed protein, which leads to a greater safety profile of this therapeutic strategy.
The data presented here add to the growing body of evidence supporting AAV9-mediated delivery of therapeutic transgenes for a variety of diseases of both peripheral and CNS origin. Because of its robust tropism for peripheral tissues, AAV9 has been used to deliver therapeutics in animal models of muscular dystrophy,42 cardiac hypertrophy,43 peripheral arterial disease,44 and very long-chain acyl Co-A dehydrogenase deficiency.45 Additionally, AAV9 has shown to be a potent tool to deliver therapeutics that lead to robust symptom improvement in models of CNS diseases with widespread pathology, including spinal muscular atrophy,46 the lysosomal storage disorder mucopolysaccharidosis III,28 Rett syndrome,47,48 and amyotrophic lateral sclerosis.49 As a vascular delivery approach for AAVs that bear disease-modifying genes moves from mice into larger animal models and potentially into the clinic, scalability becomes paramount. In the current study, the highest dose delivered was 7.5 × 1010 vg/gram bw. The equivalent amount of vector needed for a 150-pound human (68 kg) would be ~5 × 1015 total vgs, a substantial amount of vector. Scaling up AAV production can be achieved by moving away from the use of adherent mammalian cells (human embryonic kidney 293) grown as a monolayer to using non-adherent insect cell (Sf9) suspension cultures via baculovirus expression vectors.50
Effective neuroprotective therapies for HD are lacking. Pharmacotherapies currently used in the clinic, such as the dopaminergic antagonist tetrabenezine for chorea, and selective serotonin reuptake inhibitors for depression and anxiety, provide mild to moderate symptom reduction but do not slow the progression of widespread mHTT-associated pathology. A key advantage of RNAi therapy for HD, compared with existing pharmacotherapies, is the specific targeting of the disease-causing gene, as opposed to treating systems that manifest due to mHTT toxicity. While AAV-RNAi therapeutics have shown efficacy in reducing motor symptoms in HD mice using a direct striatal injection approach, a more comprehensive delivery method is warranted. The findings presented here suggest that systemic, AAV9-mediated RNAi may be a viable strategy that more comprehensively addresses the widespread pathology and devastating symptoms of HD.
Materials and Methods
Animals. All animals were group housed with littermates under controlled conditions of temperature and light (12-hour light/dark cycle). Food and water were provided ad libitum. WT C57BL/6, BACHD (stock no. 008197), and N171-82Q mice (stock no. 003627) were obtained from Jackson Laboratories (Bar Harbor, ME) and bred in the vivarium at the Oregon National Primate Research Center (ONPRC). BACHD mice were maintained on a FVB background, and N171-82Q mice were maintained on a B6C3F1/J background. Transgenic mice were genotyped using primers specific for the mutant human HTT transgene, and age-matched WT littermates were used for the indicated experiments. Body weights of all animals were recorded weekly. All experimental procedures were performed according to ONPRC and Oregon Health and Science University Institutional Animal Care and Use Committee and Institutional Biosafety Committee approved protocols.
RNAi constructs and viral vector production. siRNA sequences targeting either a sequence in exon 2 human HTT mRNA (but not mouse Htt mRNA) or a control siRNA were embedded into an artificial miRNA scaffold comparable with human miR-30 to generate mi2.1 (pri: 5′agugagcgAAGAAAGAACUUUCAGCUACCacuguaaa gccacagauggguGGUAGCUGAAAGUUCUUUCUUcgccuacu-3′, antisense sequence in bold) or miSCM (pri: 5′-AGUGAGCGCAGCGAACGACUUACGCGUU UACUGUAAAGCCACAGAUGGGUAAACGCGUAAGUCGUUCGCUACGCCUACU-3′, antisense sequence in bold). Artificial miRNA stem loops were cloned into a mouse U6 expression vector, and the expression cassettes were subsequently cloned into pFBGR-derived plasmids that coexpress cytomegalovirus-driven eGFP. Shuttle plasmids (pAAVmi2.1-GFP and pAAVmiSCM-GFP) contain the respective transcriptional units, which are flanked at each end by AAV serotype 2 145-bp inverted terminal repeat sequences. Recombinant AAV2/9 (inverted terminal repeats of AAV2 and capsid proteins of AAV9) production was performed using the Baculovirus AAV System as previously described.50 Vectors were generated by the Gene Transfer Vector Core at the University of Iowa. Twelve hours before surgery, all viral vector preps were dialyzed using dialysis spin columns (Pierce, Rockford, IL) to remove salts and diluted with Formulation Buffer 18 (Hyclone, Logan, UT) to a final working titer.
Intravenous injection of viral vectors. All mice received a single injection of either self-complimentary AAV9-eGFP, AAV9-mi2.1-eGFP, AAV9-miSCM-eGFP, or saline into the right jugular vein. Briefly, mice were anesthetized with a mixture of ketamine and xylazine and placed in a ventral recumbent position. A small incision was made above the neck, and the jugular vein was exposed. Viral vectors or saline were injected by passing a 30-gauge Hamilton needle through the pectoral muscle and into the lumen of the vein (Figure 1). In experiment 1 (biodistribution), six adult C57BL6/J mice were injected with 300 µl of either AAV9-eGFP at a titer of 1 × 1013 vg/ml (3 × 1012 total viral genomes) or 0.9% saline. In experiment 2, twenty adult BACHD transgenic mice were injected into the jugular vein with ~200 µl of mannitol (25%, 2 mg/gram bw) and, after a 6-minute wait, they were injected with 250 µl of either AAV9-mi2.1 or AAV9-miSCM at a titer of 2.5 × 1012 vg/ml (6.25 × 1011 total viral genomes). Ten WT littermates were injected into the jugular vein with ~200 µl of mannitol (25%; 2 mg/gram body weight), and after a 6-minute wait, they were injected with 250 µl of 0.9% saline. In experiment 3, eight adult N171-82Q transgenic mice were injected into the jugular vein with ~50–90 µl of mannitol (25%; 2 mg/gram bw), and after a 6-minute wait, they injected with 110–185 µl of either AAV9-mi2.1 or AAV9-eGFP at a titer of 4.46 × 1012 vg/ml (7.5 × 1010 vg/gram bw). Five WT littermates were injected into the jugular vein with ~70–100 µl of mannitol (25%; 2 mg/g bw), and after a 6-minute wait, they injected with 70–100 µl of 0.9% saline. All injections were manually performed over a 2-minute period.
Rotorod performance testing. Mice were tested on an accelerating rotorod apparatus (model 47600; Ugo Basile, Comerio, Italy) every 2 weeks, from 5 to 15 weeks of age. At week 4, mice were first habituated on the rotorod with three 4-minute sessions. For each test week, mice were tested on 2 days (spaced 3 days apart). Mice were tested three trials per test day (with at least 30 minutes of rest between trials). For each trial, acceleration was from 4 to 40 rpm over 4 minutes. Latency to fall (or if mice hung on for three consecutive rotations without running) was recorded for each mouse per trial. The trials were stopped at 240 seconds, and mice remaining on the rotorod at that time were scored as 240 seconds. Data from the six trials for each group on each test week are presented as means ± SEM.
Necropsy and tissue processing. Mice used in histological and molecular analyses were anesthetized with a ketamine/xylazine mix and transcardially perfused with 15 ml of cold 0.9% saline. Brains and peripheral tissues were removed, and brains bisected with a mid-sagittal cut. The right hemisphere was blocked into 1-mm-thick coronal slices, to be used for molecular analyses. Specific brain regions and samples from peripheral tissues for molecular analyses were dissected out in cold 0.9% saline, flash frozen in liquid nitrogen and stored at −80 °C until used. The left brain hemisphere was left intact and used for histological analyses. Left hemispheres and peripheral tissues for histology were postfixed overnight in 4% paraformaldehylde and placed in 30% sucrose until cut. All brains were cut on a microtome at a thickness of 40 µm, and peripheral tissues were cut on a cryostat at a thickness of 16 µm.
Quantitative real-time PCR. RNA was isolated from brain and peripheral tissues using 1 ml of Trizol (Invitrogen, Carlsbad, CA), per the manufacturer's instructions, and reverse transcribed with random primers and Multiscribe reverse transcriptase (Applied Biosystems, Foster City, CA). Relative human mHTT mRNA expression was assessed via qPCR. Primers were designed to flank the mi2.1 binding site in Exon 2 using Primer Express (Applied Biosystems): forward: 5′- GCCGCTGCACCGACCAAAGAA-3′, reverse: 5′- AGTTCCATAGCGATGCCCAGAAGTT-3′ and expression was assessed using SYBR Green detection. Relative gene expression was determined by using the ΔΔCT method, normalizing to 18S mRNA levels.
Immunohistochemical analyses. Brain sections were stained using an anti-eGFP antibody (Invitrogen, 1:1,000, made in rabbit) followed by a goat anti-rabbit secondary antibody conjugated to Alexa Fluor 488 (Invitrogen, 1:500). Additional sections were stained using an anti- human huntingtin antibody (aa1-82, Millipore, Billerica, MA; 1:1,000, mouse) followed by a biotinylated donkey-anti mouse secondary antibody and developed using diaminobenzidine with nickel intensification. For double-labeling of brain tissues with eGFP and cell type–specific markers, brain sections were additionally stained (in parallel) with antibodies against NeuN (Millipore, 1:500, mouse) to label neurons, glial fibrillary acidic protein (Dako, Carpinteria, CA; 1:1,000, rabbit) to label astrocytes, Iba1 (Wako, Richmond, VA; 1:500, rabbit) to label microglia, CD31 (Abcam, Cambridge, MA; 1:50, rabbit) to label vascular endothelial cells, and EM48 (Millipore, 1:100, mouse) to label mutant huntingtin inclusion bodies, followed by a goat anti-mouse or goat anti-rabbit Alexa Fluor 568 (Invitrogen 1:500). For those double-labeled tissues, for which the second (non-eGFP) primary antibody was made in rabbit, eGFP was stained using an anti-eGFP antibody made in chicken (Aves, Tigard, OR; 1:1,000) followed by a goat anti-chicken secondary antibody conjugated to Alexa Fluor 488. All sections were counterstained using Hoechst (Invitrogen). For labeling of specific peripheral tissues, sections through the testis were stained with an anti-17-α hydroxylase antibody (gift from Kevin Grove, 1:500) to label testosterone-producing Leydig cells. Sections through the pancreas were stained with anti-insulin and anti-glucagon antibodies (ImmunoStar, 1:500) to label insulin- and glucagon-producing cells in the islets of the pancreas followed by goat anti-rabbit secondary antibodies conjugated to Alexa Fluor 568. eGFP in all peripheral tissues is inherent expression from the eGFP transgene and not stained with an anti-eGFP antibody, except for the double-labeling studies mentioned previously. In all staining procedures, deletion of the primary antibody served as a control. Sections were mounted onto Super Frost Plus slides (Fisher, Waltham, MA) and cover-slipped with SlowFade gold mounting media (fluorescence) or Cytoseal (diaminobenzidine-labeled sections). Images were captured with a Leica SP5 confocal microscope (Buffalo Grove, IL) or an Olympus BX51 light microscope (Center Valley, PA). Figures were created using Adobe Photoshop and Illustrator.
Plasma LDH analysis. Blood was collected from the right ventricle of the heart immediately prior to necropsy and placed in lithium heparin green-top Vacutainer blood tubes (BACHD mice). Tubes were spun down using a tabletop centrifuge at 2,500 rpm for 20 minutes at 4 °C, and plasma was sent to the IDEXX RADIL animal diagnostic laboratory for multiplex fluorescent immunoassay analysis of LDH concentrations.
Stereological determination of vector distribution. The Optical Fractionator (Microbrightfield; MBF Bioscience, Williston, VT) was used to quantify the number of eGFP-positive neurons and astrocytes in the striatum and cortex (left hemisphere only) in experiments 3 (N171-82Q mice) and 4 (mannitol study). For striatal analysis, every 12th coronal section (1/2 series, 40-µm-thick sections) through the striatum was analyzed, from +1.42 mm (anterior) to −1.46 mm (posterior) to bregma (Mouse Brain Atlas, Paxinos & Franklin, New York, NY). For cortical cell counts in experiment 3, every 24th section (1/4 series) of the entire cortex from +2.34 to −4.48 was assessed. For experiment 4, the motor cortex was also assessed for the number of eGFP-positive cells—every sixth section was quantified from +2.46 to +0.74 mm. All regions were outlined at ×4 magnification, overlaid with a grid, and eGFP+ cells were counted at ×40 magnification. Two separate markers were used to count neurons and astrocytes (cell types were identified morphologically). Grid and counting frame sizes were piloted for each region and set to provide a reliable estimate with a small coefficient of error (Gundersen value (m = 1) ≤ 0.10). For all analyses, the counting frame was set to 130 × 130 µm. For experiment 3, the grid size was set to 400 × 400 µm in the striatum and 650 × 650 µm for the cortex. The optical fractionator was also used to quantify the number of NeuN-positive striatal neurons in experiment 3. Estimates were also taken from every 12th section, from +1.42 to −1.46 with a grid size of 425 × 425 µm and a counting frame of 24 × 24 µm. Regional brain volume for cortex (+2.34 to −4.48), striatum (+1.2 to −2.7), hypothalamus (0.74 to −2.7), hippocampus (−0.94 to −4.48), and thalamus (−0.22 to −3.16) were estimated in mice from experiment 3 using the Cavalieri method (Microbrightfield) on NeuN-stained tissue. For all regions, every sixth section was counted with the exception of cortex, for which every 12th section was counted. The grid size was 250 × 250 µm, and for all animals, the Gundersen (m = 1) coefficient of error was ≤0.10.
HTT densitometry analysis. Coronal brain sections of N171-82Q mice were stained with an anti-huntingtin antibody (aa1-82) antibody and used to quantify human mHTT protein expression levels. mHTT protein staining intensity was assessed in striatum, frontal cortex, hypothalamus, hippocampus, and thalamus. Images were captured at ×10 magnification using an Olympus BX51 light microscope, and optical density was analyzed using Image J software (National Institutes of Health, Bethesda, MD). Images from a series of three serial sections, 12 sections (480 µm) apart, were utilized for each brain region analyzed. For each section, optical density (mean pixel intensity) was measured in three equally sized region of interests (ROIs), which were randomly placed within anatomical boundaries of each brain region. Sections containing brain ROIs were matched across all mice analyzed. Three background intensity measures were also taken from within each ROI and were subtracted from each ROI mean pixel intensity measure. Optical measures were averaged across the three ROIs.
Statistical analysis. All statistical analyses were performed by using SigmaStat statistical software (SYSTAT, Chicago, IL) or JMP Version 10 (SAS Institute, Cary, NC). SigmaStat was used for all single timepoint analyses, while a repeated measures mixed model was used for all repeated measures data in JMP. Post hoc analyses were performed when statistically significant differences were detected. In all cases, P < 0.05 was considered statistically significant. Quantitative reverse transcription PCR analyses for mHTT, striatal NeuN cell counts, and volumetric data were all assessed using a two-way analysis of variance, including sex and treatment as predictors, and a Tukey's comparison was used for post hoc tests. Body weight analyses were run separately for males and females, assessed using a repeated measures mixed model for both treatment and timepoint, and post hoc comparisons were made using a Student's t-test. LDH was assessed for females only, using a repeated measures mixed model with timepoint and treatment as predictors, and post hoc comparisons were made with a Tukey's comparison. Optical fractionator GFP+ cell counts were assessed separately for each brain region using a repeated measures mixed model with cell type, sex, and treatment as predictors, and cell-type effects were compared post hoc with a t-test. For all models for which two or more predictors were included, all two-way interactions were also included in the model.
SUPPLEMENTARY MATERIAL Figure S1. AAV9-eGFP transduces specific cell types in testis and pancreas. Figure S2. AAV9 reduces mutant huntingtin protein expression. Figure S3. Pre-treatment of mannitol does not improve AAV9 transduction efficiency.
Acknowledgments
We thank Anda Cornea at the Oregon National Primate Research Center (ONPRC) for confocal microscopy expertise, the Small Lab Animals Unit at the ONPRC for continued care of the animals and the University of Iowa Gene Transfer Vector core for production of the viral vectors. We additionally thank Nikki Leila Herndon for technical assistance with the surgical and immunohistochemical aspects of each study. This research was supported by a research grant from the Hereditary Disease Foundation (J.L.M.), ONPRC Core Grants RR000163 (J.L.M.) and P51OD011092 (J.L.M.), and a T32 Neuroscience training grant NS7466-14 (B.D.D.). Confocal microscopy was supported by grants S10RR024585 and P30-NS061800.
Supplementary Material
AAV9-eGFP transduces specific cell types in testis and pancreas.
AAV9 reduces mutant huntingtin protein expression.
Pre-treatment of mannitol does not improve AAV9 transduction efficiency.
References
- The Huntington Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Gourfinkel-An I, Cancel G, Duyckaerts C, Faucheux B, Hauw JJ, Trottier Y, et al. Neuronal distribution of intranuclear inclusions in Huntington's disease with adult onset. Neuroreport. 1998;9:1823–1826. doi: 10.1097/00001756-199806010-00028. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Heinsen H, Strik M, Bauer M, Luther K, Ulmar G, Gangnus D, et al. Cortical and striatal neurone number in Huntington's disease. Acta Neuropathol. 1994;88:320–333. doi: 10.1007/BF00310376. [DOI] [PubMed] [Google Scholar]
- Yanagisawa N. The spectrum of motor disorders in Huntington's disease. Clin Neurol Neurosurg. 1992;94 Suppl:S182–S184. doi: 10.1016/0303-8467(92)90064-a. [DOI] [PubMed] [Google Scholar]
- Spargo E, Everall IP, Lantos PL. Neuronal loss in the hippocampus in Huntington's disease: a comparison with HIV infection. J Neurol Neurosurg Psychiatr. 1993;56:487–491. doi: 10.1136/jnnp.56.5.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabery S, Murphy K, Schultz K, Loy CT, McCusker E, Kirik D, et al. Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropathological analyses. Acta Neuropathol. 2010;120:777–788. doi: 10.1007/s00401-010-0742-6. [DOI] [PubMed] [Google Scholar]
- Rodda RA. Cerebellar atrophy in Huntington's disease. J Neurol Sci. 1981;50:147–157. doi: 10.1016/0022-510x(81)90049-6. [DOI] [PubMed] [Google Scholar]
- Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, Roos RA, EHDI Study Group Weight loss in Huntington disease increases with higher CAG repeat number. Neurology. 2008;71:1506–1513. doi: 10.1212/01.wnl.0000334276.09729.0e. [DOI] [PubMed] [Google Scholar]
- Trejo A, Tarrats RM, Alonso ME, Boll MC, Ochoa A, Velásquez L. Assessment of the nutrition status of patients with Huntington's disease. Nutrition. 2004;20:192–196. doi: 10.1016/j.nut.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, et al. TRACK-HD investigators Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8:791–801. doi: 10.1016/S1474-4422(09)70170-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 2009;8:765–774. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc Natl Acad Sci USA. 2005;102:5820–5825. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington's disease transgenic mice. Mol Ther. 2005;12:618–633. doi: 10.1016/j.ymthe.2005.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franich NR, Fitzsimons HL, Fong DM, Klugmann M, During MJ, Young D. AAV vector-mediated RNAi of mutant huntingtin expression is neuroprotective in a novel genetic rat model of Huntington's disease. Mol Ther. 2008;16:947–956. doi: 10.1038/mt.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice. Mol Ther. 2009;17:1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouet V, Perrin V, Hassig R, Dufour N, Auregan G, Alves S, et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann Neurol. 2009;65:276–285. doi: 10.1002/ana.21569. [DOI] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, et al. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington's disease. Mol Ther. 2011;19:2152–2162. doi: 10.1038/mt.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Kordower JH. Gene therapy for Huntington's disease. Neurobiol Dis. 2012;48:243–254. doi: 10.1016/j.nbd.2011.12.030. [DOI] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh N, Moutereau S, Durr A, Krystkowiak P, Azulay JP, Tranchant C, et al. Neuroendocrine disturbances in Huntington's disease. PLoS ONE. 2009;4:e4962. doi: 10.1371/journal.pone.0004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong TV, Tagle DA, Valdes JM, Elmer LW, Boehm K, Swaroop M, et al. Widespread expression of the human and rat Huntington's disease gene in brain and nonneural tissues. Nat Genet. 1993;5:259–265. doi: 10.1038/ng1193-259. [DOI] [PubMed] [Google Scholar]
- McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, Spengler RM, Davidson BL. Rational design of therapeutic siRNAs: minimizing off-targeting potential to improve the safety of RNAi therapy for Huntington's disease. Mol Ther. 2011;19:2169–2177. doi: 10.1038/mt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Muenzer J, Samulski RJ, Breese G, Sifford J, Zeng X, et al. Self-complementary adeno-associated virus serotype 2 vector: global distribution and broad dispersion of AAV-mediated transgene expression in mouse brain. Mol Ther. 2003;8:911–917. doi: 10.1016/j.ymthe.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Fu H, Dirosario J, Killedar S, Zaraspe K, McCarty DM. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol Ther. 2011;19:1025–1033. doi: 10.1038/mt.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty DM, DiRosario J, Gulaid K, Muenzer J, Fu H. Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther. 2009;16:1340–1352. doi: 10.1038/gt.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasset I, Sánchez-López F, Agüera E, Fernández-Bolaños R, Sánchez FM, Cruz-Guerrero A, et al. NGF and nitrosative stress in patients with Huntington's disease. J Neurol Sci. 2012;315:133–136. doi: 10.1016/j.jns.2011.12.014. [DOI] [PubMed] [Google Scholar]
- Hult S, Soylu R, Björklund T, Belgardt BF, Mauer J, Brüning JC, et al. Mutant huntingtin causes metabolic imbalance by disruption of hypothalamic neurocircuits. Cell Metab. 2011;13:428–439. doi: 10.1016/j.cmet.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- McBride JL, Ramaswamy S, Gasmi M, Bartus RT, Herzog CD, Brandon EP, et al. Viral delivery of glial cell line-derived neurotrophic factor improves behavior and protects striatal neurons in a mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2006;103:9345–9350. doi: 10.1073/pnas.0508875103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Peng Q, Hou Z, Aggarwal M, Zhang J, Mori S, et al. Structural MRI detects progressive regional brain atrophy and neuroprotective effects in N171-82Q Huntington's disease mouse model. Neuroimage. 2011;56:1027–1034. doi: 10.1016/j.neuroimage.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, et al. Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis. 2001;8:479–491. doi: 10.1006/nbdi.2001.0406. [DOI] [PubMed] [Google Scholar]
- Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranch L, Salegio EA, San Sebastian W, Kells AP, Foust KD, Bringas JR, et al. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum Gene Ther. 2012;23:382–389. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C, et al. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesielska A, Hadaczek P, Mittermeyer G, Zhou S, Wright JF, Bankiewicz KS, et al. Cerebral infusion of AAV9 vector-encoding non-self proteins can elicit cell-mediated immune responses. Mol Ther. 2013;21:158–166. doi: 10.1038/mt.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Lu PJ, Wang CH, Keramaris E, Qiao C, Xiao B, et al. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of alpha-dystroglycan and improves muscle functions. Mol Ther. 2013;21:1832–1840. doi: 10.1038/mt.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, et al. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. 2013;2:e000078. doi: 10.1161/JAHA.113.000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katwal AB, Konkalmatt PR, Piras BA, Hazarika S, Li SS, John Lye R, et al. Adeno-associated virus serotype 9 efficiently targets ischemic skeletal muscle following systemic delivery. Gene Ther. 2013;20:930–938. doi: 10.1038/gt.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino DE, Lange AM, Altynova ES, Sarkar R, Zhou S, Merricks EP, et al. Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther. 2011;19:442–449. doi: 10.1038/mt.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkhelifa-Ziyyat S, Besse A, Roda M, Duque S, Astord S, Carcenac R, et al. Intramuscular scAAV9-SMN injection mediates widespread gene delivery to the spinal cord and decreases disease severity in SMA mice. Mol Ther. 2013;21:282–290. doi: 10.1038/mt.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla KK, Bailey ME, Spike RC, Ross PD, Woodard KT, Kalburgi SN, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther. 2013;21:18–30. doi: 10.1038/mt.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. 2013;33:13612–13620. doi: 10.1523/JNEUROSCI.1854-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Salazar DL, Likhite S, Ferraiuolo L, Ditsworth D, Ilieva H, et al. Therapeutic AAV9-mediated Suppression of Mutant SOD1 Slows Disease Progression and Extends Survival in Models of Inherited ALS. Mol Ther. 2013;21:2148–2159. doi: 10.1038/mt.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RH, Levy JR, Kotin RM. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol Ther. 2009;17:1888–1896. doi: 10.1038/mt.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AAV9-eGFP transduces specific cell types in testis and pancreas.
AAV9 reduces mutant huntingtin protein expression.
Pre-treatment of mannitol does not improve AAV9 transduction efficiency.