

Abstract
Inflammation and its consequent fibrosis are two main features of diabetic nephropathy (DN), but target therapy on these processes for DN remains yet ineffective. We report here that miR-29b is a novel therapeutic agent capable of inhibiting progressive renal inflammation and fibrosis in type 2 diabetes in db/db mice. Under diabetic conditions, miR-29b was largely downregulated in response to advanced glycation end (AGE) product, which was associated with upregulation of collagen matrix in mesangial cells via the transforming growth factor-β (TGF-β)/Smad3-dependent mechanism. These pathological changes were reversed by overexpressing miR-29b, but enhanced by knocking-down miR-29b. Similarly, loss of renal miR-29b was associated with progressive diabetic kidney injury, including microalbuminuria, renal fibrosis, and inflammation. Restored renal miR-29b by the ultrasound-based gene therapy was capable of attenuating diabetic kidney disease. Further studies revealed that inhibition of Sp1 expression, TGF-β/Smad3-dependent renal fibrosis, NF-κB–driven renal inflammation, and T-bet/Th1-mediated immune response may be mechanisms associated with miR-29b treatment in db/db mice. In conclusion, miR-29b may play a protective role in diabetic kidney disease and may have therapeutic potential for diabetic kidney complication.
Introduction
Diabetes mellitus affects 346 million people worldwide, and 10–20% of people with diabetes die of kidney failure (Centers for Disease Control and Prevention 2011). Increasing evidence shows that diabetic nephropathy (DN) is a leading cause of kidney failure, accounting for 45% of end-stage renal disease. Morphological alterations in DN are manifested as glomerular hypertrophy, loss of podocytes, thickening of the glomerular basement membrane, and matrix expansion in both mesangium and tubulointerstitium. These structural abnormalities result in the leakage of albumin from glomeruli and progressive decline in renal function with the development of end-stage renal disease accompanying by glomerulosclerosis and tubulointerstitial fibrosis.1 Transforming growth factor-β (TGF-β)/Smad signaling is a well recognized pathway leading to the fibrotic change of DN.2,3,4,5 In addition, TGF-β1 has also been considered as an important regulator in immune response in diabetes.6,7 Emerging evidence demonstrates that activation of innate immunity and inflammation are highly relevant to the pathogenesis of diabetes.8,9,10,11,12,13 Inflammatory cytokines such as IL-1β and TNF-α, chemokines including MCP-1 and ICAM-1 have been shown to contribute to the development of DN.14 Activation of NF-κB signaling, the classic inflammation pathway, is a key mechanism involving the complex inflammatory cytokine regulation in DN.14 Therefore, in addition to the tight hyperglycemia control, anti-fibrosis and anti-inflammation have been considered to be a therapeutic approach to minimize the diabetic complications.
Advances in recent molecular biology reveal that microRNAs (miRNA) are potentially important in both regulation and treatment for many diseases particularly in cancer. miRNAs are single-stranded noncoding RNA molecules of ~22 nucleotides in length, which functions as regulators of gene expression by binding to the 3′UTR region of messenger RNA (mRNA) molecules and then destabilizing them or inhibiting their translation. Many studies implicate that downregulation of the miR-29 family (miR-29 a-c) contributes to various fibrotic diseases including cardiac fibrosis,15 systemic sclerosis,16 pulmonary fibrosis,17,18 hepatic fibrosis,19 and unilateral ureteral obstruction–induced renal fibrosis.20 In patients with type 2 diabetes or in cultured cells under high glucose conditions, levels of miR-29 are downregulated.21,22 In contrast, one study shows that miR-29c is upregulated in the diabetic kidney of db/db mice and in endothelial cells and podocytes under high glucose conditions,23 indicating functional differences among the miR-29 family members. Recent studies have also demonstrated that miR-29 regulates inflammation cytokine expression by T cells,24 which is mediated via the Sp1/NF-κB/miR-29b regulatory network,25 suggesting an additional role for miR-29 in inflammation and immune response. Among the miR-29 family, decreased miR-29b, but not miR-29a and c, is significantly correlated with the degree of liver fibrosis,19 suggesting miR-29b as a sensitive regulatory miRNA in fibrosis. Moreover, we recently revealed that miR-29b is downregulated by TGF-β1 via the Smad3-dependent mechanism and overexpression of miR-29b has a therapeutic effect on obstructive nephropathy and bleomycin-induced pulmonary fibrosis.18,20 However, the functional role and therapeutic potential of miR-29b for DN remain unexplored. Thus, this study investigated into the role and therapeutic potential of miR-29b for DN in vitro and in a db/db mouse model of diabetes.
Results
Overexpression of miR-29b attenuates, but knockdown of miR-29b enhances fibrosis in response to advanced glycation end stimulation in vitro
To study the role of miR-29b in diabetic conditions, rat mesangial cells (MC) with Dox-inducible miR-29b overexpression or knockdown were treated with advanced glycation end (AGE). Results showed that addition of AGE significantly decreased miR-29b with a marked upregulation of collagen I (Figure 1b,e), IV (Figure 1c,e), and fibronectin (Figure 1d,e) by MC, which became worsen when miR-29b was knocked down (Figure 2). In contrast, Dox-induced overexpression of miR-29b significantly inhibited the fibrotic effect of AGE on collagen I and IV, and fibronectin expression (Figure 1). Furthermore, the protective role of miR-29b in AGE-induced fibrosis was associated with inhibition of TGF-β1 expression and activation of Smad3 because knockdown of miR-29b enhanced but overexpression of miR-29b blocked AGE-induced upregulation of TGF-β1 mRNA and phosphorylation of Smad3 in MC (Figures 1e,f and 2e,f). In addition, miR-29b expression was also significantly decreased under high glucose conditions (Supplementary Figure S1a).
Figure 1.
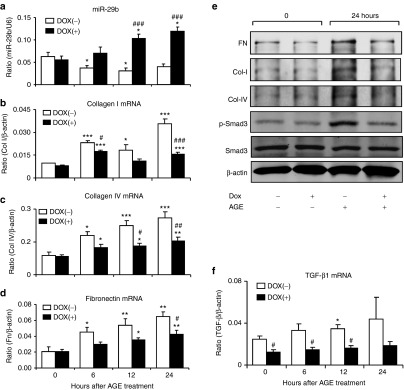
Overexpression of miR-29b attenuates advanced glycation end (AGE)-induced TGF-β/Smad3-mediated fibrosis in mesangial cells in vitro. (a) Real-time PCR detects miR-29b expression in response to AGE (50 µg/ml) stimulation by mesangial cells in the presence or absence of Dox (2 µg/ml)-induced miR-29b overexpression. (b–d) Real-time PCR shows that Dox-induced miR-29b overexpression inhibits AGE-induced collagen I, collagen IV, and fibronectin mRNA expression in mesangial cells. (e) Western blot analysis shows that Dox-induced miR-29b overexpression inhibits AGE-induced upregulation of fibronectin, collagen I, collagen IV, and phosphorylation of Smad3 in mesangial cells. (f) Real-time PCR shows that Dox-induced overexpression of miR-29b inhibits AGE-induced TGF-β mRNA expression by mesangial cells. Data represent the mean ± SE for at least four independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 compared to normal medium (0 hour). #P < 0.05, ##P < 0.01, ###P < 0.001 compared to Dox(−). Dox(+), Doxycycline induced-miR-29b overexpression.
Figure 2.

Knockdown of miR-29b enhances advanced glycation end (AGE)-induced TGF-β/Smad3-mediated fibrosis in mesangial cells. (a) Real-time PCR detects miR-29b expression in mesangial cells with/without miR-29b knockdown. (b–d) Real-time PCR detects that knockdown of miR-29b enhances AGE (50 µg/ml) induced collagen I, collagen IV, and fibronectin mRNA expression by mesangial cells. (e) Western blot analysis reveals that knockdown of miR-29b enhances AGE (50 µg/ml) induced collagen I, collagen IV, and fibronectin protein expression, and phosphorylation of Smad3. (f) knockdown of miR-29b enhances AGE (50 µg/ml) induced TGF-β expression in mesangial cells. Data represent the mean ± SE for at least four independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 compared to normal medium control (0 hour). #P < 0.05, ##P < 0.01, ###P < 0.001 compared to VC. VC, Vector control, KD, miR-29 knockdown.
Loss of intrarenal miR-29b is associated with an increase in microalbuminuria, TGF-β/Smad3-mediated renal fibrosis, NF-κB–driven renal inflammation, and T-bet–dependent immune injury in db/db mice
We then examined whether miR-29b expression is associated with the progression of diabetic kidney disease in db/db mice. Real-time PCR and in situ hybridizations showed that intrarenal levels of the miR-29b were largely decreased in db/db mice over the 10- to 20-week period, resulting in 60% reduction at week 20 when compared with age-matched db/m mice (Figure 3a,b). This was associated with a significant increase in microalbuminuria (Figure 3c), mesangial matrix expansion (Figure 3d,e), upregulation of collagen I, IV, fibronectin (Figures 4 and 5), and activation of TGF-β/Smad3 signaling (Figure 6). Interestingly, loss of renal miR-29b was also associated with upregulation of Sp1 and activation of the NF-κB pathway (Figure 7a,b), resulting in upregulation of renal TNF-α and MCP-1, and an increase in F4/80+ macrophage accumulation (Figure 7b,c). Importantly, we also found that decreased renal miR-29b contributed to upregulation of T-bet and interferon (IFN)-γ in both mRNA and protein levels (Figure 8a–d) and promoted CD4+IFN-γ+ T-cell infiltration (Figure 8e) over the 10- to 20-week period.
Figure 3.

miR-29b therapy attenuates microalbuminuria and histological damage in db/db mice. (a,b) Real-time PCR and in situ hybridization show that miR-29b expression is significantly reduced in the diabetic kidney of db/db mice, which is restored by miR-29b gene therapy using the ultrasound-microbubble gene transfer technique. (c) miR-29b treatment attenuates microalbuminuria in db/db mice. (d,e) miR-29 therapy inhibits histological injury including mesangial matrix index (PAS staining). (i) week 10 db/m mice, (ii) week 10 db/db mice, (iii) week 20 db/m mice, (iv) week 20 db/db mice, (v) week 20 db/db mice treated with empty vector (VC), and (vi) week 20 db/db mice treated with miR-29b. Scr, scramble probe as negative control for in situ hybridization (ISH). Data represent the mean ± SE for at groups of eight mice. **P < 0.01, ***P < 0.001 compared to week 20 db/m mice. #P < 0.05, ###P < 0.001 compared to week 20 db/db mice. †††P < 0.001 compared to week 10 db/m. g, glomerulus; m+, db/m; db, db/db, VC, db/db mice received empty vector control treatment; miR-29, db/db mice received miR-29b treatment. Original magnification: (b) ×200, (d) ×400.
Figure 4.

Immunohistochemistry and renal-time PCR detect that miR-29b therapy inhibits collagen I and collagen IV expression in the diabetic kidney of db/db mice. (a) Collagen I expression by immunohistochemistry and quantitative data in glomerular and tubulointerstitial areas. (b) Collagen I mRNA expression by real-time PCR. (c) Collagen IV expression by immunohistochemistry. (d) Collagen IV mRNA expression by real-time PCR. (i) week 10 db/m mice, (ii) week 10 db/db mice, (iii) week 20 db/m mice, (iv) week 20 db/db mice, (v) week 20 db/db mice treated with empty vector (VC), and (vi) week 20 db/db mice treated with miR-29b. Data represent the mean ± SE for at groups of eight mice. *P < 0.05, **P < 0.01, ***P < 0.001 compared to week 20 db/m mice. ##P < 0.01, ###P < 0.001 compared to week 20 db/db mice. ††P < 0.01 compared to week 10 db/m. m+, db/m; db, db/db, VC, db/db mice received empty vector control treatment; miR-29, db/db mice received miR-29b treatment. Original magnification: (a) ×200, (b) ×400.
Figure 5.

Western blot, immunohistochemistry, and real-time PCR reveal that miR-29b therapy inhibits renal fibrosis in db/db mice. (a) Western blot analysis shows that miR-29b therapy significantly reduces protein levels of collagen I, collagen IV and fibronectin. (b) Fibronectin immunostaining and quantitative data in the glomerular and tubulointerstitial areas. (c) Fibronectin mRNA expression by real-time PCR. (i) week 10 db/m mice, (ii) week 10 db/db mice, (iii) week 20 db/m mice, (iv) week 20 db/db mice, (v) week 20 db/db mice treated with empty vector (VC), and (vi) week 20 db/db mice treated with miR-29b. Data represent the mean ± SE for at groups of 8 mice. *P < 0.05, **P < 0.01, ***P < 0.001 compared to week 20 db/m mice; #P < 0.05, ##P < 0.01, ###P < 0.001 compared to week 20 db/db mice. †P < 0.05, †††P < 0.001 compared to week 10 db/m. m+, db/m; db, db/db, VC, db/db mice received empty vector control treatment; miR-29, db/db mice received miR-29b treatment. Original magnification: (b) ×400.
Figure 6.

miR-29b therapy blocks activation of TGF-β/Smad3 signaling in the diabetic kidney of db/db mice. (a) Immunohistochemistry for TGF-β1 expression. (b) Western blot analysis for phosphorylation of Smad3. (c) Real-time PCR for TGF-β1 mRNA expression. (i) week 10 db/m mice, (ii) week 10 db/db mice, (iii) week 20 db/m mice, (iv) week 20 db/db mice, (v) week 20 db/db mice treated with empty vector (VC), and (vi) week 20 db/db mice treated with miR-29b. Data represent the mean ± SE for at groups of 8 mice. *P < 0.05, **P < 0.01, ***P < 0.001 compared to week 20 db/m mice. #P < 0.05, ##P < 0.01 compared to week 20 db/db mice. ††P < 0.01 compared to week 10 db/m. m+, db/m; db, db/db, VC, db/db mice received empty vector control treatment; miR-29, db/db mice received miR-29b treatment. Magnification ×100.
Figure 7.

miR-29b therapy inhibits Sp1/NF-κB–driven renal inflammation in the diabetic kidney of db/db mice. (a) Western blot analysis of Sp1 expression and NF-κB/p65 phosphorylation. (b) Real-time PCR analysis of mRNA expression of Sp1, NF-κB/p65, TNF-α, and MCP-1. (c) F4/80+ macrophages infiltrating the glomerulus and tubulointerstitium. Data represent the mean ± SE for at groups of eight mice. *P < 0.05, **P < 0.01, ***P < 0.001 compared to week 20 db/m mice. #P < 0.05, ##P < 0.01, ###P < 0.001 compared to week 20 db/db mice. †††P < 0.01 compared to week 10 db/m. m+, db/m; db, db/db, VC, db/db mice received empty vector control treatment; miR-29, db/db mice received miR-29b treatment.
Figure 8.

miR-29b treatment inhibits T-bet–dependent Th1-mediated immune response in the diabetic kidney of db/db mice. (a)Western blot analysis of T-bet expression. (b,c) Real-time PCR analysis of T-bet and IFN-γ mRNA expression. (d) ELISA analysis of plasma IFN-γ levels. (e) CD4+IFN-γ+ T cells by two-color immunofluorescence. CD4 (green), IFN-γ (red), nuclei were counterstained with DAPI (blue). Results reveal that miR-29 treatment blocks Th1-mediated immune response as evidenced by inhibition of T-bet and IFN-γ expression and CD4+IFN-γ+ T helper cell infiltration. Data represent the mean ± SE for at groups of 8 mice. *P < 0.05, **P < 0.01 compared to week 20 db/m mice. #P < 0.05, ##P < 0.01 compared to week 20 db/db mice. ††P < 0.01 compared to week 10 db/m. m+, db/m; db, db/db, VC, db/db mice received empty vector control treatment; miR-29, db/db mice received miR-29b treatment. Original magnification ×800.
miR-29b therapy inhibits renal injury in db/db mice
We next studied if restored renal miR-29b is able to inhibit diabetic kidney injury. As shown in Figure 3a,b, both real-time PCR and in situ hybridization detected ultrasound-microbubble–mediated gene transfer of miR-29b restored normal levels of miR-29b over the 10- to 20-week period. This resulted in a significant inhibition of diabetic kidney disease as evidenced by normalizing microalbuminuria (Figure 3c) and histological injury such as mesangial matrix expansion over the 10- to 20-week period in db/db mice (Figure 3d,e).
Mechanisms of miR-29b protect against DN
Renal fibrosis is a hallmark of DN and has been shown to be TGF-β/Smad-dependent.5 We thus examined if the blockade of diabetic kidney injury by miR-29b is associated with inhibition of TGF-β/Smad3-mediated renal fibrosis. As shown in Figure 4, immunohistochemistry and real-time PCR analysis demonstrated that restored intrarenal miR-29b to normal levels in the diabetic kidney by ultrasound-microbubble–mediated miR-29b gene transfer protected against renal fibrosis as evidenced by inhibition of collagen I and IV (Figure 4), and fibronectin expression (Figure 5) at both mRNA and protein levels. Further studies showed that blockade of renal fibrosis by miR-29b was largely attributed to inhibition of TGF-β1 expression and phosphorylation of Smad3 (Figure 6), revealing the counter-regulatory role of miR-29b in TGF-β/Smad3-dependent renal fibrosis in diabetes.
DN is also regarded as a low grade inflammation disease. Therefore, we examined the therapeutic effect of miR-29b on NF-κB–driven renal inflammation in db/db mice. As shown in Figure 7, miR-29b therapy significantly inhibited upregulation of TNF-α and MCP-1, and F4/80+ macrophage infiltration. The anti-inflammatory effect of miR-29b was attributed to inhibition of Sp1 expression, resulting in the blockade of the activation of NF-κB/p65 pathway (Figure 7a,b). This finding revealed a possible counter-regulatory mechanism of miR-29b in Sp1/NF-κB–driven renal inflammation in DN.
Interestingly, we also found that restored renal miR-29b was capable of inhibiting T-bet–dependent Th1 immune response in the diabetic kidney of db/db mice. As shown in Figure 8, real-time PCR and western blot analysis detected that miR-29b therapy largely inhibited T-bet mRNA and protein expression, thereby inhibiting Th1 signature cytokine IFN-γ expression at both mRNA and protein levels (Figure 8a–d), resulting in inhibition of CD4+IFN-γ+ Th1 cell infiltration (Figure 8e).
Discussion
Increasing evidence shows that renal inflammation and its consequent fibrosis are a critical process leading to end-stage of DN. This study demonstrated that loss of miR-29b in the diabetic kidney enhanced this process with progressive renal injury in db/db mice, which was inhibited by miR-29b therapy. Findings from this study demonstrated a protective role and therapeutic potential of miR-29b for DN. The therapeutic effect of miR-29b on DN may be associated with the suppressive effect of miR-29b on Sp1-dependent, TGF-β/Smad3-mediated renal fibrosis, NF-κB–driven renal inflammation, and Th1-associated immune injury.
In this study, we found that renal miR-29b was largely decreased in association with the upregulation of collagen matrix and fibronectin in the db/db mouse kidney and in MC under high AGE and high glucose conditions. These findings indicated that loss of miR-29b in diabetic kidney may be attributed to the development of DN. Findings from this study that knockdown of miR-29b enhanced but overexpression of miR-29b inhibited renal fibrosis and inflammation provided evidence for a protective role of miR-29b in DN. As illustrated in Supplementary Figure S2, inhibition of TGF-β/Smad3-mediated renal fibrosis may be a mechanism associated with a protective effect of miR-29b on DN. It is well established that miR-29 directly targets the 3′UTRs of collagen I/III/IV, fibrillin, and elastin.15,23 Therefore, overexpression of miR-29 may repress the expression of collagens I and IV directly.26 Interestingly, miR-29b treatment substantially reduced levels of fibronectin expression in vivo and in vitro. Although fibronectin is not the predicted target gene of miR-29b as indicated by Targetscan (http://www.targetscan.org/) and PicTar (http://www.pictar.org/), the inhibitory effect of miR-29b on fibronection expression may be associated with the ability of miR-29b to block TGF-β/Smad3 signaling. We have previously demonstrated that Smad3 can physically interact with the miR-29b promoter and negatively regulates its expression in response to TGF-β1 in vitro and in obstructive nephropathy.20 miR-29 has also been shown to directly inhibit TGF-β1 transcription.28 Because TGF-β/Smad3 plays a pivotal role in renal fibrosis in diabetes,5,27,29 findings from this study added new evidence for TGF-β/Smad3 as a potential target of miR-29b in DN. Therefore, miR-29b may exert its protective effect on DN by negatively regulating TGF-β/Smad3-mediated renal fibrosis. In addition, cooperation between Smad3 and Sp1 has been shown to enhance the TGF-β-induced fibrotic response.30 Thus, inhibition of Sp1 expression, thereby blocking the Sp1-Smad3 interaction, may be another mechanism by which miR-29b suppresses TGF-β/Smad3-mediated renal fibrosisin the diabetic kidney of db/db mice.
Inhibition of Sp1/NF-κB–driven renal inflammation could be another mechanism whereby miR-29b inhibited diabetic kidney disease (Supplementary Figure S2). It is now well accepted that diabetes mellitus is a low grade inflammatory disease.3,22 Upregulation of proinflammatory cytokines such as TNF-α and MCP-1, and macrophage infiltration are critical in renal injury in type 2 diabetes.5,29,31,32 Consistent with these findings, this study revealed that upregulation of these proinflammatory cytokines was associated with a loss of miR-29b and activation of the Sp1/NF-κB pathway. Because miR-29 is negatively regulated by the NF-κB-YY1-miR-29 regulatory circuit,33 downregulation of miR-29b in the diabetic kidney may also be NF-κB–dependent, in addition to the TGF-β/Smad3-dependent mechanism.20 It has been shown that Sp1 functions as a key transcriptional factor to activate the NF-κB pathway.34 As shown in Supplementary Figure S3a, Sp1 is also a target of miR-29b. Thus, this study detected that the ability of miR-29b treatment to suppress renal inflammation may be associated with the blockade of the Sp1/NF-κB pathway (Supplementary Figure S2). This is supported by a recent finding of the Sp1/NF-κB/miR-29b autoregulatory loop in acute myeloid leukemia.25
We also found that inhibition of Th1-mediated immune response may be a new mechanism whereby miR-29b protects against DN (Supplementary Figure S2). Although leptin receptor deficiency can impair T-cell activation and proliferation in db/db mice,34 T cells do participate in immune response in diabetic kidneys of db/db mice as demonstrated by an increase in Th1 and Th17 subsets and a decrease in the Treg subset.35 T-bet is a Th1-specific T-box transcription factor that contributes to the induction of the hallmark Th1 cytokine, IFN-γ.12,36 Studies have reported that IFN-γ expression is significantly increased in DN.37,38 In this study, we also found upregulation of T-bet and an increase in IFN-γ in the diabetic kidney of db/db mice. Importantly, enhanced Th1 immune response in the diabetic kidney of db/db mice was associated with downregulation of miR-29b. Whereas, overexpression of renal miR-29b attenuated Th1-mediated immune response by inhibiting the T-bet-IFN-γ pathway. Indeed, miR-29b can bind T-box transcription factor (Supplementary Figure S3b), thereby inhibiting IFN-γ production.24,39,40 Thus, the therapeutic effect of miR-29b on diabetic kidney disease in db/db mice may be attributed to its inhibitory effect on T-bet–dependent Th1-mediated renal injury.
It should be pointed out that although miR-29b is reported to be able to target Sp1 in mononuclear cells and T-box in T cells,24,25,40 and both mRNA and protein levels of Sp1 and T-bet in the diabetic kidney in this study were found to be tightly associated with the expression of miR-29b, the direct regulatory mechanisms of miR-29b on Sp1 and T-bet–dependent inflammatory responses in diabetes remain largely unclear.
In summary, as shown in the Supplementary Figure S2, this study reveals that loss of renal miR-29b may contribute to progressive renal injury in db/db mice, which may be regulated by both TGF-β/Smad3 and NF-κB signaling pathways. Restored intrarenal levels of miR-29b by ultrasound-mediated gene therapy is capable of inhibiting DN by blocking TGF-β/Smad3-mediated renal fibrosis and NF-κB–driven renal inflammation. Moreover, miR-29b may also suppress T-bet–/IFN-γ–dependent immune response in db/db mice. Taken together, miR-29b may represent a novel therapeutic approach for DN.
Materials and Methods
Animal model and ultrasound-microbubble–mediated-Mir-29b gene transfer. Male db/db mice and their normal littermates (db/m) at the age of week 10 were purchased from Laboratory Animal Services Centre, the Chinese University of Hong Kong. Groups of eight mice were used: (i) groups of db/m or db/db mice were sacrificed at the age of week 10 as basic level control and (ii) groups of db/m or db/db mice were treated with/without miR-29b or empty vector control from the age of week 10 and sacrificed at week 20. All mice were maintained in a standard animal house with 12 hour/12 hour light/dark cycle and were euthanized with intraperitoneal injection of ketamine/xylene. The renal cortexes were collected by carefully removing the renal pelvis and medullar tissues. Cortexes were used for real-time PCR and western blot analysis or fixed in methyl Carnoy's for histological and immunohistochemical analysis.
A noninvasive ultrasound-microbubble–mediated gene transfer technique was used to deliver doxycline (Dox)-inducible pre-miR-29b or empty control plasmids into both kidneys following the protocol as previously described.20 Briefly, a murine miR-29b cDNA was amplified by PCR with the forward primer 5′-GAC ACG GAT CCA ATG TAA GCC TCG TGC TCA CTG-3′ and the reverse primer 5′-GAC TTC TCG AGT CGA CAA GGT CAT GTG CAC TGG GAA G-3′. The PCR-amplified product was subsequently cloned into a tetracycline-inducible vector, pTRE-hygro (Clontech, Mountain View, CA) to obtain pTRE-miR-29b. 200 μl of the mixture of pTRE-miR-29b/Tet-on plasmids/Sonovue (Bracco Diagnostics, NJ) or the control empty pTRE/Tet-on plasmids/sonovue in a 1:1 ratio (volume:volume) were intravenously injected to mouse via tail vein, followed immediately by 4 minutes of ultrasound treatment (2 W/cm2) by placing the ultrasound probe on the skin opposite to both kidney as described previously.20 Based on our previous finding,20 a dose of 100 μg/25 g body weight of pTRE-miR-29b plasmid or empty vector control was used. According to the preliminary results that ultrasound-mediated miR-29b expression in the kidney peaked at days 3–7 and declined at day 14, four treatments of miR-29b at every 18 days over the 10-week period (from week 10 to week 20) were performed by using ultrasound-microbubble–mediated pTRE2-miR-29b or pTRE2 empty vector. Levels of renal miR-29b transgene expression were tightly controlled with additional Dox in the drinking water (200 μg/ml) over the entire study period. Both db/m and db/db control mice were also given with the same concentration of Dox in the drinking water over the entire period. The experimental protocol was approved by the Animal Experimentation Ethics Committee at The Chinese University of Hong Kong.
Real-time polymerase chain reaction. Total RNA from renal cortex and cultured cells was isolated by Trizol (Invitrogen, CA). Expression of mRNAs for collagen I, IV, fibronectin, TGF-β1, IL-1β, TNF-α, MCP-1, and miR-29b was measured by real-time PCR with primers as previously described,5,18,20 while mRNA levels of IFN-γ and T-bet were measured by using the primers as described below: IFN-γ (forward 5′-AGCAACAGCAAGGCGAAAAA-3′, reverse 5′-AGCTCATTGAATGCTTGGCG-3′); T-bet (forward 5′-CCAGGAAGTTTCATTTGGGAAGC-3′, reverse 5′-ACGTGTGTGTTAGAAGCACTG-3′); Sp1 (forward 5′-GCTGCCACCATGAGCGACCAA-3′, reverse 5′-CACCGCCACCATTGCCGCTA-3′); and NF-κB/p65 (forward 5′-AGATCTTCTTGCTGTGCGACAA-3′, reverse 5′-GTGCCTCCCAGCCTGGT-3′). The housekeeping genes β-actin or U6 was used as an internal control. The ratio for the specific gene of interest over the internal control was expressed as mean ± SE.
Western blot. Protein from renal cortex and harvested cells was extracted by RIPA, and western blot analysis was performed as previously described.41 Primary antibodies used in this study included those against phospho-NF-κB/p65 (ser276), phospho-Smad3 (Cell Signaling Technology, MA), fibronectin (Dako, Glostrup, Denmark), Smad3 (Upstate Biotechnology), collagen I, collagen IV (Southern Biotech, Birmingham, AL), T-bet (N-19), NF-κB/p65 (H-286), Sp1 (H-225), and β-actin (Santa Cruz Biotechnology, CA). After being incubated with the primary antibody at 4 °C for over night, the membrane was stained with the LI-COR IRDye 800-labeled secondary antibodies (Rockland Immunochemicals, PA). The signals were detected with Odyssey Infrared Imaging System (Li-COR Biosciences, NE) and quantitated with ImageJ program (NIH). The ratio for the protein examined was normalized against β-actin or total proteins as stated in the studies and expressed as the mean ± SE.
Urinary albumin excretion. Urinary samples (24 hours) were collected in metabolic cages every 2 weeks. Urinary albumin excretion was measured by competitive ELISA according to the manufacturer's instructions (Exocell, PA). Urinary creatinine was measured by an enzymatic kit (Stanbio Labs, TX). Urinary albumin excretion was expressed as total urinary albumin/creatinine ratio.
Histology and immunohistochemistry. Both histological and immunohistochemical analyses were performed on the methyl Carnoy's fixed, paraffin-embedded tissue sections (3 μm) as previously described.42,43 Mesangial matrix expansion was measured by staining with periodic acid Schiff reagent. Tissue sections were incubated with primary antibodies against fibronectin (Dako, CA), TGF-β1, monocyte chemotactic protein-1 (MCP-1) (Cell Signaling Technology), Collagen I and IV (Southern Tech, AL), F4/80 (AbD Serotec, Kidlington, UK) for overnight at 4 °C, followed by incubation with appropriate HRP-labeled secondary antibodies. After being developed with diaminobenzidine to produce a brown color, sections were counterstained with hematoxylin. T-cell infiltration study was performed on the PLP fixative, frozen tissue sections (4 μm). To detect the Th1 cells, sections were incubated with the FITC-conjugated antibody against CD4 (eBioSciences, CA) and the rabbit anti-mouse IFN-γ (AbD Serotec, Kidlington, UK) for overnight at 4 °C, followed by the goat anti-rabbit Alexa Fluor 555 (Invitrogen) for 1 hour. Nuclei were counterstained with DAPI.
Changes in both histology and immunohistochemistry were quantitatively analyzed using the quantitative image program (Image-Pro Plus 7.0, Media Cybernetics, Bethesda, MD) as previously described.5 Briefly, the area in both glomeruli and tubulointerstitium was outlined, and the positive signals were quantitatively measured and expressed as the percentage of the area examined. For quantitative analysis F4/80+ macrophages, positive cells in 20 consecutive glomeruli were counted and expressed as cells/glomerular cross-section, whereas positive cells in the tubulointerstitium were counted under high-power fields (×40) by means of a 0.0625-mm2 graticule fitted in the eyepiece of the microscope and expressed as cells per millimeters squared.
In situ hybridization. miR-29b expression in the mouse kidney was detected by in situ hybridization as previously described.18,20 Briefly, paraformaldehyde fixed, paraffin-embedded kidney sections (6 μm) were treated with 5 μg/ml protein kinase solution at room temperature for 10 minutes, followed by pretreatment with hybridization buffer. Sections were then incubated with LNA-digoxigenin labeled mmu-miR-29b probe (5′-AACACTGATTTCAAATGGTGCTA-3′, Exiqon, Vedbaek, Denmark) or negative control scramble probe (5′-GTGTAACACGTCTATACGCCCA-3′, Exiqon) in a humid chamber overnight at 54 °C. After washing, the sections were incubated with anti-digoxigenin antibody conjugated to alkaline phosphatase (Roche Diagnostics, Indianapolis, IN) for overnight at 4 °C, and developed with 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (Sigma-Aldrich, MO) to produce positive signals.
Cell culture. To study the role of miR-29 in vitro, an established Dox-inducible miR-29b expressing rat MC line and tubular epithelial cell line (NRK52E) were used.20 Cells were treated with AGE products at a dose of 50 µg/ml for 0, 6, 12, and 24 hours. AGE was purchased from Sigma-Aldrich (Cat# A8426) and was dissolved in endotoxin free 1XPBS (10 mg/ml) and sterilized with 0.22 µm filter before use. Dox at concentrations of 0, 0.25, 0.5, 1, and 2 μg/ml was added into the culture, and an optimal dose of Dox at 2 μg/ml for induction of miR-29b was defined. Four to five independent experiments were performed. In addition, we also examined miR-29b expression under high glucose conditions. Rat MC were cultured with high glucose (30 mmol/l) or control mannitol (5.5 mmol/l glucose plus 24.5 mmol/l mannitol) for or 0, 6, 12, and 24 hours and examined for miR-29b expression by real-time PCR.
To knock down miR-29b from MC, the gene-specific insert sequences for miR-29b1 (sense: TAGTGATTGTCTAGCACCATT and antisense: AATGGTGCTAGACAATCACTA) and miR-29b2 (sense: ATCTTTGTATCTAGCACCATT and antisense: AATGGTGCTAGATACAAAGAT) were subcloned into pSuper-puro vectors (OligoEngine, Seattle, WA) to generate pSuper-puro-miR-29b RNA interference constructs as previously described.20 pSuper-puro-miR-29b was transfected into rat MC. Cells were treated with AGE at a dose of 50 µg/ml for 0, 6, 12, and 24 hours. Four independent experiments were performed.
Statistical analysis. Data from this study were expressed as mean ± SE. Statistical analyses were performed using one-way analysis of variance, followed by Newman–Keuls Post Test from the Prism Program (Prism 5.0 GraphPad Software, San Diego, CA) except stated otherwise. In addition, a repeated analysis of variance was used for albumin excretion analysis.
SUPPLEMENTARY MATERIAL Figure S1. Expression of miR-29 family members by mesangial cells and tubular epithelial cells in response to high glucose or AGEs in vitro. Figure S2. miR-29b inhibits diabetic nephropathy by blocking the TGF-β/Smad-Sp1/NF-κB-Th1 pathways. Figure S3. miR-29b binds to 3′ UTR of Sp1 and Tbx21 (T-bet) mRNA to repress their expression (http://www.targetscan.org/).
Acknowledgments
This work was supported by grants from Major State Basic Research Development Program of China (973 program, No. 2012CB517700); The Shenzhen Basic Research Program (SZSITC, JC201104220290A); the Research Grant Council of Hong Kong (GRF469110, CUHK5/CRF09, and CUHK3/CRF/12R); NSFC/RGC Joint Research Scheme (N_CUHK404/10); and the Focused Investment Scheme A and B from the Chinese University of Hong Kong. The authors declare no conflict of interest.
Supplementary Material
References
- Declèves AE, Sharma K. New pharmacological treatments for improving renal outcomes in diabetes. Nat Rev Nephrol. 2010;6:371–380. doi: 10.1038/nrneph.2010.57. [DOI] [PubMed] [Google Scholar]
- Reeves WB, Andreoli TE. Transforming growth factor beta contributes to progressive diabetic nephropathy. Proc Natl Acad Sci USA. 2000;97:7667–7669. doi: 10.1073/pnas.97.14.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb S, Ziyadeh FN. TGF-beta: a crucial component of the pathogenesis of diabetic nephropathy. Trans Am Clin Climatol Assoc. 2001;112:27–32; discussion 33. [PMC free article] [PubMed] [Google Scholar]
- Huang C, Kim Y, Caramori ML, Fish AJ, Rich SS, Miller ME, et al. Cellular basis of diabetic nephropathy: II. The transforming growth factor-beta system and diabetic nephropathy lesions in type 1 diabetes. Diabetes. 2002;51:3577–3581. doi: 10.2337/diabetes.51.12.3577. [DOI] [PubMed] [Google Scholar]
- Chen HY, Huang XR, Wang W, Li JH, Heuchel RL, Chung AC, et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes. 2011;60:590–601. doi: 10.2337/db10-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belghith M, Bluestone JA, Barriot S, Mégret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- Filippi CM, Juedes AE, Oldham JE, Ling E, Togher L, Peng Y, et al. Transforming growth factor-beta suppresses the activation of CD8+ T-cells when naive but promotes their survival and function once antigen experienced: a two-faced impact on autoimmunity. Diabetes. 2008;57:2684–2692. doi: 10.2337/db08-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, et al. Atherosclerosis Risk in Communities Study Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes. 2003;52:1799–1805. doi: 10.2337/diabetes.52.7.1799. [DOI] [PubMed] [Google Scholar]
- Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813–823. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- Crook M. Type 2 diabetes mellitus: a disease of the innate immune system? An update. Diabet Med. 2004;21:203–207. doi: 10.1046/j.1464-5491.2003.01030.x. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Ihara K, Matsuura N, Kohno H, Nagafuchi S, Kuromaru R, et al. Identification of a novel type 1 diabetes susceptibility gene, T-bet. Hum Genet. 2004;115:177–184. doi: 10.1007/s00439-004-1146-2. [DOI] [PubMed] [Google Scholar]
- Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. 2011;186:1162–1172. doi: 10.4049/jimmunol.1002615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer B, Stanczyk J, Jüngel A, Akhmetshina A, Trenkmann M, Brock M, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010;62:1733–1743. doi: 10.1002/art.27443. [DOI] [PubMed] [Google Scholar]
- Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, et al. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45:287–294. doi: 10.1165/rcmb.2010-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Meng XM, Huang XR, Chung AC, Feng YL, Hui DS, et al. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol Ther. 2012;20:1251–1260. doi: 10.1038/mt.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM, et al. TGF-ß/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol. 2011;22:1462–1474. doi: 10.1681/ASN.2010121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du B, Ma LM, Huang MB, Zhou H, Huang HL, Shao P, et al. High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 2010;584:811–816. doi: 10.1016/j.febslet.2009.12.053. [DOI] [PubMed] [Google Scholar]
- Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res. 2010;107:810–817. doi: 10.1161/CIRCRESAHA.110.226357. [DOI] [PubMed] [Google Scholar]
- Long J, Wang Y, Wang W, Chang BH, Danesh FR. MicroRNA-29c is a signature microRNA under high glucose conditions that targets Sprouty homolog 1, and its in vivo knockdown prevents progression of diabetic nephropathy. J Biol Chem. 2011;286:11837–11848. doi: 10.1074/jbc.M110.194969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner DF, Thomas MF, Hu JK, Yang Z, Babiarz JE, Allen CD, et al. MicroRNA-29 regulates T-box transcription factors and interferon-? production in helper T cells. Immunity. 2011;35:169–181. doi: 10.1016/j.immuni.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wu LC, Pang J, Santhanam R, Schwind S, Wu YZ, et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell. 2010;17:333–347. doi: 10.1016/j.ccr.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, et al. Suppression of microRNA-29 expression by TGF-ß1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol. 2012;23:252–265. doi: 10.1681/ASN.2011010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AC, Dong Y, Yang W, Zhong X, Li R, Lan HY. Smad7 suppresses renal fibrosis via altering expression of TGF-ß/Smad3-regulated microRNAs. Mol Ther. 2013;21:388–398. doi: 10.1038/mt.2012.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna C, Li G, Qiu J, Epstein DL, Gonzalez P. Cross-talk between miR-29 and transforming growth factor-betas in trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2011;52:3567–3572. doi: 10.1167/iovs.10-6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Chen HY, Huang XR, Chung AC, Zhou L, Fu P, et al. C-reactive protein promotes diabetic kidney disease in a mouse model of type 1 diabetes. Diabetologia. 2011;54:2713–2723. doi: 10.1007/s00125-011-2237-y. [DOI] [PubMed] [Google Scholar]
- Poncelet AC, Schnaper HW. Sp1 and Smad proteins cooperate to mediate transforming growth factor-beta 1-induced alpha 2(I) collagen expression in human glomerular mesangial cells. J Biol Chem. 2001;276:6983–6992. doi: 10.1074/jbc.M006442200. [DOI] [PubMed] [Google Scholar]
- Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols E, Rollins BJ, Tesch GH. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. 2007;50:471–480. doi: 10.1007/s00125-006-0497-8. [DOI] [PubMed] [Google Scholar]
- Ninichuk V, Khandoga AG, Segerer S, Loetscher P, Schlapbach A, Revesz L, et al. The role of interstitial macrophages in nephropathy of type 2 diabetic db/db mice. Am J Pathol. 2007;170:1267–1276. doi: 10.2353/ajpath.2007.060937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell. 2008;14:369–381. doi: 10.1016/j.ccr.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Cui J, Duan X, Chen H, Fan F. Suppression of type I collagen expression by miR-29b via PI3K, Akt, and Sp1 pathway in human Tenon's fibroblasts. Invest Ophthalmol Vis Sci. 2012;53:1670–1678. doi: 10.1167/iovs.11-8670. [DOI] [PubMed] [Google Scholar]
- Lago R, Gómez R, Lago F, Gómez-Reino J, Gualillo O. Leptin beyond body weight regulation–current concepts concerning its role in immune function and inflammation. Cell Immunol. 2008;252:139–145. doi: 10.1016/j.cellimm.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes. 2011;60:2954–2962. doi: 10.2337/db11-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- Mensah-Brown EP, Obineche EN, Galadari S, Chandranath E, Shahin A, Ahmed I, et al. Streptozotocin-induced diabetic nephropathy in rats: the role of inflammatory cytokines. Cytokine. 2005;31:180–190. doi: 10.1016/j.cyto.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Moon JY, Jeong KH, Lee TW, Ihm CG, Lim SJ, Lee SH. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am J Nephrol. 2012;35:164–174. doi: 10.1159/000334928. [DOI] [PubMed] [Google Scholar]
- Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon- Nat Immunol. 2011;12:861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- Chung AC, Huang XR, Zhou L, Heuchel R, Lai KN, Lan HY. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol Dial Transplant. 2009;24:1443–1454. doi: 10.1093/ndt/gfn699. [DOI] [PubMed] [Google Scholar]
- Wang W, Huang XR, Li AG, Liu F, Li JH, Truong LD, et al. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: role of Smad7. J Am Soc Nephrol. 2005;16:1371–1383. doi: 10.1681/ASN.2004121070. [DOI] [PubMed] [Google Scholar]
- Lan HY, Mu W, Nikolic-Paterson DJ, Atkins RC. A novel, simple, reliable, and sensitive method for multiple immunoenzyme staining: use of microwave oven heating to block antibody crossreactivity and retrieve antigens. J Histochem Cytochem. 1995;43:97–102. doi: 10.1177/43.1.7822770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.