

Abstract
RNA interference has become a ubiquitous biological tool, and is being harnessed for therapeutic purposes as well. Therapeutic posttranscriptional gene silencing takes advantage of the endogenous RNAi pathway through delivery of either chemically synthesized siRNAs, or transgenes expressing hairpin-based inhibitory RNAs (e.g., shRNAs and artificial miRNAs). RNAi has expanded the field of viral gene therapy from gene replacement to gene knockdown. Here, we review various noncoding RNAs such as shRNAs, miRNAs, and miRNA decoys which can be utilized for therapeutic applications when expressed from recombinant adeno-associated vectors (AAV), and present examples of their basic design. In addition the basis of exploiting cellular miRNA profiles for detargeting AAV expression from specific cells is described. Finally, an overview of AAV-mediated RNAi preclinical studies is presented, and current RNAi-based clinical trials are reviewed.
Introduction
Since the first reports of the phenomenon in Petunia hybrida L. in the 1990's,1,2 its description in C. elegans in 1998,3 and its identification in mammalian cells in 20014 and mice in 2002,5 RNA interference, or RNAi, has sustained the interest of the scientific community. Not only does it offer a powerful biological tool, but also new therapeutic possibilities for nondruggable targets. Similarly, RNAi expanded the field of viral gene therapy from gene replacement to gene knockdown, quickly replacing other strategies such as ribozymes and antisense transcripts. Importantly, it has also opened up the field to tackle genetic disorders that result from both a loss-of-function and toxic gain-of-function.6,7 Since the awarding of a 2006 Nobel Prize for the description of RNAi, academic groups as well as the pharmaceutical industry have shown interest in its application. This has grown into a reported 162 companies involved in the development of RNAi technologies, with 33 developing RNAi therapeutics and 35 developing microRNA (miRNA) therapeutics.8
The Cellular Machinery
RNAi resulting from the endogenous miRNA pathway regulates gene expression by controlling the synthesis of protein through posttranscriptional gene silencing.9 The miRNA biogenesis will only be briefly described here; for more details on this topic refer to a recent review.10 Aside from a few exceptions,11,12 the miRNA gene is usually transcribed by RNA polymerase II (RNA Pol II) from independent transcription units or from the intron of protein coding genes into a ~1–4 kb13 primary transcript called a pri-miRNA (Figure 1). In mammals the pri-miRNA is cleaved by the microprocessor complex consisting of RNAse III Drosha and the RNA-binding protein DiGeorge syndrome critical region gene 8 (DGRC8) into a ~60–80 nt-long precursor miRNA (pre-miRNA). The pre-miRNA di-nucleotide 3′ overhang and stem-loop get recognized by Exportin 5 which exports it from the nucleus to the cytoplasm via the nuclear pore (Figure 1). In the cytoplasm, the pre-miRNA is bound by a second RNAse III enzyme, known as Dicer. Much like the microprocessor complex, Dicer and its partner proteins, protein kinase R activator (PACT) and TAR RNA-binding protein (TRBP), recognize the base of the hairpin stem and cleave at a fixed distance. This measured interaction results in cleavage of the loop and the production of ~22nt-long miRNA-miRNA* duplex (Figure 1). Importantly, at this point in the pathway, RNAi can also be triggered by exogenous double-stranded RNA (dsRNA) which can be processed by Dicer to give a ~22 nt-long siRNA-siRNA*duplex.
Figure 1.
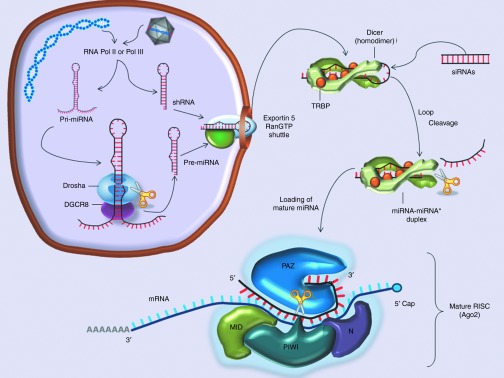
RNAi and miRNA Biogenesis. Pri-miRNA are transcribed from either endogenous or vector-derived genes by RNA polymerase II or polymerase III. The pri-miRNA transcript has a 7-methylguanosine cap and poly-A tail, as it enters the microprocessor complex (Drosha & DGCR8) for its first cleavage event. The resulting pre-miRNA as well as vector-derived shRNAs can then be bound by the Exportin 5 RanGTP shuttle and exported though a nuclear pore into the cytoplasm. After export to the cytoplasm, pre-miRNAs and shRNAs associate with Dicer and the double-stranded RNA-binding protein TRBP. This cleavage event results in the production of ~22-nt-long miRNA/miRNA* duplex. The guide strand of the duplex is then preferentially loaded into Ago, whereas the passenger strand is usually degraded. The catalytic activity of the RISC is imparted by the Ago proteins 1–4, shown here is Ago2 which contains a PIWI domain with “slicer” activity.
After pre-miRNA processing, the mature miRNA or functional siRNA or miRNA* strand (i.e., the antisense guide strand which is complementary to the messenger RNA (mRNA)) is incorporated into the “RNA-induced silencing complex” (RISC), while the opposite strand (i.e., the passenger strand) is degraded. At the core of the catalytic domain of the RISC are the Ago proteins. Ago proteins consist of four distinct domains: the N-terminal, PAZ, Mid, and PIWI domains. The PIWI domain resembles a RNase H-like enzyme. In some Ago family members (e.g., Ago2) this domain retains a functional catalytic center allowing for the cleavage of a target mRNA (Figure 1). The seed sequence (6–8 nt of the 5′ end) of the guide strand leads the RISC to bind to the mRNA by sequence complementarity. This leads to translational repression, enhanced mRNA degradation or site-specific mRNA cleavage. The later event is the most efficient inhibitory mechanism and only occurs if there is complete or near complete complementarity between the mRNA and the mi/siRNA.
Limitations of Oligonucleotide-Mediated RNAi
The earliest attempts to harness RNAi therapeutically were to deliver dsRNA molecules to feed into the RNAi pathway at the level of Dicer. For this approach, efficient delivery of RNAi effectors has been the greatest hurdle for clinical translation but there have been improvements in lipid-based siRNA delivery in the last few years.14 Another impediment has been that the direct administration of dsRNA in the form of siRNAs complexed to lipids has been shown to activate innate inflammatory pathways. Specifically, activation of the interferon response by the small RNA can trigger toxicity. Also, toll-like receptor 3 (TLR3), TLR7, and TLR8 can recognize siRNA in a sequence-dependent (TLR7 and TLR8) or -independent (TLR3) fashion, and induce interferon, interleukin 6, and tumor necrosis factor α.15,16,17 Owing to the fact that siRNAs degrade over time, these responses can be limiting when one is faced with the notion of lifelong repeated administration in some cases. Fortunately, improved lipid chemistries, chemical modifications, and recent advances in eliminating specific motifs have reduced, but not abolished, these potential serious side-effects.14
Thus, the main limitations of siRNA therapeutics today are efficient delivery to organs other than the liver, longevity and the associated innate immune response with each dose. In cases where sustained expression of RNAi effectors is needed, a vector-derived approach such as the ones described below may be more adequate.
AAV-Mediated RNAi Delivery
Vector-derived RNAi is where a vector is used to express RNA transcripts (e.g., short-hairpin RNAs (shRNAs) or micro RNAs (miRNAs) that are ultimately processed to produce siRNAs in the target cells. In this review, we focus on the use of recombinant adeno-associated vectors (rAAV) to achieve this goal. Adeno-associated virus currently is one of the most attractive gene therapy vectors. First, the virus naturally infects primates and is nonpathogenic. Second, the recombinant form used for therapy, where the rep and cap genes have been removed, remains almost completely episomal with a reported 0.05% integration in neonatal mice18 and between 10E−4 and 10E−5 in liver and muscle of nonhuman primates19 and humans.20 The relatively limited packaging capacity of AAV of 4.7 kb can be a disadvantage when designing vectors for gene replacement but not for RNAi-based applications, which typically employ smaller-sized expression cassettes. In recent times, rAAV vectors have continued to add to their safety profile by their evidence of therapeutic success in clinical trials (in particular for Leber's congenital amaurosis,21 hemophilia B,22 and lipoprotein lipase deficiency).23 Below, we describe the use of AAV-based strategies for the expression of noncoding inhibitory RNAs as well as the use of noncoding RNAs to detarget AAV expression. We conclude with an overview of AAV-mediated RNAi preclinical studies, and current RNAi-based clinical trials.
shRNA- and Artificial miRNA-Mediated Knockdown
To date most of the AAV-RNAi approaches takes either the form of a shRNA or pri-miRNA as the effector molecule. While the design differences are subtle, and they both converge to silence targets via the RISC, they are processed differently, which as described below, may have important consequences. For guidance on how to design and clone either of these into AAV vectors refer to the following protocol.24
ShRNAs are stem-loop RNAs that bypass Drosha processing and are incorporated in the pathway directly at the point of Exportin 5 followed by Dicer cleavage (Figure 1) to generate siRNAs. An example of a shRNA with its typical structure is presented in Figure 2a. shRNAs have been widely used for gene knockdown applications because of their simple design and, when expressed from an AAV vector for their long-term expression and stability.
Figure 2.
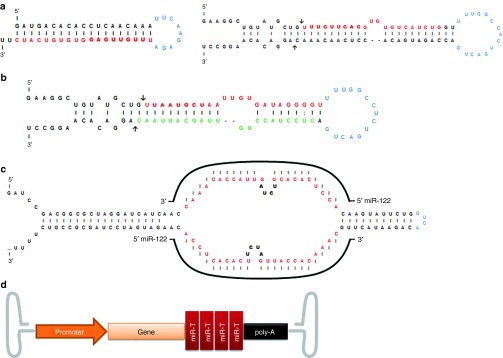
RNAi tools that can be expressed from an adeno-associated vectors (AAV): main features and structure. (a) Structure of shRNAs and artificial miRNAs. The mature shRNA or miRNA guide strand is represented in red with the seed sequence in bold. The loop is represented in blue, in the miRNA the arrows mark the Drosha cleavage sites, adapted from Borel et al., 2011.31 (b) Representation of the endogenous mouse miR-122, where mature miRNA-122-5p is represented in red with the seed sequence in bold, and the mature miRNA-122-3p is represented in green; the arrows mark the Drosha cleavage sites. (Note in this case the guide strand is in 5p and the passenger strand is in 3p, depending on the thermostability of the strands this can be reversed in other miRNAs.) (c) Representative sequence and structure of a miRNA tough decoy (TuD). In red is the sequence that is complementary to the target miRNA (e.g., miR-122) adapted from Xie et al., 2012.52 (d) Illustration of a vector construct with four copies of target sequence for miR-142 at the 3′UTR of the cDNA, adapted from Brown et al., 2006.62
However, in 2006, concerns about toxicity of shRNA molecules were raised by the report of severe liver injury, and in some cases death, in mice injected with high dose AAV8-shRNAs.25 The hepatocellular toxicity appeared to be sequence-independent as it was observed with 36 out of 49 tested shRNAs, ultimately resulting in morbidity within 2 months with 23 different shRNA constructs. At the time it was hypothesized that high levels of shRNAs competed with endogenous miRNAs for intracellular processing and to the extent that it impeded vital cellular processes. This was partially corroborated by the fact that overexpression of Exportin 5, which exports pre-miRNAs (Figure 1), increased knockdown but also toxicity which suggested saturation of downstream factors as well.26 In fact, argonaute 2 was later shown to be another saturable factor in the RNAi pathway.26,27,28 Many groups using AAV vectors to deliver RNAi constructs have since then reproducibly demonstrated that high levels of shRNAs, such as those generated by the classic RNA polymerase III promoters (RNA Pol III), will compete with the endogenous RNAi species, eventually leading to saturation of the cellular machinery.29 shRNAs are usually expressed from RNA Pol III promoters because of their well-defined initiation and termination sites; the most commonly used are the human U6 small nuclear RNA promoter and the human H1-RNA gene promoter,30 U6 being stronger than H1. Toxicity appears not to be U6-specific, as was evident from a study that used H1-driven shRNAs in the liver.31 In these studies with AAV-H1-shRNA, a viral dose-dependent hepatotoxicity was reported in mice, characterized by elevated transaminases levels, signs of stress and ultimately death, which appeared to be paralleled by a decrease in some endogenous miRNA levels, including miR-122.31
ShRNA toxicity is neither organ-specific nor species-specific. In the brain, Elhert et al. observed toxicities in the red nucleus leading to neuronal degeneration and showed that reducing the viral dose or using a less efficient serotype could alleviate this toxicity,32 confirming the original work of Grimm et al.25 described above that first reported this toxicity. Cardiac toxicity was reported in dogs following injection of AAV-shRNA, which also appeared to be paralleled by a decrease in endogenous miRNA levels.33 A majority of reported shRNA-associated toxicities occurred in the CNS, including the mouse striatum,34,35 mouse cerebellum,36 rat substantia nigra37,38 and rat red nucleus neurons.32 In those studies, the authors observed neurotoxicity with several shRNAs including control shRNAs. In general, with shRNAs, there is a risk of high expression that has to be balanced with AAV serotype and promoter choice as well as vector dose. In summary, most of these studies indicate that supraphysiologic expression of these RNA species may be at the root of their toxicity. In fact, in some cases, switching shRNA expression from a RNA Pol III to a RNA Pol II promoter has been showed to alleviate toxicity, pointing to the fact that RNA Pol II promoters may be a safer approach for shRNA-mediated silencing.36,39
Another potential source of toxicity is off-targeting, i.e., the downregulation of an undesired target containing a partial or complete homology to the si/miRNA being expressed. In fact unintended seed sequence homology as determined by a bioinformatics analysis has been shown to correlate with off-target transcriptional changes.40 Off-targeting remains a major issue as it is difficult to predict and to identify. Moreover, preclinical studies are of limited use in this respect because off-targeting can be species-specific.41 Following rational design rules can help reduce off-targeting, in particular selecting artificial RNAi effectors whose seeds have a low incidence in 3′UTRs,42 the predominant location of off-target events.
Another cause of off-targeting is Dicer “slippage,” or noncanonical shRNA cleavage by Dicer, leading to different 5′ and 3′ cleavage sites and siRNA length,43 which by creating a heterogeneous siRNA pool increases off-target multiplicity. It is not yet known if these “by-products” contribute to the toxicity. Nevertheless, to circumvent this, a new generation of shRNAs have been designed to have an internal 3-nt bulge, 2 nt away from the cleavage site of 29 bp-stem shRNAs which may be recognized by the Dicer helicase domain, and as a result imprecise cleavages are abrogated,44 reducing off-targeting. Other methods to reduce shRNA toxicity involve bypassing Dicer all together. The 17–19 nt-long shRNAs named AgoshRNAs, which are shorter than the conventional 21 nt-long ones, appear to be directly incorporated into RISC and processed by AGO.45 By avoiding imprecise Dicer cleavage, these shRNAs not only lead to more specific and more predictable siRNAs but also do not compete with this step of the pathway. Designing shRNA constructs which follow these additional rules will help prevent off-target effects and reduce toxicity. As mentioned above, another concept for reducing the extent of the off-targeting is using the lowest siRNA dose for sufficient knockdown of the desired target.46 Employing low concentrations of multiple siRNAs directed against the same target can maintain a sufficient knockdown while dissipating nonspecific effects, due to the differences between sequences.47
As mentioned earlier, AAV-RNAi is either achieved by delivery of a shRNA or an artificial miRNA. Early on, most of the studies centered on the use of shRNAs, probably due to their inherently simple design. As the field matured and some of above-mentioned evidence of shRNA-associated toxicity was reported, the use of artificial miRNAs (i.e., a cellular miRNA whose stem has been modified to be partially complementary to a mRNA of interest, Figure 2a) gained more popularity. Interestingly enough, many of the observed toxicities with shRNA have been alleviated by shifting to artificial miRNAs. However, it should be noted that using an artificial miRNA over a shRNA adds another potentially saturable step, the Drosha cleavage of the pri-miRNA into pre-miRNA (Figure 1), although to this date no such toxicity has been described. Like shRNAs, artificial miRNAs can be expressed from RNA Pol III but also RNA Pol II promoters, which have lower rate of expression but offer many possibilities in terms of tissue-specific and regulatable expression, all desirable options to avoid potential toxicity and off-target effects. The inherently reduced abundance and more efficient processing of artificial miRNAs over shRNAs48 may be due to the rate limiting cleavage of Drosha. Although this has not been formally proven, an elegant study points in that direction. The study used a fair comparison method in which the artificial miRNA and shRNA were both expressed from U6 promoters and were designed to yield the same siRNA after Dicer processing.36 Thus, the only known difference was that the shRNA bypassed Drosha while the artificial miRNA was dependent on Drosha cleavage. Interestingly, the findings suggest that the shRNA disrupted endogenous miRNA biogenesis, whereas the designed artificial miRNA did not. This was attributed to the higher abundance of unprocessed shRNAs despite being expressed from the same promoter as the artificial miRNA.36 Further testing in vivo showed that the shRNA-expressing vectors led to overt neurotoxicity in cerebellar Purkinje neurons.36 The authors conclude that AAV-artificial miRNA vectors where just as efficient at silencing, but more importantly they resulted in reduced toxicity when compared to AAV-shRNAs.35,36 For knockdown of most endogenous gene targets, RNA Pol II driven artificial miRNAs may be sufficient—however, for some conditions like viral infection where one has to reduce viral loads by many orders of magnitude—RNA Pol III driven shRNAs may have some advantages.
Our group also showed that artificial miRNAs can be expressed from cassettes with therapeutic transgenes. This approach allows to create a single vector to knockdown a mutant mRNA while complementing expression of the normal mRNA.7 This strategy is particularly useful for disorders in which there is both a toxic gain-of-function and a loss-of-function associated with the disease as is the case for most patients with alpha-1 antitrypsin deficiency. We termed these vectors “dual function” vectors which express an artificial miRNA along with a therapeutic normal copy of cDNA. This approach works either by using allele-specific miRNA or altering the cDNA of the gene to be replaced at the nucleotide level to detarget the nonallele specific miRNA. As part of this study, we also performed a thorough microarray study looking at the endogenous liver miRNA profile. The results indicate that the expression of artificial miRNAs after systemic AAV9 delivery did not alter the endogenous miRNAs significantly. Thus, for conditions in which the downregulation of target genes is likely to be necessary for prolonged periods of time the emergence of the rAAV-based miRNA platform holds great promise.
Cellular miRNA Modulation
Another application which has gained interest in the field is the use of rAAV vectors to create somatic models, to study miRNA function, by either overexpressing or antagonizing endogenous miRNAs. The former scenario can be accomplished by using a miRNA expression cassette (Figure 2b), leading to downregulation of the miRNA target genes. Inversely, reduction of miRNAs levels would induce upregulation of their target genes, which can be achieved by a sponge-like mechanism using either synthetic or vector-encoded49 miRNA inhibitors. The initial miRNA bulged sponges described in 2007 by Ebert et al. express 6–10 tandemly arranged miRNA targets with a central bulge from a U6 promoter.50 But the latest downregulation technology are the tough decoys (TuD), a hairpin containing a large internal loop presenting two imperfect miRNA target sites (Figure 2c), which have been shown to be more efficient than sponges in vitro.51 Interestingly, rather than sequestering the mature miRNA, scAAV9-delivered fully complementary TuDs induce miRNA destruction,52 supposedly via the target RNA directed tailing and trimming pathway.53
The advantage of targeting cellular miRNAs is that one will not achieve modulation of a single gene but of a pool of genes, as a single miRNA may target several hundred genes.54,55 It will more likely modulate a pool of potentially functionally related genes, e.g., a group of oncogenes. An example of such a gene network is well described in hepatocellular carcinoma, where downregulation of tumor-suppressor miR-122 upregulates, among others, antiapoptotic BCL2L2,56 metastasis-associated metallopeptidases ADAM1057 and ADAM17,58 tumorigenesis-promoting SRF and IGF1R,57 and cell cycle modulator CCNG1.59 Target multiplicity can also have negative aspects, for instance the promising perspective of therapeutically targeting miR-122 (e.g., to inhibit HCV infection60 or modulate serum cholesterol levels)61 might enhance the risk of tumorigenicity in the liver due to upregulation of the above-mentioned oncogenes. Tumor formation has been observed over time in animals containing an embryonic knockout of the miR-122 locus but has yet to be described in animals60 or humans treated with a miR-122 antisense drug. Nevertheless, TuDs and other methods of miRNA knockdown remain useful tools to study miRNA knockdown and its consequences in vivo.
Detargeting Vector Gene Expression with miRNAs
In addition to the use of rAAV vectors as a vehicle to direct RNAi to specific tissues, the gene therapy field has also been using the RNAi pathway to specifically affect the rAAV-mediated gene expression. This novel way to regulate transgene expression utilizes the endogenous cellular miRNA expression patterns to detarget vector-derived mRNAs by silencing transgene expression in specific cell types where unwanted transgene expression might be detrimental. This method was initially developed by the group of Luigi Naldini who showed that adding target sites for miR-142-3p to the 3′UTR of their lentivirus-delivered transgene (as shown in Figure 2d) efficiently suppressed transgene expression in hematopoietic lineages, hence reducing the need for immunosuppression upon therapeutic gene delivery.62 The same group later showed that this method was applicable to more than just hematopoietic cells and allows control over tissue, lineage, and even differentiation state.63 This elegant method has successfully been used to suppress the liver expression of a peripherally delivered AAV9 in order to restrict transgene expression to the heart.64 It was also used to detarget transgene expression from the liver, heart, and skeletal muscle to restrict it to the CNS.65 Recently, the concept of miR-142-3p to detrage expression as describe above has also been shown to function in the context of AAV vector.66
AAV-Mediated RNAi Therapeutics: Translational Studies
Most RNAi therapeutics are still in preclinical stages, and because a viral delivery renders the toxicology studies of a product more complex than a classic formulation, there currently are no AAV-mediated RNAi vectors in clinical development. However, after the recent approval of the first gene therapy product (alipogene tiparvovec (Glybera), UniQure) by the European Medicine Agency (EMA), an AAV-based product is finally going to reach the market. That places this viral vector as a prime candidate to translate RNAi therapies from the bench to the bedside. Table 1 lists translational and preclinical studies using AAV-mediated RNAi therapeutics, which include various applications in the liver, CNS, eye, heart, lungs, muscle, and prostate. These studies generally fall into either gene knockdown or miRNA modulation. An example of the first approach was recently published by our group, where we knocked-down mutant alpha-1 antitrypsin (SERPINA1) using a single-stranded (ss)AAV9-delivered triple artificial miRNA cluster7 without any signs of toxicity. Importantly, the rAAV-mediated miRNA expression did not alter endogenous cellular miRNA profiles. An example of a miRNA modulation study is the self-complementary (sc)AAV8-mediated delivery of endogenous mmu-miR-26a after which the authors showed a reduction of liver tumor incidence from 75% to 20%.67 Another application that has shown early promise for clinical translation is the use of miRNA or shRNA to target viruses. In fact, rapidly mutating targets such as RNA viruses are an ideal target for RNAi therapeutics. This approach allows targeting of highly conserved sequences, and is desirable due to the relative simplicity of the RNAi design and the ability to multiplex various targets is less laborious than the development of several small molecules against various viral proteins. In the field of HCV, one group is using three individual shRNA expression cassettes each driven by a different promoter targeting three highly conserved regions of the genome.68 Other efforts have focused on creating an artificial miRNA cluster targeting HCV based on the structure of the endogenous miR-17–92 cluster. The artificial cluster contains five miRNAs, three of them targeting the conserved regions of the 5′UTR of HCV along with a miRNA targeting the structural (Core) and one targeting the nonstructural (NS5B) gene.69 Both of these methods have generated promising preclinical data.68,69 A similar combinatorial RNAi approach was shown to be efficient in preventing viral escape in HIV infection.70
Table 1. Preclinical studies of AAV-mediated RNAi therapeutics.

The field has learned a lot from the preclinical AAV-RNAi studies including problems with toxicity, shRNA and miRNA design and as a whole it has responded and adapted quickly generating a couple of candidates that are approaching the early clinical stages. However, if one includes nonviral RNAi clinical trials, there are quite a few ongoing or planned trials using this relatively new discovery (Table 2). Trials employing RNAi in various delivery modalities include a wide array of applications such as the eye, liver, lung, kidney, intestine, infections, skin, and cancer. Out of 25 clinical trials, 20 are siRNA-based with various formulations (naked, SNALP, liposomal, chemically modified, RONDEL, LODER), while 5 only are shRNA-based. The delivery methods are variable (oral bacteria, ex vivo lentivirus, plasmid electroporation, intratumoral lipoplex) with only one being AAV-based. The first AAV-RNAi clinical trial is on schedule to be initiated this year by Tacere Therapeutics, a subsidiary of Benitec Biopharma. According to David Suhy, Sr VP of R&D at Tacere, 14 HCV-infected patients will receive an intravenous infusion of TT-034, a scAAV8-U6-3xshRNA targeting three highly conserved regions across multiple genotypes of the (+) RNA HCV genome, and a liver biopsy will be taken at 3 weeks after injection. Doses of TT-034 will start at 4E10 vg/kg and range up to 4E12 vg/kg. Besides safety and tolerability, the objectives of this Phase I/II study include determining the percentage of hepatocytes that have been transduced and quantifying HCV viral load and shRNA expression.
Table 2. RNAi therapeutics currently in clinical studies.

Conclusion
The unprecedented pace at which RNAi has transformed the field of gene therapy and biology as a whole is impressive. This has resulted in the relatively fast translation of basic research to clinical applications as evidenced by the RNAi clinical trial pipeline. As discussed above various strategies using AAV exist to knockdown either a target mRNA or endogenous miRNAs. There are inherent advantages of using viral vectors such as AAV for RNAi. The most obvious is the potential of lifelong persistence of RNAi, but also the ability to achieve systemic delivery, and in the case of polymerase II promoters driving shRNA or miRNA expression, the tissue specificity that can be achieved. While there is great cause for excitement in the field one must consider the combined toxicity imparted by the viral vector itself as well as issues related to RNAi. Thus, toxicity still remains an issue, and should therefore be carefully considered on a case-by-case basis; however, the large number of successful preclinical studies is encouraging. With regards to RNAi it seems that for the most part toxicity is dependent on both the dose and stem-loop design. However, our current understanding of its safety is still limited due to the lack of long-term preclinical data. Also, some applications would benefit from the flexibility of turning on and off the induction of RNAi. In this regard, one area of research that is of utmost importance is the further development of conditional promoters composed of human proteins, so as to evade immune responses to them. Importantly, new research shows that shRNA and artificial miRNA expression can also be controlled by RNA aptamers, a class of structured RNA designed to bind specific ligands such as small molecules, cell surface markers, or intra- or extra-cellular components, with high specificity and affinity. Incorporating an aptamer into the loop or the basal segment of a shRNA was shown to inhibit Dicer cleavage of the shRNA hence restricting shRNA expression in presence of the aptamer ligand.71,72 RNA aptamers could also help target a specific subpopulation of cells.73 Altering the miRNA biogenesis pathway74 could potentially be used for therapeutic modulation of miRNA levels as well.
The preclinical and clinical trial experience with rAAV is extensive and it has already yielded an approved product in Europe. While immune responses to the capsid remain a possible concern, recent data from several clinical trials has demonstrated that these can be managed and/or tolerated. In addition, transgene immune responses are not much of a factor unless a conditional promoter is used. Thus, it is only a matter of time before the idea of utilizing recombinant AAV as a platform for RNAi translates into a successful clinical product.
References
- Napoli C, Lemieux C, Jorgensen R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell. 1990;2:279–289. doi: 10.1105/tpc.2.4.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Krol AR, Mur LA, Beld M, Mol JN, Stuitje AR. Flavonoid genes in petunia: addition of a limited number of gene copies may lead to a suppression of gene expression. Plant Cell. 1990;2:291–299. doi: 10.1105/tpc.2.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- McCaffrey AP, Meuse L, Pham TT, Conklin DS, Hannon GJ, Kay MA. RNA interference in adult mice. Nature. 2002;418:38–39. doi: 10.1038/418038a. [DOI] [PubMed] [Google Scholar]
- Mao H, Gorbatyuk MS, Rossmiller B, Hauswirth WW, Lewin AS. Long-term rescue of retinal structure and function by rhodopsin RNA replacement with a single adeno-associated viral vector in P23H RHO transgenic mice. Hum Gene Ther. 2012;23:356–366. doi: 10.1089/hum.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller C, Tang Q, Gruntman A, Blomenkamp K, Teckman J, Song L, et al. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol Ther. 2012;20:590–600. doi: 10.1038/mt.2011.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain KK. Jain PharmaBiotech: Basel, Switzerland; 2013. RNAi: Technologies, markets and companies. [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13:1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- Canella D, Praz V, Reina JH, Cousin P, Hernandez N. Defining the RNA polymerase III transcriptome: Genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 2010;20:710–721. doi: 10.1101/gr.101337.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini HK, Griffiths-Jones S, Enright AJ. Genomic analysis of human microRNA transcripts. Proc Natl Acad Sci USA. 2007;104:17719–17724. doi: 10.1073/pnas.0703890104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Satterlee A, Huang L. In vivo gene delivery by nonviral vectors: overcoming hurdles. Mol Ther. 2012;20:1298–1304. doi: 10.1038/mt.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- Karikó K, Bhuyan P, Capodici J, Weissman D. Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J Immunol. 2004;172:6545–6549. doi: 10.4049/jimmunol.172.11.6545. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Piao C, Kotchey NM, Wu X, Nakai H. Frequency and spectrum of genomic integration of recombinant adeno-associated virus serotype 8 vector in neonatal mouse liver. J Virol. 2008;82:9513–9524. doi: 10.1128/JVI.01001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowrouzi A, Penaud-Budloo M, Kaeppel C, Appelt U, Le Guiner C, Moullier P, et al. Integration frequency and intermolecular recombination of rAAV vectors in non-human primate skeletal muscle and liver. Mol Ther. 2012;20:1177–1186. doi: 10.1038/mt.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeppel C, Beattie SG, Fronza R, van Logtenstein R, Salmon F, Schmidt S, et al. A largely random AAV integration profile after LPLD gene therapy. Nat Med. 2013;19:889–891. doi: 10.1038/nm.3230. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, et al. Vision 1 year after gene therapy for Leber's congenital amaurosis. N Engl J Med. 2009;361:725–727. doi: 10.1056/NEJMc0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Davidoff AM, Hanawa H, Hu Y, Hoffer FA, Nikanorov A, et al. Sustained high-level expression of human factor IX (hFIX) after liver-targeted delivery of recombinant adeno-associated virus encoding the hFIX gene in rhesus macaques. Blood. 2002;100:1662–1669. doi: 10.1182/blood-2002-02-0589. [DOI] [PubMed] [Google Scholar]
- Gaudet D, Méthot J, Déry S, Brisson D, Essiembre C, Tremblay G, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20:361–369. doi: 10.1038/gt.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, Davidson BL. Generation of hairpin-based RNAi vectors for biological and therapeutic application. Meth Enzymol. 2012;507:275–296. doi: 10.1016/B978-0-12-386509-0.00014-4. [DOI] [PubMed] [Google Scholar]
- Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- Grimm D, Wang L, Lee JS, Schürmann N, Gu S, Börner K, et al. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J Clin Invest. 2010;120:3106–3119. doi: 10.1172/JCI43565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L, et al. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 2007;35:5154–5164. doi: 10.1093/nar/gkm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs S, Jung S, Rothenberg SM, Smolen GA, Mlody BG, Haber DA. Coexpression of Argonaute-2 enhances RNA interference toward perfect match binding sites. Proc Natl Acad Sci USA. 2008;105:9284–9289. doi: 10.1073/pnas.0800803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D. The dose can make the poison: lessons learned from adverse in vivo toxicities caused by RNAi overexpression. Silence. 2011;2:8. doi: 10.1186/1758-907X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer M, Nilsen TW, Costigan C, Altman S. Structure and transcription of a human gene for H1 RNA, the RNA component of human RNase P. Nucleic Acids Res. 1990;18:97–103. doi: 10.1093/nar/18.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel F, van Logtenstein R, Koornneef A, Maczuga P, Ritsema T, Petry H, et al. In vivo knock-down of multidrug resistance transporters ABCC1 and ABCC2 by AAV-delivered shRNAs and by artificial miRNAs. J RNAi Gene Silencing. 2011;7:434–442. [PMC free article] [PubMed] [Google Scholar]
- Ehlert EM, Eggers R, Niclou SP, Verhaagen J. Cellular toxicity following application of adeno-associated viral vector-mediated RNA interference in the nervous system. BMC Neurosci. 2010;11:20. doi: 10.1186/1471-2202-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bish LT, Sleeper MM, Reynolds C, Gazzara J, Withnall E, Singletary GE, et al. Cardiac gene transfer of short hairpin RNA directed against phospholamban effectively knocks down gene expression but causes cellular toxicity in canines. Hum Gene Ther. 2011;22:969–977. doi: 10.1089/hum.2011.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JN, Wolken N, Brown T, Dauer WT, Ehrlich ME, Gonzalez-Alegre P. Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther. 2011;18:666–673. doi: 10.1038/gt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride JL, Boudreau RL, Harper SQ, Staber PD, Monteys AM, Martins I, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, Martins I, Davidson BL. Artificial microRNAs as siRNA shuttles: improved safety as compared to shRNAs in vitro and in vivo. Mol Ther. 2009;17:169–175. doi: 10.1038/mt.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodr CE, Sapru MK, Pedapati J, Han Y, West NC, Kells AP, et al. An a-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson's disease, but displays toxicity in dopamine neurons. Brain Res. 2011;1395:94–107. doi: 10.1016/j.brainres.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A, Sahin G, Björklund T, Aebischer P, Kirik D. Dose optimization for long-term rAAV-mediated RNA interference in the nigrostriatal projection neurons. Mol Ther. 2009;17:1574–1584. doi: 10.1038/mt.2009.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giering JC, Grimm D, Storm TA, Kay MA. Expression of shRNA from a tissue-specific pol II promoter is an effective and safe RNAi therapeutic. Mol Ther. 2008;16:1630–1636. doi: 10.1038/mt.2008.144. [DOI] [PubMed] [Google Scholar]
- Anderson EM, Birmingham A, Baskerville S, Reynolds A, Maksimova E, Leake D, et al. Experimental validation of the importance of seed complement frequency to siRNA specificity. RNA. 2008;14:853–861. doi: 10.1261/rna.704708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchard J, Jackson AL, Malkov V, Needham RH, Tan Y, Bartz SR, et al. MicroRNA-like off-target transcript regulation by siRNAs is species specific. RNA. 2009;15:308–315. doi: 10.1261/rna.1326809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, Spengler RM, Davidson BL. Rational design of therapeutic siRNAs: minimizing off-targeting potential to improve the safety of RNAi therapy for Huntington's disease. Mol Ther. 2011;19:2169–2177. doi: 10.1038/mt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maczuga P, Lubelski J, van Logtenstein R, Borel F, Blits B, Fakkert E, et al. Embedding siRNA sequences targeting apolipoprotein B100 in shRNA and miRNA scaffolds results in differential processing and in vivo efficacy. Mol Ther. 2013;21:217–227. doi: 10.1038/mt.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Jin L, Zhang Y, Huang Y, Zhang F, Valdmanis PN, et al. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 2012;151:900–911. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YP, Schopman NC, Berkhout B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. 2013;41:3723–3733. doi: 10.1093/nar/gkt036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler R, Surendranath V, Heninger AK, Slabicki M, Theis M, Putz G, et al. Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nat Methods. 2007;4:337–344. doi: 10.1038/nmeth1025. [DOI] [PubMed] [Google Scholar]
- Boudreau RL, Monteys AM, Davidson BL. Minimizing variables among hairpin-based RNAi vectors reveals the potency of shRNAs. RNA. 2008;14:1834–1844. doi: 10.1261/rna.1062908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak RO, Hollensen AK, Mikkelsen JG. Managing microRNAs with vector-encoded decoy-type inhibitors. Mol Ther. 2013;21:1478–1485. doi: 10.1038/mt.2013.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak RO, Hollensen AK, Primo MN, Sørensen CD, Mikkelsen JG. Potent microRNA suppression by RNA Pol II-transcribed ‘Tough Decoy' inhibitors. RNA. 2013;19:280–293. doi: 10.1261/rna.034850.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Ameres SL, Friedline R, Hung JH, Zhang Y, Xie Q, et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods. 2012;9:403–409. doi: 10.1038/nmeth.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung JH, Xu J, Ghildiyal M, Weng Z, et al. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Gong HY, Tseng HC, Wang WL, Wu JL. miR-122 targets an anti-apoptotic gene, Bcl-w, in human hepatocellular carcinoma cell lines. Biochem Biophys Res Commun. 2008;375:315–320. doi: 10.1016/j.bbrc.2008.07.154. [DOI] [PubMed] [Google Scholar]
- Bai S, Nasser MW, Wang B, Hsu SH, Datta J, Kutay H, et al. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem. 2009;284:32015–32027. doi: 10.1074/jbc.M109.016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WC, Hsu PW, Lai TC, Chau GY, Lin CW, Chen CM, et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49:1571–1582. doi: 10.1002/hep.22806. [DOI] [PubMed] [Google Scholar]
- Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–6099. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Brown BD, Venneri MA, Zingale A, Sergi Sergi L, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. 2006;12:585–591. doi: 10.1038/nm1398. [DOI] [PubMed] [Google Scholar]
- Brown BD, Gentner B, Cantore A, Colleoni S, Amendola M, Zingale A, et al. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol. 2007;25:1457–1467. doi: 10.1038/nbt1372. [DOI] [PubMed] [Google Scholar]
- Geisler A, Jungmann A, Kurreck J, Poller W, Katus HA, Vetter R, et al. microRNA122-regulated transgene expression increases specificity of cardiac gene transfer upon intravenous delivery of AAV9 vectors. Gene Ther. 2011;18:199–209. doi: 10.1038/gt.2010.141. [DOI] [PubMed] [Google Scholar]
- Xie J, Xie Q, Zhang H, Ameres SL, Hung JH, Su Q, et al. MicroRNA-regulated, systemically delivered rAAV9: a step closer to CNS-restricted transgene expression. Mol Ther. 2011;19:526–535. doi: 10.1038/mt.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majowicz A, Maczuga P, Kwikkers KL, van der Marel S, van Logtenstein R, Petry H, et al. Mir-142-3p target sequences reduce transgene-directed immunogenicity following intramuscular adeno-associated virus 1 vector-mediated gene delivery. J Gene Med. 2013;15:219–232. doi: 10.1002/jgm.2712. [DOI] [PubMed] [Google Scholar]
- Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhy DA, Kao SC, Mao T, Whiteley L, Denise H, Souberbielle B, et al. Safe, long-term hepatic expression of anti-HCV shRNA in a nonhuman primate model. Mol Ther. 2012;20:1737–1749. doi: 10.1038/mt.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Marcucci K, Anguela X, Couto LB. Preclinical evaluation of an anti-HCV miRNA cluster for treatment of HCV infection. Mol Ther. 2013;21:588–601. doi: 10.1038/mt.2012.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutimah F, Eekels JJ, Liu YP, Berkhout B. Antiviral strategies combining antiretroviral drugs with RNAi-mediated attack on HIV-1 and cellular co-factors. Antiviral Res. 2013;98:121–129. doi: 10.1016/j.antiviral.2013.02.011. [DOI] [PubMed] [Google Scholar]
- An CI, Trinh VB, Yokobayashi Y. Artificial control of gene expression in mammalian cells by modulating RNA interference through aptamer-small molecule interaction. RNA. 2006;12:710–716. doi: 10.1261/rna.2299306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel CL, Chen YY, Culler SJ, Hoff KG, Smolke CD. Design of small molecule-responsive microRNAs based on structural requirements for Drosha processing. Nucleic Acids Res. 2011;39:2981–2994. doi: 10.1093/nar/gkq954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Zhang Y, Ribas J, Chowdhury WH, Castanares M, Zhang Z, et al. Prostate-targeted radiosensitization via aptamer-shRNA chimeras in human tumor xenografts. J Clin Invest. 2011;121:2383–2390. doi: 10.1172/JCI45109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MA, Hammond SM. Emerging paradigms of regulated microRNA processing. Genes Dev. 2010;24:1086–1092. doi: 10.1101/gad.1919710. [DOI] [PMC free article] [PubMed] [Google Scholar]