

Abstract
Clinicians rely upon the severity of liver fibrosis to segregate patients with well-compensated nonalcoholic fatty liver disease (NAFLD) into sub-populations at high versus low-risk for eventual liver-related morbidity and mortality. We compared hepatic gene expression profiles in high- and low-risk NAFLD patients to identify processes that distinguish the two groups and hence, might be novel biomarkers or treatment targets. Microarray analysis was used to characterize gene expression in percutaneous liver biopsies from low-risk, “mild” NAFLD patients (fibrosis stage 0–1, n=40) and high risk, “severe” NAFLD patients (fibrosis stage 3–4, n=32). Findings were validated in a second, independent cohort and confirmed by real time PCR and immunohistochemistry. As a group, patients at risk for bad NAFLD outcomes had significantly worse liver injury and more advanced fibrosis (severe NAFLD) than clinically-indistinguishable NAFLD patients with a good prognosis (mild NAFLD). A 64 gene profile reproducibly differentiated severe NAFLD from mild NAFLD, and a 20 gene subset within this profile correlated with NAFLD severity, independent of other factors known to influence NAFLD progression. Multiple genes involved with tissue repair/regeneration and certain metabolism-related genes were induced in severe NAFLD. Ingenuity Pathway Analysis and immunohistochemistry confirmed deregulation of metabolic and regenerative pathways in severe NAFLD, and revealed overlap among the gene expression patterns of severe NAFLD, cardiovascular disease, and cancer.
Conclusion
By demonstrating specific metabolic and repair pathways that are differentially activated in livers with severe NAFLD, gene profiling identified novel targets that can be exploited to improve diagnosis and treatment of patients who are at greatest risk for NAFLD-related morbidity and mortality.
Keywords: fibrosis, liver disease, biopsy, humans, microarray
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is one of the most common types of liver disease in the world. Most patients with NAFLD do not develop clinically significant liver disease, but cirrhosis and/or liver cancer emerge in a subset.(1) The molecular mechanisms underlying the heterogeneous outcomes of NAFLD remain unclear, and this knowledge gap has made it challenging to diagnose and treat NAFLD patients before symptomatic cirrhosis or liver cancer ensue. Liver biopsy studies have provided some help by demonstrating that bad liver outcomes are much more likely in fatty livers with coincident hepatocyte injury and liver inflammation (i.e., nonalcoholic steatohepatitis, NASH) than in livers with simple steatosis.(2, 3) NASH encompasses a spectrum of liver injury and inflammation (4), however, and not all individuals with NASH ultimately develop cirrhosis or liver cancer.(5) Also, although the severity of NASH generally correlates with the severity of fibrosis, some individuals with advanced fibrosis have relatively little NASH at the time liver tissue is sampled.(6) Moreover, when advanced fibrosis is present, absence of NASH is no longer prognostic. Therefore, while a diagnosis of NASH provides some evidence for a worse prognosis than non-NASH NAFLD, it is of relatively limited help in predicting the ultimate outcome of NAFLD in an individual patient.(3, 5) On the other hand, the stage of fibrosis on liver biopsy independently associates with liver-related mortality and generally correlates with the severity of portal hypertension.(7) The latter is an excellent predictor of eventual liver-related morbidity, liver cancer, and death.(8, 9) Hence, clinicians typically rely upon fibrosis staging to approximate risk for NAFLD-related morbidity and mortality. The need for early and accurate risk stratification, as well as effective risk-appropriate therapies, is particularly pressing in NAFLD because the disease has become epidemic, imposing a potential public health burden.(10)
Tissue gene expression profiling has been valuable for developing diagnostic and predictive biomarkers, as well as for targeting therapy, in cancer and other diseases in which tissue biopsies provide the basis for estimating prognosis and guiding treatment recommendations.(11–13) Therefore, the aim of this study was to use microarray analysis to characterize a liver gene expression profile that reliably differentiated clinically similar and relatively asymptomatic individuals who were at opposite extremes of the risk spectrum for bad NAFLD-related liver outcomes based on their histologic stage of liver fibrosis. This profile was validated in a second, independent cohort, and confirmed by quantitative RT-PCR (QRT-PCR) analysis. After adjusting for the effect of known clinical correlates of liver fibrosis, a subset of the differentially expressed genes that independently correlated with NAFLD severity emerged. Pathway analysis and immunohistochemistry revealed that certain metabolic and repair-related processes were selectively induced in livers with severe NAFLD. These findings will facilitate development of novel diagnostic tests and treatments that target the subgroup of “pre-symptomatic” NAFLD patients who are at greatest risk for bad NAFLD outcomes.
MATERIALS AND METHODS
Detailed methods for each section provided in the Supplementary Materials.
Patient Selection and Clinical Variables
We conducted a cross-sectional study utilizing prospectively collected data from NAFLD subjects in the Duke University Health System NAFLD Biorepository. This biorepository was approved by our Institutional Review Board and contains frozen liver biopsies and clinical data from NAFLD patients who underwent a diagnostic liver biopsy to grade and stage severity of disease as part of standard of care. For the present study, NAFLD was defined as: (1) presence of ≥ 5% hepatic steatosis on liver biopsy; (2) absence of histologic and serologic evidence for other chronic liver disease in a patient with risk factors for the metabolic syndrome. Patients were selected for inclusion based on histologically-defined liver fibrosis stage, a key determinant of clinical outcome.(5) Two groups at the extremes of NAFLD formed the discovery cohort: ”mild” NAFLD, defined as fibrosis stages 0 or 1(n=53) and thus, little probability of developing clinically significant liver disease over the next one to two decades, and “severe” NAFLD, defined as fibrosis stage 3 or 4 (n=56) and thus, significant likelihood of developing liver-related morbidity and mortality over the same time period (i.e. bad NAFLD outcomes). The groups were matched for gender, age (± 5 years) and body mass index (BMI, kg/m2) (± 3 points). Frozen liver biopsies and clinical data from a second, independent cohort of patients (n=40) with biopsy-proven NAFLD were identified in the same manner in order to evaluate the predictive performance of the gene profile (validation cohort). Demographic data (i.e., height, weight, BMI, age, gender, race, ethnicity, smoking status and comorbid illnesses) and laboratory studies (i.e. lipids, glucose, hemoglobin A1c (HbA1c), liver aminotransferases, and measures of liver synthetic function) were obtained within 6 months of liver biopsy in all patients. Rigorous quality control procedures resulted in final analyses of 72 patients in the discovery cohort and 17 patients in the validation cohort (Supplementary Figure 1 and Supplementary Tables 1–6).
Liver biopsy and Histopathological Analysis
Biorepository liver samples are remnants from clinically-indicated liver biopsies. Samples were snap frozen in liquid nitrogen and stored at −80°C. The bulk of each liver biopsy had been processed for routine histology. For the present study, liver slides were re-reviewed and scored by a liver pathologist blinded to the clinical and laboratory data. The severity of NAFLD-related injury and fibrosis were graded and scored according to published criteria.(4)
RNA Preparation
Microarray hybridization and gene expression analysis
Microarray hybridization was performed on Affymetrix Human Genome U133 Plus 2.0 GeneChip arrays (Affymetrix, Santa Clara, CA), using MessageAmp Premier (Applied Biosystems, Foster City, CA) for RNA amplification and hybridization. Data are publically available through NCBI (GSE31803). Differential gene expression was determined using limma (R/Bioconductor statistical package).(14) Results were corrected for multiple testing via the Benjamini-Hochberg method to control the false discovery rate (FDR) at 5%. We built and performed validation of a gene expression profile associated with advanced NAFLD using Support Vector Machines (SVM).
Quantitative Real-Time RT-PCR
TaqMan QRT-PCR was used to validate the differential expression of eight randomly selected genes identified in the gene profile. Using the available remaining total RNA from selected liver biopsy samples, the reverse transcription reaction was performed using the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) using random hexamer priming according to the manufacturer’s protocol.
Pathway and functional enrichment analysis
We used the Ingenuity Pathways Analysis (IPA, Ingenuity systems, Inc., Redwood City, CA, www.ingenuity.com) tool to examine biological functions and disease as well as functional relationships between genes and gene networks.
Immunohistochemistry
Formalin-fixed, paraffin-embedded liver biopsy samples from a subset of patients (n = 24; 13 mild NAFLD and 11 advanced NAFLD) were available for immunohistochemical (IHC) staining. The primary antibodies used were Sonic Hedgehog (SHH), glioblastoma 2 (GLI2), keratin 7 (CK7), alpha-smooth muscle actin (α-SMA), and sex determining region Y-box 9 (SOX9).
Statistical analysis
Demographic, laboratory, histologic and IHC data were compared between groups using t-tests or Wilcoxon rank sum tests for continuous predictors and chi squared or Fisher’s exact tests for categorical variables. All tests of significance were 2-sided, with p-value ≤ 0.05 considered significant. Multiple logistic regression analysis was used to assess gene associations with severe NAFLD while controlling for HbA1c, BMI, age and gender (P < 0.0005 considered significant). All analyses were done using R statistical packages (www.r-project.org) or JMP7 statistical software (SAS Institute Inc., Cary, North Carolina).
RESULTS
Patient characteristics
The 72 patients in the discovery cohort included 40 with mild NAFLD and 32 with severe NAFLD (Table 1). As others have reported, (15), patients with mild NAFLD had a lower prevalence of diabetes mellitus (DM) than those with severe NAFLD, but did not differ significantly in other components of the metabolic syndrome, such as obesity, hypertension, or hyperlipidemia, or medication use that might impact NASH. In contrast, histologic characteristics reflecting disease severity differed among severe NAFLD patients and those with mild NAFLD: the severe NAFLD group had significantly more lobular inflammation, portal inflammation, hepatocyte ballooning, and included more patients with a NAFLD Activity Score (NAS) ≥ 5. The findings also demonstrate that fibrosis was an excellent predictor of global liver damage at the time of gene expression analysis in the present study. Clinical and histologic characteristics for the 10 mild NAFLD and 7 severe NAFLD patients in the validation cohort were comparable to those of the discovery cohort (Table 1).
Table 1. Characteristics of the discovery cohort and the validation cohort utilized in the NAFLD gene expression analyses.
| Discovery Cohort | Comparison | |||||
|---|---|---|---|---|---|---|
| Patient demographics and clinical characteristics | Mild NAFLD (stage 0 and 1) (n=40) | Severe NAFLD (stage 3 and 4) (n=32) | P Value | Discovery Cohort (n=72) | Validation Cohort (n=17) | P Value |
| Male sex, n (%) | 16 (40) | 9 (28.1) | 0.3 | 25 (34.7) | 3 (17.6) | 0.25 |
| Age in years at Biopsy (Mean ± SD) | 49.9 ± 10.6 | 51.4 ± 11.7 | 0.57 | 50.54 ± 11 | 52.8 ± 8.7 | 0.44 |
| Ethnicity | 1 | 1 | ||||
| Non-Hispanic | 39 (97.5) | 28 (87.5) | 67 (93.1) | 14 (82.4) | ||
| Hispanic | 1 (2.5) | 1 (3.1) | 2 (2.8) | 0 (0) | ||
| Unknown | 0 (0) | 3 (9.4) | 3 (4.1) | 3 (17.6) | ||
| Race | 0.55 | 0.25 | ||||
| White | 35 (87.5) | 28 (87.5) | 63 (87.5) | 17 (100) | ||
| Black | 3 (7.5) | 3 (9.4) | 6 (8.3) | 0 (0) | ||
| Asian | 2 (5) | 0 (0) | 2 (2.8) | 0 (0) | ||
| Hawaiian Pacific Islander | 0 (0) | 1 (3.1) | 1 (1.4) | 0 (0) | ||
| BMI, median kg/m2 (IQR)a | 32.5 (29.2–40.1) | 33.8 (31.3–41.9) | 0.23 | 33.7 (29.5–40.4) | 37.2 (32.2–45.8) | 0.31 |
| Hypertension, n (%) | 20 (50) | 23 (72) | 0.06 | 43 (59.7) | 12 (70.6) | 0.58 |
| Diabetes Mellitus, n (%) | 8 (20) | 19 (59.4) | <0.01 | 27 (37.5) | 10 (58.8) | 0.17 |
| Hemoglobin A1c, median % (IQR)b | 5.9 (5.4–6.4) | 6.6 (5.9–7.2) | 0.003 | 6 (5.6–6.7) | 6.3 (5.7–7.2) | 0.36 |
| Hyperlipidemia, n (%) | 25 (62.5) | 18 (56.3) | 0.59 | 43 (59.7) | 14 (82.4) | 0.1 |
| Current Smoking, n (%) c | 1 (2.6) | 3 (10.7) | 0.3 | 4 (6.2) | 2 (11.8) | 0.6 |
| Medications | ||||||
| Vitamin E, n (%) | 3 (7.5) | 0 (100) | 0.25 | 3 (4.2) | 0 (0) | 1 |
| Fish Oil, n (%) | 9 (22.5) | 5 (15.6) | 0.46 | 14 (19.4) | 5 (29.4) | 0.37 |
| Statins, n (%) | 7 (17.5) | 6 (19.4) | 0.89 | 13 (18.1) | 5 (29.4) | 0.30 |
| Laboratory measures, median (IQR)c | ||||||
| Serum AST, U/L | 38 (26.8–64.3) | 58 (37–72.5) | 0.12 | 44 (32–72) | 50 (21–74.5) | 0.98 |
| Serum ALT, U/L | 50.5 (32.2–93.3) | 71 (34.5–90.5) | 0.84 | 51 (33–91) | 53 (26.3–97.8) | 0.84 |
| AST/ALT | 0.75 (0.58–1) | 0.98 (0.64–1.26) | 0.1 | 0.79 (0.62–1.10) | 0.92 (0.77–1.08) | 0.25 |
| Histologic characteristics, n (%) | ||||||
| Steatosis (% ≥ 34%) | 23 (57.5) | 19 (59.4) | 0.87 | 42 (58.3) | 5 (29.4) | 0.06 |
| Lobular inflammation (% ≥ grade 2)d | 7 (18.9) | 13 (43.3) | 0.03 | 20 (29.9) | 5 (29.4) | 1 |
| Portal inflammation (% > mild)d | 9 (24.3) | 21 (67.7) | <0.01 | 30 (44.1) | 5 (29.4) | 0.41 |
| Ballooning (% any)a | 26 (65) | 30 (93.8) | 0.004 | 56 (77.8) | 13 (81.3) | 1 |
| NAFLD Activity Score (% ≥ 5)* | 10 (25) | 18 (56) | 0.007 | 28 (38.9) | 7 (41.2) | 1 |
| Fibrosis (% ≥ stage 3) | n/a | n/a | 32 (44.4) | 7 (41.2) | 1 | |
NAFLD Activity Score (range 0–8) is a sum of scores for steatosis, lobular inflammation, and ballooning.
missing data for 1 patient;
missing data for 14 patients;
missing data for 6 patients;
missing data for 4 patients
Gene expression differs between mild and severe NAFLD
In the discovery cohort, a total of 1132 genes were significantly differentially expressed in patients with severe versus mild NAFLD. Unsupervised hierarchical clustering analysis of the top 100 differentially expressed probes revealed two distinct groups with minimal overlap (Figure 1). No significant differences in gene expression were detected between patients with no fibrosis (n=17) and stage 1 fibrosis (n=23) using the same methodology. Gene ontology analysis demonstrated that the top up-regulated genes in severe NAFLD included genes associated with biological functions such as such as cell adhesion and migration (THBS2, EFEMP1 and DPT), development and extracellular matrix organization (COL1A2, COL4A1, COL3A1, LUM, FBN1, DKK3) and regulation of development, transcription and signal transduction (IGFBP7, ID4, EPHA3, PDGFRA). Interestingly, many of the other top up-regulated genes were markers of adult liver progenitor cells, such as JAG1, EPCAM, SOX9, PROM1, SPP1. The top down-regulated genes in severe NAFLD were generally involved in metabolism, and included CYP2C19, DHRS2, OAT, MAT1A, GNMT, and DGAT2. Additional differentially expressed genes are shown in Supplementary Table 7.
Figure 1. Hierarchical clustering analysis.
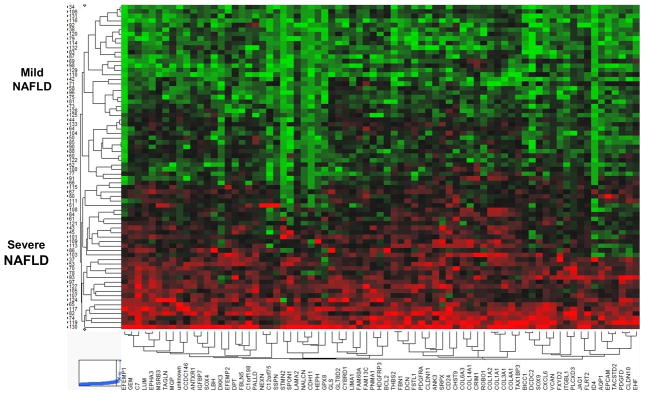
Hierarchical clustering of the top 100 differentially expressed probes from the 72 NAFLD patients separated the samples into two main groups: mild NAFLD and severe NAFLD. Data are presented in heat map format in which patient samples are shown in rows and genes (probes) in columns. Red color corresponds to genes that are up-regulated in severe NAFLD as compared to the mean, and green color corresponds to genes that are down-regulated in severe NAFLD as compared to the mean.
Ingenuity Pathway Analysis identifies dysregulation of cancer-, cardiovascular disease-, and metabolism-associated genes in severe NAFLD
Ingenuity pathway analysis (IPA) is useful for revealing similarities between a poorly-understood disease and other biologic processes that have been better characterized. This approach identified biological processes that were over-represented among patients with severe NAFLD relative to mild NAFLD. A core IPA of the set of 1132 differentially expressed genes revealed overlap with several biological processes, with three of the top five identified being cancer, genetic disorders, and cardiovascular disease (Table 2). Closer inspection of the cancer category demonstrated over-representation of genes associated with liver cancer (p= 3.49 × 10−4) and colorectal carcinoma (p= 4.03 × 10−1), two cancers associated with NAFLD and the metabolic syndrome.(16, 17) For example, patients in our severe NAFLD cohort exhibited significant down-regulation of certain metabolic genes that IPA identified as having significant correlations with liver cancer and fibrosis, namely MAT1A, GNMT and DGAT2. In mouse models, inhibiting these gene products causes steatohepatitis (MAT1A, GNMT, DGAT2), advanced liver fibrosis (GNMT, DGAT2) and hepatocellular carcinoma (HCC) (MAT1A and GNMT).(18–20) Cardiovascular disease (CVD) pathways were also overrepresented in our severe NAFLD cohort, consistent with the known association between NASH and CVD, (21) and corroborating a recent publication describing increased cardiovascular mortality in NAFLD patients with noninvasive evidence of advanced liver fibrosis.(22)
Table 2. 64-Gene profile of severe NAFLD.
Genes are listed according to the percentage they appear in the 1000 iterations used to generate the gene profile. Genes in bold are also found in the 20-gene profile of severe NAFLD.
| Affymetrix Probe ID | Gene Symbol | Gene Title | Gene Ontology and Function | Percentage appears 1000 iterations* |
|---|---|---|---|---|
| 224694_at | ANTXR1 | anthrax toxin receptor 1 | cell adhesion, tumor specific endothelial cell marker | 100% |
| 209047_at | AQP1 | aquaporin 1 | transepithelial water transport, positive regulation of fibroblast proliferation | 100% |
| 207542_s_at | 71% | |||
| 213429_at | BICC1 | bicaudal C homolog 1 | modulates protein translation during embryonic development | 100% |
| 202992_at | C7 | complement component 7 | response to wounding, complement and coagulation cascades | 100% |
| 208651_x_at | CD24 | CD24 molecule | cell adhesion, regulation of epithelial cell differentiation, Wnt signaling, hypoxia response | 100% |
| 209771_x_at | 100% | |||
| 216379_x_at | 100% | |||
| 266_s_at | 100% | |||
| 208650_s_at | 98% | |||
| 228335_at | CLDN11 | claudin 11 | cell adhesion, tight junctions | 100% |
| 202404_s_at | COL1A2 | collagen, type I, alpha 2 | extracellular matrix organization, TGF-β signaling, focal adhesion, PDGF signaling | 100% |
| 202403_s_at | 96% | |||
| 211161_s_at | COL3A1 | collagen, type III, alpha 1 | extracellular matrix organization, focal adhesion, integrin and PDGF signaling, TGF-β signaling | 100% |
| 215076_s_at | 100% | |||
| 201852_x_at | 94% | |||
| 211980_at | COL4A1 | collagen, type IV, alpha 1 | extracellular matrix organization, focal adhesion, signaling, epithelial cell differentiation | 100% |
| 206336_at | CXCL6 | chemokine (C-X-C motif) ligand 6 | response to wounding | 100% |
| 209335_at | DCN | Decorin | extracellular matrix organization, organ morphogenesis, TGF-β signaling | 100% |
| 214247_s_at | DKK3 | dickkopf homolog 3 | Wnt signaling, embryonic development | 100% |
| 221127_s_at | 68% | |||
| 202196_s_at | 63% | |||
| 213068_at | DPT | dermatopontin | cell adhesion, extracellular matrix organization and interactions | 100% |
| 213071_at | ||||
| 201842_s_at | EFEMP1 | EGF-containing fibulin-like extracellular matrix protein 1 | extracellular matrix organization, cell adhesion, cell migration | 100% |
| 201843_s_at | ||||
| 201839_s_at | EPCAM | epithelial cell adhesion molecule | cell adhesion, embryonic stem cell proliferation and differentiation | 100% |
| 206070_s_at | EPHA3 | EPH receptor A3 | signal transduction, response to cytokine stimulus | 100% |
| 202766_s_at | FBN1 | fibrillin 1 | extracellular matrix organization and structure, integrin interactions | 100% |
| 204472_at | GEM | GTP binding protein overexpressed in skeletal muscle | immune response, receptor mediated signal transduction regulatory protein | 100% |
| 209291_at | ID4 | inhibitor of DNA binding 4, dominant negative helix-loop-helix protein | Regulation of DNA binding and transcription, TGF-β signaling | 100% |
| 209292_at | 59% | |||
| 201163_s_at | IGFBP7 | insulin-like growth factor binding protein 7 | cell adhesion, cell growth and proliferation | 100% |
| 205422_s_at | ITGBL1 | integrin, beta-like 1 | cell adhesion, Integrin signaling | 100% |
| 214927_at | 100% | |||
| 231993_at | 100% | |||
| 1557080_s_at | 80% | |||
| 209099_x_at | JAG1 | jagged 1 | Notch signaling, cell migration and proliferation, morphogenesis | 100% |
| 216268_s_at | ||||
| 201744_s_at | LUM | lumican | extracellular matrix organization, epithelial cell migration | 100% |
| 225782_at | MSRB3 | methionine sulfoxide reductase B3 | oxidation-reduction, stress response | 100% |
| 230081_at | PLCXD3 | phosphatidylinositol-specific phospholipase C, X domain containing 3 | Lipid metabolism, signal transduction | 100% |
| 203083_at | THBS2 | thrombospondin 2 | extracellular matrix interactions, focal adhesion, TGF-β signaling | 100% |
| 204359_at | FLRT2 | fibronectin leucine rich transmembrane protein 2 | cell adhesion, receptor signaling, extracellular matrix organization | 99% |
| 203001_s_at | STMN2 | stathmin-like 2 | microtubule dynamics, cell motility, signal transduction | 99% 98% |
| 203000_at | ||||
| 213519_s_at | LAMA2 | laminin, alpha 2 | extracellular matrix interactions, focal adhesion, embryonic development | 98% |
| 223063_at | C1orf198 | chromosome 1 open reading frame 198 | unknown | 98% |
| 201417_at | SOX4 | SRY (sex determining region Y)-box 4 | Wnt signaling, embryonic development, determination of cell fate, transcription factor | 98% |
| 202291_s_at | MGP | matrix Gla protein | multicellular organismal development, bone formation, cell adhesion | 97% |
| 205547_s_at | TAGLN | transgelin | cytoskeleton organization, actin cross-linking | 97% |
| 222925_at | DCDC2 | doublecortin domain containing 2 | microtubule dynamics, cell migration | 95% |
| 206385_s_at | ANK3 | ankyrin 3, node of Ranvier | extracellular matrix organization, cell motility and activation | 94% |
| 208782_at | FSTL1 | follistatin-like 1 | BMP signaling, cell proliferation and differentiation | 93% |
| 219304_s_at | PDGFD | platelet derived growth factor D | Focal adhesion, embryonic development, angiogenesis, PDGF signaling | 91% |
| 221510_s_at | GLS | glutaminase | amino acid metabolism, metabolic pathways | 90% |
| 222453_at | CYBRD1 | cytochrome b reductase 1 | oxidoreductase, dietary iron absorption | 88% |
| 206580_s_at | EFEMP2 | EGF-containing fibulin-like extracellular matrix protein 2 | extracellular maxtrix, coagulation, activation of complement, connective tissue development | 88% |
| 221731_x_at | VCAN | versican | extracellular maxtrix, cell adhesion, tissue morphogenesis | 87% |
| 227070_at | GLT8D2 | glycosyltransferase 8 domain containing 2 | transferase activity | 86% |
|
205674_x_at 207434_s_at |
FXYD2 | FXYD domain containing ion transport regulator 2 | ion transport, regulation of sodium/potassium ATP transport | 85% |
| 223737_x_at 224400_s_at |
CHST9 | carbohydrate (N-acetylgalactosamine 4-0) sulfotransferase 9 | cell-cell interaction, signal transduction, hormone and carbohydrate biosynthesis | 84% 83% |
| 201438_at | COL6A3 | collagen, type VI, alpha 3 | extracellular matrix receptor interaction, focal adhesion, PDGF signaling | 84% |
| 218224_at | PNMA1 | paraneoplastic antigen MA1 | apoptosis, inflammatory response, focal adhesion | 84% |
| 228608_at | NALCN | sodium leak channel, non-selective | voltage-independent cation channel activity | 79% |
| 203685_at | BCL2 | B-cell CLL/lymphoma 2 | focal adhesion, apoptosis signaling | 77% |
| 226103_at | NEXN | nexilin | cell adhesion and migration, cytoskeleton organization | 75% |
| 202936_s_at | SOX9 | SRY (sex determining region Y)-box 9 | cell fate determination, skeletal development, regulation of apoptosis, cell proliferation | 75% |
| 205328_at | CLDN10 | claudin 10 | cell adhesion, tight junctions | 70% |
| 203088_at | FBLN5 | fibulin 5 | extracellular matrix organization, cell adhesion, endothelial cell adhesion | 65% |
| 212865_s_at | COL14A1 | collagen, type XIV, alpha 1 | extracellular matrix organization, cell adhesion | 63% |
| 209154_at | TAX1BP3 | Tax1 (human T-cell leukemia virus type I) binding protein 3 | transcription regulator, Rho signaling, negative regulation of Wnt signaling | 63% |
| 1556499_s_at | COL1A1 | collagen, type I, alpha 1 | extracellular matrix organization, cell adhesion, focal adhesion | 61% |
| 227091_at | CCDC146 | coiled-coil domain containing 146 | unknown | 58% |
| 221011_s_at | LBH | limb bud and heart development homolog | multicellular organismal development, regulation of transcription | 56% |
| 217892_s_at | LIMA1 | LIM domain and actin binding 1 | focal adhesion, actin cytoskeleton, angiogenesis | 54% |
| 204955_at | SRPX | sushi-repeat-containing protein, X-linked | cell adhesion | 54% |
| 225015_at | C12orf75 | chromosome 12 open reading frame 75 | unknown | 53% |
| 225645_at | EHF | Ets homologous factor | regulation of DNA transcription, epithelial cell differentiation, cell proliferation | 52% |
| 212233_at | MAP1B | microtubule-associated protein 1B | microtubule assembly, cytoskeleton | 51% |
Percentage appears over 1000 iterations signifies the percentage that the Affymetrix probe was included in the gene profile of advanced non-alcoholic fatty liver disease over 1000 iterations of SVM model building.
Gene profile associated with severe NAFLD
Using the hepatic gene expression data from the discovery cohort of 72 patients, we identified a 64 gene profile that was reproducibly associated with severe NAFLD (Table 2). On average, 7% of patients were misclassified in a test set from the discovery cohort using the SVM repeated hold-out method over 1000 training and testing iterations. This 64 gene profile had a corresponding area under the receiver operating characteristic (ROC) curve of 0.978 (Supplementary Figure 2). All 64 genes in the profile were up-regulated in severe NAFLD relative to mild NAFLD. As determined by IPA, prominent genes were associated with biological functions such as tissue remodeling/regeneration (COL1A2, COL3A1, COL4A1, LUM), progenitor cells (EPCAM, JAG1, SOX9), cancer (AQP1, CD24, EFEMP1, PDGFRA, SRPX, SOX4) and cardiovascular disease (DCN, LAMA2, THBS2, BCL2). QRT-PCR was used to validate the expression of eight randomly selected genes using total liver RNA from seven patients with mild NAFLD and seven patients with severe NAFLD from the discovery cohort. In each case, the microarray results were confirmed, with all genes showing significantly increased expression in patients with severe NAFLD (Figure 2).
Figure 2. Quantitative RT-PCR of human NAFLD RNA samples.

Bar graphs represent the fold difference in gene expression for mild NAFLD as compared to severe NAFLD. Dark bars represent the average gene expression in mild NAFLD from 7 unique patients while the light bars represent the average gene expression in severe NAFLD from 7 unique patients. RPL35 was used as the control gene for all analyses. Results are means ± SE.
Validation of severe NAFLD gene profile
The 64 gene profile was tested in an independent validation cohort consisting of 17 NAFLD patients (10 mild and 7 severe). It predicted disease status in the validation cohort with a misclassification error rate of 17.6% (i.e., accuracy of 82.4%), confirming that the differentially-expressed genes reproducibly characterized two distinct sub-populations of NAFLD patients. Clinical parameters (e.g., age, gender, BMI, and diabetes mellitus), (23) blood tests (e.g., AST platelet ratio index, APRI) (24), or combinations of clinical and laboratory variables (e.g., the NAFLD Fibrosis Score, NFS) (25) are often used to estimate the severity of liver fibrosis. These approaches were able to rule out F3–4 fibrosis in our discovery and validation cohorts with high specificity, but demonstrated only limited sensitivity for detecting advanced fibrosis (Supplementary Tables 8 and 9). Thus, the predictive values of standard noninvasive tools for staging liver fibrosis in our study populations were similar to those reported by others, (24, 25) underscoring the unmet need for more informative noninvasive biomarkers of NAFLD severity.
To identify genes that enhance accurate characterization of severe NAFLD, we used logistic regression to control for known factors associated with NAFLD-fibrosis (i.e., age, gender, BMI, HbA1c), and identified a 20 gene subset of the 64 gene profile that was independently associated with severe NAFLD (p<0.0005) in the discovery cohort. These 20 genes (Table 2) were then used to build a new SVM model that improved the misclassification rate to 5.9% in the validation cohort. Analysis of these genes again revealed up-regulation of genes associated with cell adhesion and tissue remodeling/regeneration.
Immunohistochemistry confirms activation of pathways that promote the ductular reaction in severe NAFLD
Closer inspection of the data acquired via our unbiased gene profiling approaches indicated that severe NAFLD is characterized by increased expression of Hedgehog (HH) target genes that promote the ductular reaction (DR) (e.g., SOX9, SPP1, and JAG1). Because the DR strongly correlates with fibrosis stage in NAFLD, (26) the present results independently corroborate earlier studies which reported a strong correlation between the level of HH pathway activity and fibrosis severity in other NAFLD patients.(27) Therefore, we felt it was critical to assure that the observed changes in liver mRNAs were accompanied by changes in expression of the respective proteins in pertinent cell types in the present NAFLD cohort. We used immunohistochemistry to mark and quantify accumulation of HH ligand-producing cells (expressing sonic hedgehog (SHH)), HH-responsive cells (expressing glioblastoma 2(GLI2)), myofibroblasts (expressing α-smooth muscle actin (α-SMA)), and progenitors (expressing keratin 7 (K7) and SOX9). The number of cells expressing SHH was greater in severe NAFLD (Figure 3; p<0.001) compared to mild NAFLD, as were GLI2-positive and K7-positive cells (Figure 3; p=0.035 and p=0.004). Similarly, the number of cells co-expressing GLI2 and SOX9 was greater in severe NAFLD relative to mild NAFLD, and paralleled the increase in myofibroblastic cells (α-SMA+ cells) (Figure 3; p<0.001). Thus, the current IHC data support the microarray data, with both confirming that severe NAFLD is characterized by activation of signaling pathways (such as HH) that promote the DR, a repair response that correlates with fibrosis severity (and thus, liver outcomes) in NAFLD.(26, 27)
Figure 3.

Hepatic accumulation of markers of tissue repair and regeneration and liver progenitor cells is greater in severe NAFLD compared to mild NAFLD. Photomicrographs of SHH, GLI2, K7, SOX9 and α-SMA IHC in patients with mild and advanced NAFLD are shown (400x magnification). Liver sections stained for SHH (brown, arrowheads) show greater numbers of positive cells in severe NAFLD (B) compared to mild NAFLD (A). Liver sections double stained for K7 (brown) and GLI2 (red, nuclear, arrow head) reveals significantly higher grade of co-staining (arrows) in livers with severe NAFLD (D) than in mild NAFLD (C). Photomicrographs illustrate parallel accumulation of liver progenitor cells co-expressing GLI2 (red, nuclear) and SOX9 (blue, nuclear), along with α-SMA-positive myofibroblastic cells (brown) in severe NAFLD (F) compared to mild NAFLD (E). Many bile ductular cells in severe NAFLD (1000x magnification) express SOX9 (blue, nuclear, arrows); such cells generally co-express GLI2 (red, nuclear, arrow heads)(G). Figure and table summarizing the semi-quantitative immunohistochemistry results are shown in the Supplementary Materials and Methods.
DISCUSSION
Improving the outcomes of NAFLD has been hampered by poor understanding of the mechanisms that control either its resolution or progression. Insight has been gleaned from careful characterization of histologic parameters that correlate with the severity of liver fibrosis which, in turn, is used to predict the likelihood of NAFLD progression.(1, 4, 5, 25) Liver biopsy findings demonstrate that the extent of liver cell injury is one of the main parameters that differentiate NASH from NAFL. Histology also shows that NASH itself is a heterogeneous disease that is variably associated with fibrosis.(4) These results suggest that therapeutic success might be improved by sub-classifying NAFLD/NASH patients into more homogeneous groups that share common factors for disease progression. Histology alone, however, is unable to reveal biological processes underlying prognostically relevant differences in hepatic morphology. Thus, alternative methodologies are necessary to further development of noninvasive diagnostic tests and optimize treatments for various NAFLD sub-groups.
Encouraged by successes in the field of oncology wherein gene profiling of malignancies has permitted disease sub-classification, (12, 13) we used microarray-based approaches to characterize the gene expression profiles of NAFLD livers at opposite extremes of the fibrosis spectrum. We studied tissues acquired by percutaneous liver biopsy of patients referred to us for NAFLD staging because we reasoned that the resultant information might guide development of novel biomarkers and/or treatments for types of NAFLD patients who are typically referred to practicing hepatologists for management advice. The resultant microarray data generated a comprehensive and unbiased “snap-shot” of the respective gene expression profiles in NAFLD livers with either severe (F3–4) or mild (F0–1) fibrosis. Further computational analysis identified sub-sets of consistently differentially expressed genes, as well as various signaling pathways that are differentially activated (or suppressed), in the two disease states.
Differential expression of various matrix molecules was demonstrated in NAFLD livers with mild and severe NAFLD. Because we used differences in histologic fibrosis stage to categorize our study cohorts, molecular evidence for fibrosis differences validates the utility of the microarray approach for identifying gene expression profiles that are truly discriminatory. Equally important is the fact that the microarray data reveal specific collagen species (e.g., collagen 1α1, 1α2, 3α1, 4α1, 6α3, 14α1) and other matrix molecules (e.g., fibrillin 1, laminin α2, fibulin 5) that are being actively generated in severe NAFLD. Matrix characteristics regulate the fates of cells that mediate fibrogenesis and fibrinolyisis (28) and thus, clinical trials are beginning to target specific matrix molecules for modification in an attempt to promote fibrosis regression (e.g., Clinical Trials.gov identifier: NCT01672866). By characterizing matrix molecules that are selectively accumulating in livers with severe NAFLD-related damage, the microarray data identify novel therapeutic targets. Our gene profiling also demonstrated differential expression of PDGFRA and one of its ligands, PDGFD, in severe versus mild NAFLD. Pharmacologic antagonists of PDGF signaling are currently available, but there are no reports of their use as NAFLD therapies. Based on our microarray findings, it might be reasonable to target PDGF signaling in pre-symptomatic patients with NAFLD because this pathway is known to be activated during fibrogenesis and in many cancers. (29)
Another noteworthy difference between severe and mild NAFLD was the up-regulation of regeneration/repair-related genes in severe NAFLD. NAFLD outcomes have been correlated with differences in the extent of hepatocyte death. Indeed, serum levels of cytoskeletal proteins that are released by dying hepatocytes (K8/18) are useful biomarkers of fibrosis severity in NASH.(30) Liver cell death triggers regenerative responses that mobilize myofibroblasts and progenitors, cell types that are involved in the DR.(26, 27) The intensity of the DR correlates with the severity of NAFLD-related fibrosis.(27) However, the mechanisms underlying this association, and why only some individuals with NASH develop a prominent DR, are unclear. By characterizing the gene profiles that occur in livers with severe NAFLD and mild NAFLD, our microarray analysis provides unbiased insight into these issues. Our results demonstrated that direct transcriptional targets of GLI proteins are prominent among the up-regulated genes in severe NAFLD. This is relevant because GLI proteins are down-stream effectors of Hedgehog (HH), a morphogenic signaling pathway that controls the fates of ductular type progenitors and myofibroblasts during adult liver regeneration.(31) Three of the HH-target genes that are activated in severe NAFLD (SOX9, SPP1, and Jagged-1) are particularly pertinent to the DR. Both ductular progenitors (32) and myofibroblastic-stellate cells (MF-HSC) (33, 34) express SOX9. SOX9 and GLI proteins interact to control the transcription of SPP1 (osteopontin).(33, 35)
Osteopontin is expressed by ductular-type progenitors and MF-HSC in injured livers, and functions as a pro-fibrogenic factor for HSC.(33, 35, 36) HH also stimulates MF-HSC to produce Jagged1(37), a ligand for the Notch pathway that stimulates ductular differentiation of liver progenitors.(37, 38) In mouse models of liver injury and fibrosis, inhibiting SPP1 or blocking HH signaling prevents accumulation of ductular-type progenitors and myofibroblasts (i.e., the DR) and reduces fibrosis.(33–35) Hence, our microarray findings provide proof-of-concept support for using unbiased gene profiling to identify molecules/pathways that can be targeted to develop new diagnostic tests and therapies for severe NAFLD. This, in turn, justifies further scrutiny of the present microarray data to identify other novel therapeutic and diagnostic targets in NAFLD. For example, genes that control discrete metabolic processes (e.g., bile acid biosynthesis, branched chain amino acid degradation, and glycine/serine/threonine metabolism) were found to be differentially expressed in livers with mild and severe NAFLD, supporting the need for research to determine if/how these processes impact NAFLD progression.
Useful non-invasive biomarkers of liver disease accurately reflect tissue pathology. Because large clinical data sets are typically enriched with individuals with mild NAFLD, serum biomarker development in NAFLD has focused on perfecting blood tests to detect NAFLD in the general population, or to differentiate NASH from simple hepatic steatosis in obese populations at high risk for NAFLD. In contrast, our study population was comprised entirely of subjects with biopsy-proven NAFLD who had either very mild fibrosis or histologically-advanced fibrosis. This permitted a more focused comparison of hepatic gene expression profiles that distinguish individuals at opposite ends of the spectrum of NAFLD severity. To further refine the specificity of gene expression differences for marking net differences in NAFLD histology, we used logistic regression to control for potential effects of age, gender, BMI, and glycemic control on liver gene expression. This approach minimized other confounding influences that are known to impact NAFLD-related fibrosis (and thus, outcomes). A smaller sub-set of 20 genes that independently correlated with NAFLD severity emerged from our initial 64 gene profile. Subsequent validation studies demonstrated that this 20 gene profile correctly classified NAFLD severity in 94% patients, inspiring confidence that the differentially-expressed molecular targets could be useful for developing new non-invasive diagnostic tests for severe NAFLD. Success would facilitate population-based screening to detect individuals with NAFLD who merit more intensive management because they are at high risk for progressive liver disease. Several of the differentially-expressed hepatic genes (e.g., C7, CXCL-6, IGFBP7, and THBS2) encode soluble proteins that are not currently being used as diagnostic/prognostic markers in NAFLD but are easily assayed and hence, could be readily tested for this purpose.
Despite its merits, our study had limitations that are important to acknowledge. First, the sample size was small, particularly in the validation cohort, and this may have limited our power to detect real differences in gene expression. Validation in a second larger, independent dataset should be performed to assure confidence in these initial results. Second, the cross-sectional design of the study precludes use of discrete data points to predict NAFLD progression and therefore, cannot prove causality. The fact that a particular gene/pathway is differentially expressed in patients with mild and severe NAFLD simply identifies it as a marker of one or the other state, and suggests that it is a plausible therapeutic target. Future studies that manipulate the activity of such target(s) and assess the impact on clinically-relevant outcomes will demonstrate which, if any, drive disease progression. Third, we dichotomized our cohort into more extreme histologic phenotypes and eliminated the intermediate level of fibrosis (i.e., F2). This increased our power to discern more significant and clinically relevant changes in gene expression, as outcomes of intermediate levels of fibrosis are more variable. Future larger studies which include NAFLD patients with F2 fibrosis will be important to further validate our findings.
Supplementary Material
Table 3. Ingenuity Pathway Analysis.
| Top Biological Functions | ||
|---|---|---|
| Diseases and Disorders | p value | Genes (n) |
| Cancer | 5.43 × 10−28 – 1.31 × 10−3 | 442 |
| Reproductive System Disease | 1.61 × 10−16 – 2.30 × 10−4 | 267 |
| Gastrointestinal Disease | 3.04 × 10−11 – 1.10 × 10−3 | 180 |
| Cardiovascular Disease | 3.49 × 10−10 – 1.28 × 10−3 | 143 |
| Genetic Disorder | 5.67 × 10−10 – 8.62 × 10−3 | 269 |
| Molecular and Cellular Functions | ||
| Cellular Movement | 1.10 × 10−14 – 1.29 × 10−3 | 219 |
| Cellular Growth and Proliferation | 9.27 × 10−13 – 1.01 × 10−3 | 337 |
| Cell Morphology | 1.62 × 10−11 – 1.16 × 10−3 | 228 |
| Cellular Assembly and Organization | 2.88 × 10−11 – 1.19 × 10−3 | 196 |
| Protein Synthesis | 7.91 × 10−10 – 2.20 × 10−4 | 47 |
| Top Canonical Pathways | Ratio | |
| Fatty Acid Metabolism | 3.18 × 10−8 | 25/184 |
| Valine, Leucine and Isoleucine Degradation | 1.25 × 10−7 | 17/107 |
| Bile Acid Biosynthesis | 2.23 × 10−7 | 15/105 |
| Glycine, Serine and Threonine Metabolism | 3.11 × 10−6 | 16/147 |
| Butanoate Metabolism | 5.65 × 10−6 | 14/128 |
| Top Toxicology Functions | ||
| Hepatotoxicity | ||
| Hepatocellular Carcinoma | 3.49 × 10−4 – 1.77 × 10−1 | 54 |
| Liver Cholestasis | 1.10 × 10−3 – 1.66 × 10−1 | 15 |
| Liver Necrosis/Cell Death | 1.36 × 10−3 – 5.71 × 10−1 | 26 |
| Liver Hepatitis | 1.43 × 10−3 – 2.60 × 10−1 | 20 |
| Liver Proliferation | 1.80 × 10−3 – 3.51 × 10−1 | 20 |
The data represents the number of genes that are either up or down-regulated in severe NAFLD relative to mild NAFLD. Biological functions, canonical pathways and toxicological functions were assigned to the overall analysis using findings that have been extracted from the scientific literature and stored in the ingenuity pathway analysis. A Fisher Exact test corrected for multiple testing via the Benjamini-Hochberg method is used to calculate a q-value determining the probability that the function or pathway assigned to the analysis is explained by chance alone.
Acknowledgments
We would like to thank all of the study participants who contributed their biospecimens and data to the Duke University Health System NAFLD Clinical Database and Biorepository and gratefully acknowledge our referring physicians, research and data management personnel, study coordinators, and clinical personnel without whom this study would not have been possible. The authors would also like to thank James L. Burchette (formerly of Duke University, Durham, NC) for performing all of the immunohistochemical staining.
Financial support: Majority of this work was supported through an American Recovery and Reinvestment Act (ARRA) grant from the NIAAA: 5RC2 AA019399 (Anna Mae Diehl, Principal Investigator). Drs. Diehl and Abdelmalek received funding support from NIH/NIDDK grant U01-DK57149. Dr. Abdelmalek was supported by a NIH/NIDDK K23 Career Development Award (K23-DK062116).
Abbreviations
- α-SMA
α-smooth muscle actin
- APRI
AST platelet ratio index
- BMI
body mass index
- CVD
cardiovascular disease
- DM
diabetes mellitus
- FDR
false discovery rate
- GLI2
glioblastoma 2
- HbA1c
hemoglobin A1c
- HH
hedgehog
- HCC
hepatocellular carcinoma
- IHC
immunohistochemical
- IPA
Ingenuity Pathways Analysis
- K7
keratin 7
- LPCs
liver progenitor cells
- MF_HSC
myofibroblastic-stellate cells
- NCBI
National Center for Biotechnology Information
- NAFLD
non-alcoholic fatty liver disease
- NAS
NAFLD Activity Score
- NFS
NAFLD Fibrosis Score
- NASH
nonalcoholic steatohepatitis
- QRT-PCR
quantitative reverse-transcription polymerase chain reaction
- ROC
receiver operating characteristic
- SHH
sonic hedgehog
- SOX9
sex determining region Y-box 9
- SVM
Support Vector Machines
Footnotes
CONFLICT OF INTEREST
Potential competing Interests: None.
Guarantor of the article: Anna Mae Diehl, MD.
Specific author contributions: All authors have contributed to data interpretation and approved final version of the manuscript. Study concept and design: Moylan, Suzuki, Abdelmalek, Diehl; data collection: Moylan, Suzuki, Choi, Michelotti, Hampton, Chen; statistical analysis: Pang, Dellinger, Suzuki, Garrett, Ashley-Koch, Hauser, Moylan; immunohistochemistry and interpretation: Guy, Suzuki, Moylan, Diehl; drafting of manuscript: Moylan, Pang, Hauser, Diehl; critical review of study design and manuscript revision: Hauser, Murphy, Tillmann, Abdelmalek, Moylan, Diehl.
Contributor Information
Cynthia A. Moylan, Email: cynthia.moylan@dm.duke.edu.
Herbert Pang, Email: herbert.pang@dm.duke.edu.
Andrew Dellinger, Email: andrew.dellinger@dm.duke.edu.
Ayako Suzuki, Email: ayako.suzuki@dm.duke.edu.
Melanie E. Garrett, Email: melanie.garrett@duke.edu.
Cynthia D. Guy, Email: cynthia.guy@dm.duke.edu.
Susan K. Murphy, Email: susan.murphy@dm.duke.edu.
Allison E. Ashley-Koch, Email: allison.ashleykoch@duke.edu.
Steve S. Choi, Email: steve.choi@dm.duke.edu.
Gregory A. Michelotti, Email: greg.michelotti@dm.duke.edu.
Daniel D. Hampton, Email: hamptdan@gmail.com.
Yuping Chen, Email: yuping.chen@dm.duke.edu.
Hans L. Tillmann, Email: hans.tillmann@dm.duke.edu.
Michael A. Hauser, Email: mike.hauser@dm.duke.edu.
Manal F. Abdelmalek, Email: manal.abdelmalek@duke.edu.
Anna Mae Diehl, Email: annamae.diehl@duke.edu.
References
- 1.Pagadala MR, McCullough AJ. The relevance of liver histology to predicting clinically meaningful outcomes in nonalcoholic steatohepatitis. Clin Liver Dis. 2012;16:487–504. doi: 10.1016/j.cld.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 3.Rafiq N, Bai C, Fang Y, Srishord M, McCullough A, Gramlich T, Younossi ZM. Long-term follow-up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol. 2009;7:234–238. doi: 10.1016/j.cgh.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 5.Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, Goodman Z. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology. 2011;53:1874–1882. doi: 10.1002/hep.24268. [DOI] [PubMed] [Google Scholar]
- 6.Caldwell SH, Lee VD, Kleiner DE, Al-Osaimi AM, Argo CK, Northup PG, Berg CL. NASH and cryptogenic cirrhosis: a histological analysis. Ann Hepatol. 2009;8:346–352. [PMC free article] [PubMed] [Google Scholar]
- 7.Mendes FD, Suzuki A, Sanderson SO, Lindor KD, Angulo P. Prevalence and indicators of portal hypertension in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2012;10:1028–1033. e1022. doi: 10.1016/j.cgh.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44:217–231. doi: 10.1016/j.jhep.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 9.Bosch J, Abraldes JG, Berzigotti A, Garcia-Pagan JC. The clinical use of HVPG measurements in chronic liver disease. Nat Rev Gastroenterol Hepatol. 2009;6:573–582. doi: 10.1038/nrgastro.2009.149. [DOI] [PubMed] [Google Scholar]
- 10.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 11.Sia D, Hoshida Y, Villanueva A, Roayaie S, Ferrer J, Tabak B, Peix J, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology. 2013;144:829–840. doi: 10.1053/j.gastro.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SM, Leem SH, Chu IS, Park YY, Kim SC, Kim SB, Park ES, et al. Sixty-five gene-based risk score classifier predicts overall survival in hepatocellular carcinoma. Hepatology. 2012;55:1443–1452. doi: 10.1002/hep.24813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang DY, Done SJ, McCready DR, Boerner S, Kulkarni S, Leong WL. A new gene expression signature, the ClinicoMolecular Triad Classification, may improve prediction and prognostication of breast cancer at the time of diagnosis. Breast Cancer Res. 2011;13:R92. doi: 10.1186/bcr3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ritchie ME, Silver J, Oshlack A, Holmes M, Diyagama D, Holloway A, Smyth GK. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23:2700–2707. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- 15.Loomba R, Abraham M, Unalp A, Wilson L, Lavine J, Doo E, Bass NM, et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology. 2012;56:943–951. doi: 10.1002/hep.25772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, McGlynn KA. Metabolic syndrome increases the risk of primary liver cancer in the United States: A study in the SEER-medicare database. Hepatology. 2011;54:463–471. doi: 10.1002/hep.24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong VW, Wong GL, Tsang SW, Fan T, Chu WC, Woo J, Chan AW, et al. High prevalence of colorectal neoplasm in patients with non-alcoholic steatohepatitis. Gut. 2011;60:829–836. doi: 10.1136/gut.2011.237974. [DOI] [PubMed] [Google Scholar]
- 18.Lu SC, Alvarez L, Huang ZZ, Chen L, An W, Corrales FJ, Avila MA, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci U S A. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, Martinez N, Varela M, Luka Z, Capdevila A, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 21.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 22.Kim D, Kim WR, Kim HJ, Therneau TM. Association between noninvasive fibrosis markers and mortality among adults with nonalcoholic fatty liver disease in the United States. Hepatology. 2013;57:1357–1365. doi: 10.1002/hep.26156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–1362. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- 24.Wai CT, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, Lok AS. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology. 2003;38:518–526. doi: 10.1053/jhep.2003.50346. [DOI] [PubMed] [Google Scholar]
- 25.Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, Enders F, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45:846–854. doi: 10.1002/hep.21496. [DOI] [PubMed] [Google Scholar]
- 26.Roskams T, Yang SQ, Koteish A, Durnez A, DeVos R, Huang X, Achten R, et al. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am J Pathol. 2003;163:1301–1311. doi: 10.1016/S0002-9440(10)63489-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, Bhathal PS, Dixon JB, et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology. 2007;133:80–90. doi: 10.1053/j.gastro.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Greenbaum LE, Wells RG. The role of stem cells in liver repair and fibrosis. Int J Biochem Cell Biol. 2011;43:222–229. doi: 10.1016/j.biocel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farooqi AA, Waseem S, Riaz AM, Dilawar BA, Mukhtar S, Minhaj S, Waseem MS, et al. PDGF: the nuts and bolts of signalling toolbox. Tumour Biol. 2011;32:1057–1070. doi: 10.1007/s13277-011-0212-3. [DOI] [PubMed] [Google Scholar]
- 30.Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN, McCullough AJ. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology. 2009;50:1072–1078. doi: 10.1002/hep.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ochoa B, Syn WK, Delgado I, Karaca GF, Jung Y, Wang J, Zubiaga AM, et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology. 2010;51:1712–1723. doi: 10.1002/hep.23525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T, Hosokawa S, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2011;43:34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- 33.Pritchett J, Harvey E, Athwal V, Berry A, Rowe C, Oakley F, Moles A, et al. Osteopontin is a novel downstream target of SOX9 with diagnostic implications for progression of liver fibrosis in humans. Hepatology. 2012 doi: 10.1002/hep.25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michelotti GA, Xie G, Swiderska M, Choi SS, Karaca G, Kruger L, Premont R, et al. Smoothened is a master regulator of adult liver repair. J Clin Invest. 2013 doi: 10.1172/JCI66904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, Mi Z, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology. 2011;53:106–115. doi: 10.1002/hep.23998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, Xie G, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. 2012;61:1323–1329. doi: 10.1136/gutjnl-2011-301857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, Ridgway RA, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18:572–579. doi: 10.1038/nm.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liew PL, Wang W, Lee YC, Huang MT, Lee WJ. Roles of hepatic progenitor cells activation, ductular reaction proliferation and Notch signaling in morbid obesity. Hepatogastroenterology. 2012;59:1921–1927. doi: 10.5754/hge10861. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.