

1. Introduction and Scope
A prior thematic issue of Chemical Reviews in 20041 provides broad coverage of the field of biomimetic inorganic chemistry. One principal objective of this field, which is a component of the continually burgeoning multidisciplinary enterprise that is bioinorganic chemistry, is the synthesis of analogues of mononuclear and polynuclear sites in proteins which convey information of significance in interpreting the physical and chemical properties of such sites. This article focuses on polynuclear analogues, specifically higher nuclearity, biomimetic metal-sulfur clusters.
Many synthetic and biological clusters can be designated as either strong-field or weak-field. Strong-field clusters are formed by first transition series elements with π-acceptor ligands and by second and third series elements regardless of ligation, and manifest properties arising from large splittings of the d-orbital manifold at individual metal sites. Such species are subsumed under a restrictive definition of clusters as containing two or more metal atoms where direct and substantial metal-metal bonding is present.2 A broader definition now normally employed leads to recognition of weak-field clusters. These species contain σ/π-donor ligands that induce smaller d-orbital splittings favoring individual metal sites with high-spin configurations, magnetic interactions among these individual sites, paramagnetic molecular ground states, and labile ligand binding. These clusters contain first transition series elements.
Prominent examples of metals in native weak-field cluster sites include (but are not limited to) Mn3–6 (catalases, photosystem II), non-heme Fe7–10 (O2 carriers, oxygenases, reductases, hydrogenase), Fe-S11–14 (electron transfer, nitrogenase, numerous nonredox functions), Ni15,16 (urease), and Ni-Fe17–19 (hydrogenase). The only exceptions to the weak-field designation are Fe sites in [FeFe]- and [NiFe]-hydrogenases, which occur as the fragments Fe(CO)(CN)(μ2-CO) and Fe(CO)(CN)2(μ2-H), respectively. We also note that the weak-field/strong field distinction does not strictly apply to zinc and copper complexes because ZnII and CuI are necessarily diamagnetic, CuII has a spin-doublet ground state, and CuIII is uniformly diamagnetic. However, all but CuIII manifest the substitutional lability associated with the weak-field case. As examples of weak-field clusters, structures of selected protein-bound sites 1–8 containing Mn, non-heme Fe, Fe-S, Ni, and Ni-Fe (Ni site) are provided in Figure 1. Because of the restricted scope of this article, a selected bibliography of summary accounts of the biomimetic chemistry of mono- and polynuclear sites is presented in Table 1. Citations are in the period 2004–2013 and do not include the contents of the previous thematic issue.1 We note in particular a book devoted to bioinorganic synthesis,20 a treatment of biosynthetic inorganic chemistry involving manipulation of protein-bound sites,21 and biomimetic research on sulfur-ligated sites.22 Other examples of weak-field metal sites in proteins are found in this issue.
Figure 1.

Schematic structures of illustrative weak-field protein-bound clusters: O2-evolving center in photosystem II (1), [FeFe]-hydrogenase (2), [NiFe]-hydrogenase (3); dinuclear (4), trinuclear cuboidal (5), and tetranuclear cubane-type (6) iron-sulfur clusters; ribonucleotide reductase (7), and urease (8). Catalytically active forms of certain of these clusters may differ in protonation and minor bonding interactions from those shown.
Table 1.
Selected Bibliography of Biomimetic Inorganic Chemisty: 2004–2013
| biometal | proteins/enzymes | refs.* |
|---|---|---|
| V | peroxidases, phosphatases | a,b |
| Mo/W | oxotransferases, hydroxylases, nitrogen fixation | b–h |
| Mo/V-Fe-S | nitrogenase | b,c |
| Mn | photosystem II, O2 evolution | i–m |
| non-heme Fe | O2-carriers, oxidases, oxygenases, reductases, hydrogenase | b,m–r |
| Fe-S | redox proteins, various enzymes | b,s |
| Ni | urease, CODH, SOD, Me-CoM reductase | b,s,t |
| Ni-Fe | hydrogenases, CODH | b,c,l,n,u–w |
| Cu | O2-carriers, redox proteins, oxidases, oxygenases | b,x,y,ac |
| Zn | peptidases, proteases, carbonic anhydrase, other mono- and multinuclear enzymes | b,z,aa,ab,ac |
Collected references are in alphabetical order.
Rehder, D. Bioinorganic Vanadium Chemistry; Wiley & Sons, Ltd.: West Sussex, England, 2008.
Kratz, H.-B.; Metzler-Nolte, N. Concepts and Models in Bioinorganic Chemistry; Wiley-VCH: New York, 2006.
Groysman, S.; Holm, R. H. Biochemistry 2009, 48, 2310–2320.
Holm, R. H.; Solomon, E. I.; Majumdar, A.; Tenderholt, A. Coord. Chem. Rev. 2011, 255, 993–1015.
Majumdar, A.; Sarkar, S. Coord. Chem. Rev. 2011, 255, 1039–1054.
Schulzke, C. Eur. J. Inorg. Chem. 2011, 1189–1199.
Dreher, A.; Stephan, G.; Tuczek, F. Adv. Inorg. Chem. 2009, 61, 367–405.
Pellei, M.; Papini, G.; Lobbia, G. G.; Santini, C. Current Bioactive Cpds 2009, 5, 321–352.
Mukhopadhyay, S.; Mandal, S. K.; Bhaduri, S.; Armstrong, W. H. Chem. Rev. 2004, 104, 3981–4026.
Mullins, C. S.; Pecoraro, V. L. Coord. Chem. Rev. 2008, 252, 416–443.
Tyagi, P.; Singh, U. P. Current Bioactive Cpds 2009, 5, 296–320.
Vrettos, J. S.; Brudvig, G. W., Bio-coordination Chemistry (Comprehensive Coordination Chemistry II); Que, L., Jr., Tolman, W. B. Eds., Chap. 8.20, Oxford: Elsevier, 2004.
Cho, J.; Sarangi, R.; Nam, W. Acc. Chem. Res. 2012, 45, 1321–1330.
Heinekey, D. M. J. Organometal. Chem. 2009, 694, 2671–2680.
Lee, D.; Lippard, S. J., in ref. l,, Chap. 8.13.
Liu, X.; Ibrahim, S. K.; Tard, C.; Pickett, C. J. Coord. Chem. Rev. 2005, 249, 1641–1652.
He, C.; Mishina, Y. Curr. Opinion Chem. Biol. 2004, 8, 201–208.
Friedle, S.; Reisner, E.; Lippard, S. J. Chem. Soc. Rev. 2010, 39, 2768–2779.
Koay, M. S.; Antonkine, M. L.; Gärtner, W.; Lubitz, W. Chem. Biodiversity 2008, 5, 1571–1587.
van der Vlugt, J. I.; Meyer, F. Met. Ions Life Sci. 2007, 2, 181–240.
Ohki, Y.; Tatsumi, K. Eur. J. Inorg. Chem. 2011, 973–985.
Tard, C.; Pickett, C. J. Chem. Rev. 2009, 109, 2245–2274.
Bouwman, E.; Reedijk, J. Coord. Chem. Rev. 2005, 249, 1555–1581.
Gennari, M.; Marchio, L. Current Bioactive Cpds 2009, 5, 244–263.
Solomon, E. I.; Hadt, R. G. Coord. Chem. Rev. 2012, 255, 774–789.
Berreau, L. M. Eur. J. Inorg. Chem. 2006, 273–283.
Parkin, G. New J. Chem. 2007, 31, 1996–2014.
Fisher, N. V.; Tuerkoglu, G.; Burzlaff, N. Current Bioactive Cpds 2009, 5, 277–295.
See articles in Bio-coordination Chemistry (Comprehensive Coordination Chemistry II).
The purview of this article is the biomimetic chemistry of metal-sulfur clusters as confined to post-2004 advances of homo- and heterometallic iron-sulfur clusters of nuclearity four and higher. The emphasis is on the synthetic approaches to such clusters, some of which in their native condition are implicated directly in numerous enzymological processes.
2. General Tactics in Cluster Synthesis
2.1. Overview
The synthesis of molecular transition element clusters of general formulation [MmQqLl]z, where m is the cluster nuclearity, Q is a bridging ligand of variable connectivity, L is a terminal ligand, [MmQq]n is the core, and z and n are the cluster and core charges, respectively, is a frequently empirical endeavor. Fortunately, in the last several decades sufficient information has been collected on diverse systems to allow descriptive classification of some synthetic procedures, affording what is hoped to be the beginning of an ultimately organized subject of expansive scope.
Here we present a classification of the primary tactics employed in the synthesis and related reactions of metal-sulfur weak-field clusters by use of illustrative cases, the majority of which are drawn from our own work. Biomimetic synthesis is an endeavor directed toward synthetic representations of protein-bound metal sites with the attendant potential benefit of the discovery of new reactions and structures regardless of biological relevance.
In a previous summary of biomimetic cluster synthesis,23 we briefly identified synthetic methodologies that are potentially useful in organizing and classifying some of the many approaches to weak-field clusters. Here we expand on this theme by examining the four methodologies we consider to be of pragmatic value in this regard. Three of these are of particular importance and are well-represented by syntheses outlined in subsequent sections.
2.2. Reaction Types
-
Self-Assembly: self-organized (i.e., spontaneous) assembly of clusters from simple mononuclear precursors; in the most elementary case separate metal, core ligand, and terminal ligand precursors are employed.
The term self-assembly, or often interchangeably self-organization, is widely used, especially in supramolecular chemistry where it is applied to spontaneous and reversible processes in which molecules (sometimes identical) are organized into larger entities stabilized by non-covalent interactions.24 Here the term applies to a spontaneous reaction in which the product synthesis is driven by formation of covalent bonds between metal and ligand possibly accompanied by electron transfer. Self-assembly applies to an individual reaction or to a system of consecutive reactions.
-
Fragment Condensation: combination of a preexisting di- or polynuclear cluster with itself or with another mononuclear or polynuclear species to form (usually) higher-nuclearity clusters; ideally, product composition and structure are predictable from precursors.
This method, often described as fragment coupling, finds analogy with organic synthesis if one reactant is mononuclear (linear syntheses) or both are di- or polynuclear (convergent synthesis). Peptide coupling exemplifies these synthesis types. The term is applicable to individual or consecutive reaction systems and may involve electron transfer. Placement of a reaction in this category does not require that the fragments remain intact over the entire reaction, only that the initial reactants are of the type(s) specified. While sometimes not recognized as such, fragment condensation can provide efficient entry to a range of new cluster types.
-
Core Rearrangement: transformation of a preexisting cluster to a different core geometry with retention of nuclearity by thermal treatment, oxidation-reduction, or ligand substitution or addition.
This method encompasses geometrical isomerization at constant cluster composition and formation of distinct new cluster types upon alteration of core composition. At its simplest, this reaction type presupposes minimal core deconstruction–as opposed to significant cluster disassembly and reassembly–over the course of reaction. This matter has not been examined experimentally, and plausible reactions for this category currently implicate reactant clusters of stability sufficient for isolation or formation in solution. Core rearrangement usually applies to discrete reactions.
-
Fragmentation: transformation of a preexisting cluster to a lower nuclearity cluster; under ideal rational circumstances, the reduced nuclearity product is a discernable substructure of the parent cluster.
This reaction type is the reverse of fragment condensation. As a synthetic method, fragmentation has limited application inasmuch as nuclearity expansion is the usual objective in cluster synthesis. Nonetheless, fragmentation has use in cases where higher nuclearity cores are readily achieved, and lower nuclearity structures can be derived therefrom. Cluster excision – the removal of intact molecular clusters from cluster-containing network solids – can be viewed as the limiting case of fragmentation from extended lattices.25–27
In the sections that follow, depicted structures have been authenticated by X-ray crystallography, redox potentials are quoted vs. the SCE, and 57Fe isomer shifts in Mössbauer spectroscopy are referenced to iron metal at room temperature. Because the primary concerns are cluster synthesis and structure types, electronic features are not treated in detail.
3. Fe4S4 Clusters
As metallobiochemistry has progressed over the last several decades, the evolutionary biological distribution and the multifarious roles of cubane-type clusters with the [Fe4(μ3-S)4]n core and variable oxidation states n have become increasingly disclosed. Functions include inter alia electron transfer, enzymatic catalysis, nitrogen fixation, photosynthesis, respiration, signaling (O2, NO, iron concentration), gene regulation, and DNA repair and replication. Indeed, in the realm of metal cofactors the Fe4S4 cluster has assumed a prominence rivaling that of the heme group. It is appropriate, therefore, in an issue devoted to bioinorganic enzymology that recent fundamental chemistry of Fe4S4 clusters, and heterometal and heteroligand versions thereof, be examined. [Fe4S4(SR)4]2− clusters were first chemically synthesized 40 years ago in classic self-assembly systems such as 4FeCl3/6RS−/4HS−/4MeO− in methanol and, 4FeCl2/10RS−/4S and 4FeCl3/14RS−/4S in acetonitrile and other solvents (Section 6.1). Balanced reactions are easily written for these and other methods of synthesis which are described elsewhere.11,28 The success of the self-assembly approach proved that proteins are unnecessary for the synthesis of this cluster type. Given their essential roles in biology, the chemistry of Fe4S4 and related clusters remains an important subject. The synthetic chemistry of these clusters through 2004 has been summarized.11,28 In the following sections, newer features of these clusters, in some cases enhancing their role as synthetic analogues of protein sites, are considered. The primary concern is with systems containing the generalized clusters [M4Q4Ll]z (l ≥4) whose existence is a consequence of research in biomimetic inorganic chemistry.1 The clusters [Fe4S4Ll]z constitute a large majority; the usual case has l = 4 with tetrahedral weak-field coordination at the iron sites.
3.1. Limiting Oxidation States
As seen in Figure 2, cubane-type clusters present five conceivable core oxidation states [Fe4S4]n (n = 0,…,4+) based on Fe2+,3+ content. Of these, three are implicated in the classic ferredoxin ([Fe4S4]2+/1+) and high-potential protein ([Fe4S4]3+/2+) redox couples, also frequently observed in proteins of higher molecular weight. In the period 2005–2010, the remaining two oxidation states have been achieved in isolated compounds.
Figure 2.

Range of oxidation levels in synthetic and protein-bound Fe4S4 clusters showing the states involved in Fd and HP redox couples, principal ligands stabilizing different oxidation levels in synthetic clusters, and ground spin states.
3.1.1. Superreduced (All-Ferrous) State
Recently, considerable interest has attended the unequivocal spectroscopic demonstration of the all-ferrous [Fe4S4]0 state formed by reaction of oxidized A. vinelandii Fe protein of nitrogenase29 and an A. fermentans dehydratase30 with the strong reductant TiIII(citrate). Although this state has been detected electrochemically as [Fe4S4(SR)4]4− (E½4−/3− ≲ −1.6 V),31 no synthetic cluster of any type had ever been isolated owing to extreme oxidative instability. Synthetic access to these clusters was achieved in 2005–2008 by reaction schemes [1] and [2] in Figure 3. In scheme [1], cluster 9, obtained by standard self-assembly (box), is transformed by ligand substitution reactions and reduction to [Fe4S4(CN)4]3−.32 This cluster is further reduced with the powerful reductant benzophenone ketyl radical anion to the all-ferrous cluster 10 which was authenticated by an X-ray structure determination and Mossbauer spectroscopy.33 It is the first [Fe4S4]0 site analogue isolated in substance. However, this cluster is extraordinarily sensitive to oxidation and is an impractical object of investigation, particularly in solution.
Figure 3.

Syntheses of the [Fe4S4]0 state stabilized by cyanide (2, scheme [1]) and carbene (3, scheme [2]) ligation and of the [Fe4S4]4+ state stabilized by bis(trimethylsilyl)amide (5, scheme [3]) ligation.
Scheme [2] was devised as an alternative method following the preparation of 10. Here an assembly system (box) containing two equiv of the N-heterocyclic carbene Pri2NHCMe2 affords all-ferrous 11. The same system but with one equiv of carbene leads to the octanuclear edge-bridged double cubane (EBDC) 12, which may be cleaved by two equiv of carbene to the single cubane (SC) 3.34 The oxidative instability of the fully reduced clusters is reflected by their low redox potentials: (10)4−/3− −1.42 V (acetonitrile), (11)0/1+ −1.30 V (THF). However, 11 is amenable to isolation and solution study. The all-ferrous state is substantiated by 57Fe isomer shifts at 4.2 or 77 K: 10, 0.65 mm/s; 11, 0.60 mm/s; Av Fe protein, 0.68 mm/s. These values are consistent with an empirical correlation of shifts with oxidation state for the tetrahedral FeS4 unit.11 Both 11 and the native protein manifest an S = 4 ground state, arising from a 3:1 iron site differentiation in C3v symmetry. The spins of the three majority sites (each S = 2) are aligned parallel to one another but antiparallel to the unique site, leading to the observed cluster spin. The electronic origin of this peculiar state has been analyzed theoretically.35 In contrast, the EBDC cluster 12 is composed of two 3:1 site-differentiated [Fe4S4]0 subclusters and has a diamagnetic ground state, a situation that arises from antiferromagnetic exchange coupling of the two identical subclusters each with S = 4.36 As will be seen, the EBDC geometry is frequently observed among higher nuclearity Fe-S clusters.
There is no clear evidence that the all-ferrous state of the iron protein plays a role in electron transfer to the MoFe protein of nitrogenase, which contains the catalytic site for the six-electron reduction of dinitrogen to ammonia. As is sometimes the case, a protein-bound metal site can be adjusted to an oxidation state that is not physiologically relevant. The principal message from this work (other than the synthesis itself) is that the core of 11 is an accurate structural and electronic analogue of the fully reduced native cluster. Further, the electronic properties, most notably the S = 4 ground state unique in biology, are intrinsic to the core itself and little influenced by protein environment.
3.1.2. Superoxidized (All-Ferric) State
The remaining oxidation level is all-ferric [Fe4S4]4+, which as yet has not been detected in proteins nor postulated to have a natural function. The only synthetic weak field example is [Fe4S4{N(SiMe3)2}4] (13), obtained in moderate yield in self-assembly systems by the reaction of [Fe{N(SiMe3)2}2] with one equiv of sulfur in toluene,37 or by the balanced reaction of [FeCl{N(SiMe3)2}2(THF)] with NaSH in THF.38 These procedures are incorporated in scheme [3]. Addition of Na2S to 13 leads to reduced clusters and the isolation of the three-membered redox series [Fe4S4{N(SiMe3)2}4]0,1−,2−. From a chemical perspective, isolation of the full suite of five [Fe4S4]n oxidation states, four of which have been stabilized by cysteinate ligation in proteins, is a matter of considerable satisfaction while simultaneously emphasizing the importance of terminal ligation on oxidation state stability (Figure 2). Cysteinate and its surrogate thiolate ligands (whose σ/π basicity and steric features are controllable by substitution) stabilize the three intermediate states (n = 1+, 2+, 3+). These ligands also bind to the n = 0 state as does the carbene, a potent neutral σ-donor that ameliorates oxidative instability by minimizing overall negative charge, resulting in a cluster of sufficient stability for isolation and study. As a strong σ/π donor anion, (Me3Si)2N− stabilizes FeIII and, unlike thiolate, is not readily oxidized. Additionally, the ligand provides steric shielding of the oxidized core against attack by solvent or other nucleophiles.
3.2. Stabilization in Partially Aqueous Media
The preparation, reactions, and physical properties of clusters, primarily as quaternary ammonium salts of the [Fe4S4(SR)4]2− type, have been extensively examined.in polar non-aqueous media such as acetonitrile, DMF, and Me2SO.11,28,39 Information in partially or wholly aqueous systems is meager by comparison. Aqueous solubility can be promoted by hydrophilic substituents such as R = -CH2CH2OH and -CH2CH2CO2−. However, clusters tend to solvolyze and deposit precipitates; excess ligand is required to maintain solution integrity. Under such conditions, a limited body of results on ligand exchange and redox potentials has been obtained.40–42 Recently, the potential use of iron-sulfur clusters in treating human diseases originating with defective cluster biogenesis and attendant mitochondrial malfunction has been raised.43 An unsubstantiated method of redress would be delivery of intact clusters to the organelle for reconstitution of scaffold proteins implicated in the biogenesis process. Meaningful testing of this idea would minimally require clusters that are sustainable in at least a partially aqueous medium, and resuscitates interest in water-stable [Fe4S4]2+ clusters.
A very recent approach to cluster stability in partially aqueous solvents employs α-cyclodextrin mono- and dithiolates44 and a β-cyclodextrin dithiolate45 as ligands. These are obtained by a multi-step synthesis; the latter is conveniently isolated as the diaryl disulfide 14. As shown in Figure 4, the disulfide is reduced by the aliphatic thiolate cluster 9 to yield the product formulated as [Fe4S4{β-CD-(1,3-NHCOC6H4S)2}2]2− (16a). This is a specific example of generalized redox reaction [4], a new cluster ligand exchange process demonstrated first with simple aryl disulfides.45 Cluster 16a has spectroscopic and redox properties consistent with those of [Fe4S4(SPh)4]2−, which is recovered in 98% in situ conversion by treatment of 16a with excess benzenethiol. In addition, this cluster shows a circular dichroism spectrum indicative of chiral ligation. When monitored by UV-visible spectra, 16a is adequately stable for up to 12 hours in 20–80% Me2SO/water solutions. Decomposition is indicated by progressive absorbance increases above ca. 500 nm. Systems based on α-cyclodextrinthiolates behave similarly but are somewhat less stable.
Figure 4.

Scheme showing the formation of β-cyclodextrin dithiolate clusters 16a and 16b by ligand substitution reactions with disulfide 14 and dithiol 15, respectively. The chelate-type binding mode 16 (adapted from ref. 46) represents a possible structure for both clusters but is unproven by X-ray methods.
| [4] |
The foregoing investigations were stimulated in part by the report in 1988 of an [Fe4S4]2+ cluster bound to two equivalents of the dianion of the β-CD dithiol 15.46 The cluster was described with the stoichiometry [Fe4S4{β-CD-(1,3-SC6H4S)2}2]2− (16b) and the chelate-type structure 16 proposed later for 16a. There is no crystallographic structure proof of either cluster. However, the isotropically shifted 1H NMR spectrum of 16a (600 MHz) indicates one type of phenylcarboxamido group, consistent with the proposed structure. Cluster 16b was reported to have a half-life of >120 hours in neutral aqueous phosphate buffer. This remarkable claim may be overestimated because spectra were not monitored beyond 500 nm where decomposition is more evident. However, it and subsequent observations on 16a and 16b and related clusters44 disclose the efficacy of cyclodextrin ligand platforms in solubilizing clusters in aqueous media under anaerobic conditions. The hydrophilicity of cyclodextrin thiolates and substantial steric screening of the cluster core from water by the voluminous ligand are favorable factors. Limited data suggest that clusters with amphiphilic dendrimer ligands may offer a similar possibility.47 Any ultimately successful cluster must permit substitution with protein ligands, which likely will place a limit on the size of the synthetic ligand used for solubility and stability.
3.3. Structural Features
Considered next are several new developments in the essential structural properties of [Fe4S4]n clusters, primarily with n = 2+. Metric data are collected elsewhere48 for synthetic Fe4Q4 (Q = S, Se, RN) and native Fe4S4 clusters and are not considered here.
3.3.1. Diastereomers and Enantiomers
Protein-bound clusters [Fe4S4(SCys)4] are rendered chiral by the binding of four L-cysteinate ligands as illustrated by 17 in Figure 5 and by their incorporation in folded protein structures. In the [Fe4S4]2+ state, these clusters manifest multi-featured circular dichroism spectra and diastereotopic splittings of cysteinate β-CH2 protons in 1H NMR spectra. In contrast, synthetic clusters with chiral ligands are nearly unknown, the few examples involving cysteinyl peptides11,13,49 and [Fe4S4(SCH2CH(OH)Me)4]2− synthesized from the racemic thiol.50 If ligands with chiral centers are employed in synthesis, diastereomeric or enantiomeric clusters will result depending upon whether the ligand is racemic or optically pure. A further issue is the detection of components of diastereomeric mixtures. These matters have been examined using substitution reactions [5] and [6] with three asymmetric ligands R*S− (18–20, Figure 5) monitored in acetonitrile or Me2SO by 1H NMR (600 MHz).51
Figure 5.

The [Fe4S4(SCys)4] cluster 17 in proteins showing diastereotopic methylene protons, synthetic clusters 18–20 with chiral substituents, and the diastereomeric pairs possible for a cluster prepared from a racemic thiol.
| [5] |
| [6] |
Intermediate clusters (n = 1–3) formed with racemic ligands were detected in mixtures with fully substituted clusters (n = 4) carrying substituents 19 and 20 owing to the sensitivity of isotropically shifted -SCH2- resonances upon cluster ligation. Diastereotopic splittings of 0.14 and 0.43 ppm were observed for fully substituted clusters containing 19 and 20, respectively. Spectra of reaction [6] with 1–4 equiv of racemic or enantiomeric 2,3-dihydroxypropane-1-thiolate are identical. Spectra of diastereomers were not identified under any conditions, although three diastereomeric pairs are possible for 18–20 (Figure 5) and a total of seven for the cluster set n = 2–4. Evidently, larger substituents with reduced conformational mobility and/or more than one chiral center leading to enhanced interligand interactions are required for NMR detection of diastereomers. Circular dichroism spectra of cluster 20 prepared from nearly optically pure ligands closely approach the mirror image relationship of enantiomers.
Ligand chirality provides several interesting consequences. (Et4N)2[18] crystallizes in a centrosymmetric cell containing an inversion-related pair of configurations [SS(RS)(RS)] and [RR(RS)(RS)], where (RS) and (SR) denote R/S disorder at two ligand sites; this crystal demonstrates binding of different chiralities to the same cluster. Further, (Et4N)2[19] crystallizes in part in chiral space group C2 in the [SSSS] configuration, an uncommon case of spontaneous resolution by crystallization. Cluster 20 is the only synthetic or native Fe4S4 cluster ever obtained as separate enantiomers. Lastly, while individual Fe4S4 cores are achiral, the cyclic combination of four such cores by edge-bridging interactions in [Fe16S16(PR3)8] (35, Figure 9) has D4 symmetry52,53 and thus is chiral. This cluster type has not been separated into optical isomers.
Figure 9.

Schematic depictions of known structures formed by the bridging of [Fe4S4] units by sulfide (32) and thiolate (33) and by intercore Fe-(μ4-S) bonds involving two (34) and four (35) cubane units.
3.3.2. 3:1 Site-Differentiated Clusters
A small fraction of native Fe4S4 clusters, often with a catalytic or electron transfer function, occur in the 3:1 site-differentiated general form [Fe4S4(SCys)3L] in which L is an exogenous or endogenous ligand. A leading example is aconitase, which binds OH−/H2O and substrate (citrate), intermediates, and product (isocitrate) in its catalytic cycle.54,55 Other cases include the cluster [Fe4S4(SCys)3(NHis)], one of three Fe-S clusters comprising the electron transfer pathway in [NiFe]-hydrogenase,17,56 and the site-differentiated cluster that provides the bidentate (N,O) binding site for S-adenosyl-L-methionine in biotin synthase.57,58 Additionally, certain constructs M-SCys-Fe4S4(SCys)3 differentiate one cluster cysteinate from the other three by bridge formation to another metal site M. Examples with M = Fe include the H-cluster of [FeFe]-hydrogenase,59 sulfite reductase,60 and, with M = Ni, the A-cluster in acetyl coenzyme A synthase.61,62 Because of these and related results, some of which preceded those cited, the synthesis of 3:1 site-differentiated clusters became an imperative in the analogue chemistry of protein-bound sites. This goal was first accomplished using the deprotonated tridentate thiol 2111 in Figure 6.
Figure 6.

Trithiols (21, 23) whose deprotonated forms stabilize 3:1 site-differentiated clusters and the structure of [Fe4S4(LS3)Cl]2− (22) as the Ph4P+ salt (adapted from ref. 63). In this structure only one p-tolyl substituent is below the central benzene ring opposite to the cluster. In other structures, two or three of these non-coordinating substituents occur on the side of the ring opposite to the three coordinating arms.
Ligand 21 is designed such that the three coordinating side chains of the central benzene platform can be displaced upward, a disposition often sterically buttressed by the p-tolyl groups in alternating positions around the ring. Ligand dimensions allow capture of an [Fe4S4]2+ core at three sites, leaving a fourth manipulable by substitution reactions. Cluster 22 illustrates the ligand stereochemistry found in (Ph4P)2[Fe4S4(LS3)Cl]63 (Figure 6). The clusters [Fe4S4(LS3)(SR)]3− are accessible by chemical reduction.64 Numerous other [Fe4S4]2+ structures with the LS3 ligand are available, as are other tridentate trithiolates whose cluster binding is based on a similar principle.11 Recently, the repertoire of tridentate thiolates stabilizing site-differentiated clusters has been augmented with the synthesis of the trithiols 23. These have been shown to form [Fe4S4]2+ clusters with monothiolates at the unique site with the methyl groups of the ethyl substituents and the coordinating arms on opposite sides of the central ring.65
Clusters of the 3:1 type are recently accessible without the necessity of trifunctional ligand synthesis. Scheme [7] of Figure 7 demonstrates a particularly interesting approach based on bis(trimethylsilyl)amido clusters 13 and 26.66 Treatment of 13 with highly hindered 2,6-bis(mesityl)benzene-1-thiol results in elimination of (Me3Si)2NH, core reduction, and thiolate binding. Addition of THF affords the tris(THF) cluster 24 which undergoes substitution at the unique site with tetramethylimidazole to form 25. Reaction of 26 with the hindered thiol leads to 27 without change in core oxidation state. However, displacement of thiolate with the imidazole forms 28 with concomitant reduction to the [Fe4S4]2+ state. The two reductions in Scheme [7] are presumably effected by thiolate which is oxidized to disulfide. It should also be recognized that 24–27 are [Fe4S4]3+ clusters with physiologically relevant thiolate ligation. As such they are analogues of the HPox site, for which there has been only one previously isolated analogue, [Fe4S4(Stip)4]1−, also stabilized by sterically protective ligands.67
Figure 7.

Reaction scheme for the formation of 3:1 site-differentiated clusters (24, 25, 28) with unidentate terminal ligands derived from bis(trimethylsilyl)amide clusters 13 and 26 (adapted from ref. 66). Three [Fe4S4]n oxidation states are represented: n = 4+ (13), 3+ (24–27), 2+ (28).
Site-differentiated clusters can also be formed by substitution reaction [8] and redox reaction [9] (X-X = RSSR, RSeSeR, I2) starting from an all-ferrous tetracubane.53,68 Other than [Fe4S4(LS3)(SR)]3−, these are the only site-differentiated clusters isolated in the [Fe4S4]1+ state, stabilized in these cases by phosphine ligation.
| [8] |
| [9] |
Analogue 3:1 clusters have been employed in a number of biologically relevant investigations, including ligand binding affinities at the unique site,11 formation of sulfide-bridged double cubanes,66,69, binding of cluster and iron porphyrins through an Fe-S-Fe bridge,69,70 construction of the H-cluster framework of [FeFe]-hydrogenases,71 and reaction of thiolate at the unique site with sulfonium cations in relation to the reductive cleavage of 71S-adenosyl-L-methionine by biotin synthase.64 Other applications dependent upon regioselective reactivity will doubtless follow, particularly because of the availability of a succeeding generation of trithiols (23) more convenient synthetically than the first (21) and the emergence of clusters (24–28) not requiring multi-step ligand synthesis. When employing site-differentiated clusters with non-zero net charge in polar media, the possibility of ligand exchange leading to mixed-ligand species must be examined if a reaction system is to be well-defined. Neutral clusters such as 24 and 25 in weakly polar or nonpolar solvents are unlikely to undergo exchange which generates charged species in low dielectric media. Cluster 24 in particular is a potential entrant to a wide range of 3:1 clusters by displacement of its THF ligands.
3.3.3. Dioxygen-Protective Fe4S3 Clusters
The cubane-type [Fe4S4]n clusters are no longer the only tetranuclear iron-sulfur clusters recognized in proteins. “Standard” [NiFe]-hydrogenases catalyze the reaction H2 ⇆ 2H+ + 2e− and are rapidly deactivated by dioxygen. These enzymes transfer electrons from dihydrogen oxidation via the chain [Fe4S4] (proximal) ↔ [Fe3S4] (medial) ↔ [Fe4S4] (distal) in which the locations of the clusters relative to the active site (5, Figure 1) are indicated. Certain membrane-bound enzymes, however, remain functional under aerobic conditions. Recent investigations have located the main source of difference in behavior. Dioxygen-tolerant [NiFe]-hydrogenases utilize a second tetranuclear structure type as the proximal electron transfer site. Structures of the proximal clusters in the dihydrogen-reduced state (29) and in the ferricyanide-oxidized state (30) of the H. marinus enzyme and the reduced cluster of the R. eutropha H16 enzyme are depicted in Figure 8.72,73 These clusters are built of an Fe4S3 core to which are bonded six cysteinate residues, one of which is doubly bridging. The iron sites assume distorted tetrahedral coordination similar to that in all other Fe-S electron transfer clusters; however, the bis(cysteinate) coordination of Fe(4) is unique to these clusters. Three oxidation states have been identified, [Fe4S3]3+,4+,5+, ranging from [Fe2+3Fe3+] to [Fe2+Fe3+3].74 The principal structural difference among them is the long Fe(2)⋯S(3) separation of 4.01 Å in oxidized 30 which results from the coordination of the deprotonated amide nitrogen of the same bound cysteinate. The Fe(2)-S(3) distance in the reduced form (2.40 Å) is the outer limit of bonded Fe-S interactions in these clusters (2.2–2.4 Å).
Figure 8.

Structures of reduced (29) and oxidized (30) proximal Fe4S3 clusters in the electron transfer chain of H. marinus and R. eutropha H16 hydrogenases; idealized representation of 29 as the hypothetical cluster [Fe4S3(SR)6]3−,2− (31).
Proximal cluster structures sustain 3+/4+ and 4+/5+ redox steps separated by about 200 mV, a property thought to arise from localized electronic structures to allow pairs of electrons to be transferred efficiently in the physiological range (ca. 50–250 mV) to and from the catalytic site. Protection from dioxygen results from its ultimate reduction to water by electrons from the proximal cluster and active site. Such would not be the case in a standard hydrogenase whose conventional electronically delocalized proximal cluster operates with a potential difference of ≳800 mV. Detailed considerations of the mechanism of protection of dioxygen-tolerant enzymes are available.72–75
Lastly, generalized depiction of the reduced clusters as 31 allows recognition of idealized mirror symmetry, replacement of sulfide in an Fe4S4 cubane with thiolate, and the inaptness of a cubane description owing to the absence of an Fe-S(R) bond in the symmetry plane (containing Fe(3,4)S(4) in 29). Further, 31 emphasizes the inverse atom population with respect to the ubiquitous Fe3S4 core found in the medial cluster of the [NiFe]-hydrogenase electron transfer chain and elsewhere. Reaction [9] is currently the only desulfurization reaction leading to isolable Fe4S3 clusters.76,77 Owing primarily to nitrosyl coordination, these are strong-field clusters whose cores of C3v symmetry are not directly comparable with the weak-field cores of 29. Structures closely resemble that of the classic Roussin’s black anion, [Fe4S3(NO)7]1−, which is prepared by self-assembly.78 The weak-field Fe4S3 core is proposed as an intermediate in the formation of an Fe8S7 cluster from two [Fe4S4{N(SiMe3)2}4] clusters and PR3.79 As yet, no synthetic weak field Fe4S3 cluster has been isolated, although it seems highly probable that establishment of the proximal cluster will encourage research toward that end.
| [9] |
3.3.4. Di- and Tetracubane Clusters
Individual cubane units can be covalently coupled to each other to form higher nuclearity cores. The resultant structures, depicted in Figure 9, include [Fe8S8(μ2-S)]2+ (32),11,28,65 and [Fe8S8(μ2-SR)]3+ (33),80 whose subclusters are connected by an unsupported sulfido or thiolato bridge, and [Fe8(μ3-S)6(μ4-S)2]0,2+ (34) and [Fe16(μ3-S)8(μ4-S)8] (35)11,28,53 whose components are bonded through uncommon Fe-(μ4-S)-Fe interactions. Clusters derived from 32 exhibit Fe-S-Fe bridge angles over a wide range (110–162°) and coupled one-electron reductions separated by ca. 200 mV. In the single example of 33, the bridge angles are 120° and 123° in independent clusters. Clusters containing the cores 34 and 35 are prepared as phosphine clusters, although the former has also been isolated in carbene-ligated form as 12 (Figure 3). Core 34 is representative of the EBDC stereochemistry as found in [Fe8S8(PPri3)6] and similar phosphine-ligated clusters.53 These clusters are prepared by one-electron reduction of [Fe4S4(PR3)4]1+ to form the putative all-ferrous clusters [Fe4S4(PR3)4]. These are susceptible to phosphine dissociation and upon standing in solution deposit crystalline [Fe8S8(PPri3)6] and/or [Fe16S16(PPri3)8] by fragment condensation. In solution, these clusters can be equilibrated with each other in the presence of added phosphine. While other high-nuclearity clusters are known,28 only those with cores 32-35 are constructed of discrete cubane units, raising the possibility that they may also be found in proteins. Several other clusters built from cubane or cuboidal units can be recognized81 but are hypothetical in the iron-sulfur context. Lastly, the EBDC core is emphasized here because of the role of this structure type in the synthesis of higher nuclearity heterometal clusters (cf. Section 5.3).
4. Heterometallic Clusters
4.1. Introduction
The vertices of cubane-type clusters can be differentiated in ways other than by alteration of terminal ligands. The most prominent, longstanding case continues the 3:1 site theme and is exemplified in the heterometallic cores 37-39 in Figure 10. These can be considered substituted versions of all-iron 36. Although MFe3S4 cores depart from the familiar and efficient idealized symmetry of the Fe4S4 cubane structure, the heterometallic cores naturally conform to the homometallic parent structures via the common Fe3S4 fragment. The attributes of these heterometal clusters parallel those of Fe4S4 clusters and include multiple oxidation states, similar overall cubane geometries with metric differences confined mainly to the heterometal environment, and substitutional lability at the tetrahedral iron sites involving thiolate and (pseudo)halide binding in particular. A significant difference is the stereochemistry at the M sites, particularly for M = MoIII,IV, WIII,IV, and VIII, which with very few exceptions follow the normal six-coordinate preference.
Figure 10.
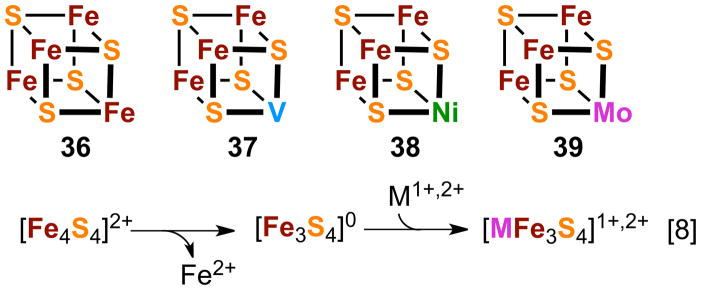
Schematic depiction of homometal and heterometal cluster cores (36-39) and the synthesis of heterometal cores by fragment condensation (reaction [8]).
The history of biological heterometallic M-Fe-S clusters extends back to several key events. These include the partial structure determination of the Mo-Fe-S cluster of the nitrogenase cofactor,82 recognition of the voided Fe3S4 motif in proteins,83,84 which can incorporate heterometals to form MFe3S4 units of probable cubane structure,85 and establishment of a NiFe3S4,5 cluster in nickel-containing carbon monoxide dehydrogenase.86–88 These examples spurred the synthesis of heterometallic clusters with the biological metals M = V, Mo, W, Co, Ni, and Cu as well as with the non-biological metals M = Ag and Tl.23,89–94 These clusters are not formed by direct substitution of a core metal atom of 36 but by other means involving self-assembly and/or fragment condensation. The latter is exemplified by the core reaction sequence [8] (Figure 10). Iron(II) is removed from the site-differentiated [Fe4S4]2+ cluster [Fe4S4(LS3)(OTf)]2− by a process of fragmentation by chelation to yield the cuboidal cluster [Fe3S4(LS3)]3− with the core structure of 5.95 This species serves as the fragment to which metal ions are bound to form the heterometallic product [(L′1-3)MFe3S4(LS3)]z whose charge depends on atom M and its terminal ligand(s) L′. Because summary accounts of synthetic23,89,96–99 and protein-bound MFe3S4 clusters (likely formed by fragment condensation)85,99 and a theoretical examination of electronic structure100 are available, only certain of these clusters useful in the synthesis of higher nuclearity species are further considered.
4.2. Heterometal Single Cubanes
Isoelectronic vanadium and molybdenum chloride clusters 39a and 39b, respectively, are accessible by the substitution reaction and assembly reactions [9] in Figure 11. Displacement of chloride with PEt3 in reaction [10] yields 40a and 40b. In the reaction of 40b (but not 40a), the phosphine serves as a one-electron reductant with the result that the product [MFe3S4]2+ cores differ by one electron. Clusters 40a and 40b are precursors to a variety of single cubanes by ligand substitution reactions [11] with L = halide, azide, cyanide, and thiolate.101–108 Product clusters are isostructural and nearly isometric. Each is oxidized and reduced by one electron in the couples 2−/1− and 3−/2−, leading to the three-membered electron transfer series [(Tp)MFe3S4L3]3−/2−/1−. 57Fe isomer shifts are consistent with the oxidation levels [MFe3S4]1+,2+,3+ in which each contains M3+ and the iron oxidation states Fe2+, Fe2.33+, or Fe2.67+ as the core is oxidized. Here and elsewhere, mixed oxidation states imply delocalized structures. The cluster set [(Tp)MFe3S4L3]z (M = V, Mo) allows for comparison of redox potentials and core electron distribution under fixed conditions. For example, at constant L and z = 2−, the cores are described as M3+Fe2+2Fe3+. For all 3−/2− and 2−/1− couples, the potential differences EV - EMo are large and positive (ca. 0.20 to 0.50 V in acetonitrile108), revealing the interesting result that molybdenum single cubanes are more easily oxidized than vanadium clusters at parity of ligand and charge.
Figure 11.

Synthesis of edge-bridged double cubanes (41, 42) and PN-type clusters (43) using self-assembly ([9], Mo), ligand substitution ([10], [11], [13]), fragment condensation [12], and core rearrangement ([14], [15]) reactions. Numbers in parentheses are values of z; L− = halide, N3−, CN−, RS−.
| [11] |
4.3. Heterometal Cubanes as Building Blocks of Higher Nuclearity Structures
4.3.1. Edge-Bridged Double Cubanes
Edge-bridged double cubanes are the next structure type of increased nuclearity built only from single cubanes. Their real or idealized centrosymmetric M2Fe6(μ3-S)6(μ4-S)2 cores with heteroatoms necessarily in transoid positions are structurally analogous to the Fe8S8 core 34. Clusters of the general type [(L−1-3)2M2Fe6S8L4}z have been prepared with M = V, Mo, and W (ligands bound to M precede it in formulas). Terminal ligands L′ complete six-coordination at M and when tridentate (such as But3tach and tris(pyrazoly)hydroborate) they function as protecting groups for the M site, directing ligand substitution to the iron positions. The first EBDC reported was [(Cl4cat)2(Et3P)2Mo2Fe6S8(PEt3)4] in 1995,109 prepared from the SC [(Cl4cat)(MeCN)MoFe3S4Cl3]2− that itself requires a multiple-step synthesis. Thereafter, efficient ligand substitution and self-assembly reactions leading to the isoelectronic chloride single cubanes 39a110 and 39b,102 respectively, have been developed.
Figure 11 depicts the reactions applicable to both the M = V and Mo systems that lead to significant higher-nuclearity clusters 41-43. We recapitulate this scheme, which has been described in detail previously.23,103,104 Chloride clusters 39 undergo ligand substitution to afford the [MFe3S4]2+ phosphine clusters 40. Thereafter, reduction of the phosphine clusters leads to the neutral isostructural EBDC clusters 41a and 41b by the reactions [12]. Two [MFe3S4]1+ clusters react with loss of one phosphine each and combine by fragment condensation to afford octanuclear products. Lability of phosphine in highly reduced clusters is anticipated by the formation of [Fe4S4(PEt3)3X] from [Fe4S4(PEt3)4]1+ (Fe2.25+) and halide, and of 34 and 35 (Figure 9) from the latter cluster and a strong reductant.53
EBDC clusters present certain characteristic properties.103–108,111 Subclusters are bridged by a planar Fe2(μ4-S)2 rhomb in which the intercubane bridge bond is normally 0.08–0.19 Å shorter than the bond within the cubane. This feature contributes to the stability of the double cubane arrangement. Phosphine ligands are displaceable in reactions [13], analogous to [11], to form [(Tp)2M2Fe6S8L4]4− with retention of the double cubane core. Molybdenum clusters support three-membered electron transfer series [(Tp)2Mo2Fe6S8L4]4−/3−/2− with the core compositions 2[MoFe3S4]1+, [MoFe3S4]1+[MoFe3S4]2+, and 2[MoFe3S4]2+ as oxidation proceeds. A similar series is likely but has not been established for vanadium clusters.
| [13] |
The clusters [(Tp)2V2Fe6S8L4]4− are readily isolated under anaerobic conditions. Although certain analogous molybdenum clusters have also been isolated in the 4− state, their E4−/3− potentials are quite low (usually ≲ −1.4 V).106,111 Oxidation of the 4− state by trace oxidants is often observed, leading to isolation of 3− clusters. This matter notwithstanding, the procedure in Figure 11 allows straightforward isolation of EBDCs 41 and 42.
4.3.2. PN-Type Clusters
Clusters 43a and 43b are the second high-nuclearity cluster type accessible by the scheme in Figure 11. Treatment of phosphine- or chloride-substituted EBDCs 41 and 42 with hydrosulfide generates the new clusters [(Tp)2M2Fe6S9(SH)2]3−,4−.103–105,107 Because nuclearity is unchanged, reactions [14] and [15] are core conversions in which an external nucleophile has been incorporated. The new core reveals the connectivity [M2Fe6(μ2-S)2(μ3-S)6(μ6-S)]0,1+ in C2v symmetry and is unusual for having three types of bridging sulfide. The core may be conceptualized as two distorted MFe3S4 cubanes with a common vertex and further supported by two sulfide bridges. The vanadium cluster is isolated as the 4− anion. Mössbauer data indicate the all-ferrous description [V3+2Fe2+6S9]0, revealing no change in oxidation state in reaction [14] or [15]. The molybdenum cluster is presumably also generated in the 4− state but is isolated in the 3− state formulated as [Mo3−2Fe2+5Fe3+S9]1+, again a consequence of a very low redox potential (E4−/3− = −1.80 V). The core supports terminal ligand replacement with ligands that maintain low cluster redox potentials111 such as RS−, CN−, and OH−.112 The signal structural feature of these clusters is a μ6-S atom, highly unusual but not unprecedented in molecular compounds, acting as a vertex in a structural element with a large external Fe-S-Fe angle of ca. 150°.
Comparison of 43a and 43b with the structurally characterized clusters 44-46 of nitrogenase in Figure 12,113–115 immediately reveals a structural relationship between the synthetic clusters and the PN cluster. The synthetic and native clusters are both composed of two distorted cubanes sharing a common vertex with a large Fe-S-Fe external angle. In the native cluster, the two cubanes are further connected by two Fe-(μ2-SCys)-Fe bridges while in the synthetic systems the corresponding feature is two sulfide bridges. Further comparison of the synthetic and native clusters by superposition of crystallographic coordinates reveals a convincingly close topological relationship between the two.23 Thus, 43a and 43b, initially reported in 2002–03, are the first topological analogues of the PN cluster 46. They are not, however, chemical analogues because of the presence of heterometals whereas the PN cluster is all-iron in content. As will be seen (Section 6.2), topologically similar all iron-analogues of 46 are accessible. So far there are no synthetic representations of the POX cluster 45, which contains a cubane-type Fe4S4 subcluster and an Fe4(μ3-S)3 unit that resembles in part the Fe4(μ3-S)4(μ4-S) fragment of the “basket” cluster [Fe6S6(PEt3)4Cl2].116 The same is true of FeMo-co (44) which contains homometallic and heterometallic cubane-type clusters with a common “vertex” atom X = C and three Fe-(μ2-S)-Fe bridges.
Figure 12.

Schematic structures of the clusters of nitrogenase: FeMo-cofactor (44), the two-electron oxidized P-cluster (POX, 45), and the P-cluster (PN, 46). During catalysis, the all-ferrous PN cluster is believed to transfer electrons to the catalytic site (FeMo-co) from the Fe4S4 cluster of the iron protein of the enzyme complex.
5. Heteroligated Cluster Cores
Beyond terminal heteroligation and heterometallic core composition, cluster vertices can also be differentiated through core heteroligation. Weak-field core heteroligated Fe-Q-S clusters were first demonstrated in reaction system [16] and the corresponding system of 2− clusters in acetonitrile.117 Five species with core compositions [Fe4S4-nSen]2+/1+ (n = 0–4) were identified from the concentration and time dependencies of highly sensitive m-H and p-Me NMR contact shifts as equilibria were approached. Clusters with n = 1 (Se) and n = 3 (S) are the initially formed heteroligated species in the two reaction systems. Isolation of mixed-ligand clusters was not attempted. This work was part of any early exploration of cluster reactivity; the mechanism of core atom exchange remains unestablished.
| [16] |
Focused interest in core heteroligated clusters is a more recent development motivated in part by the discovery of an interstitial atom X in the[MoFe7S9X] core of the FeMo-cofactor (44) of nitrogenase. X-ray diffraction data were originally consistent with X = C, N, or O,115 thus presenting the general problem of Mo-Fe-S-X cluster synthesis. Recent X-ray diffraction, spectroscopic, and radiolabeling results point to X = C,118–120 the most surprising of the three original possibilities, as the interstitial component.
Unlike heterometallic M-Fe-S cluster synthesis, for which extensive study has yielded several independent and rational preparative strategies, the inverse goal of heteroligated core construction has received only limited systematic attention to date and remains very much a standing challenge. A number of tactical issues are immediately evident. (1) Weak-field, four-coordinate iron environments are intrinsically labile, and the preservation of specific mixed ligand environments is problematic, especially under mechanistically ill-defined cluster assembly conditions. (2) The simple, polyanionic nature of the target core ligands (e.g., sulfide, oxide, nitride, carbide, or substituted variants thereof) largely precludes the use of chelate ligand designs to control core heteroligand compositions. (3) The different physical and chemical properties of the two separate ligand components raise problems of compatible reactivity and stability; these differences are particularly significant for sulfide versus light 2p anions, and they grow with increasing anion basicity, i.e., O < N < C. At this juncture, the deliberate installation of any heteroatom, whether as a discrete monoatomic ligand or as a substituted derivative, constitutes a reasonable and essential first step in synthetic discovery.
5.1. Imide-Sulfide Clusters
Cubane clusters with mixed tert-butylimide-sulfide core ligation can be obtained according to the syntheses in Figure 13.121,122 The product clusters 47-49 span all possible heteroligated cubane cores of general formulation [Fe4(NBut)n S4-nCl4]z (n = 1–3), while the n = 0, 4 endpoints are the established homoleptic all-sulfide and all-imide species. The syntheses of the Fe-NR-S clusters are selective, with each specific composition accessible by a separate and distinct reaction route, using reaction sequences that rely on previously developed iron-N-anion cluster chemistry to furnish the imide ligands in the final products.23,123
Figure 13.

Synthesis of imide-sulfide cluster series [Fe4(NBut)nS4-nCl4]z (n = 1–3, 47-49) with use of precursor species 50-53 obtained from the indicated reactions.
The mixed core species share basic physical properties and trends with the previously characterized homoleptic cores.48,122 Metal sites are well-described as independent, weakly-coupled high-spin tetrahedral centers. The cluster structures combine the specific geometric characteristics associated with imide and sulfide core ligation; e.g., Fe-Q distances are shorter, Q-Fe-Q angles are sharper, and Fe-Q-Fe angles are wider for Q = N vs Q = S.48 Multiple redox states are accessible, with oxidative redox stabilization due to strongly basic nitrogen anion donors reflected in progressive linear decreases in redox potentials (−435 mV for z = 1−/2−, −385 mV for z = 2−/3−) for each replacement of sulfide by imide in the cubane core. Under ambient conditions, individual clusters of a given mixed core composition do not disproportionate to other core heteroligand compositions, and mixtures of clusters of different core compositions do not give rise to new core compositions.
Within the heteroleptic series, monoimide trisulfide cluster 47 is particularly notable in possessing a core Fe4NS3 framework that superposes closely onto the Fe4S3X portion of FeMo-co (Figure 14).121,122a Initial reactivity studies summarized in Figure 14 reveal both similarities and differences between this heteroligated core and the well-known homoleptic all-sulfide species.122b The terminal chloride ligands in both clusters are readily replaced by thiolates, lending enhanced redox stability to the cores and allowing electrochemical observation of reversible oxidative (z = 1−/2−) and reductive (z = 2−/3−) processes, albeit with more reducing potentials (ca. −300 mV shifts) for the imide-containing core. Thiolate-ligated oxidized (z = 1−) clusters can be isolated in both instances. In contrast, equivalent reduced (z = 3−) species, while well-known in Fe4S4 chemistry, have proven elusive to chemical isolation for the Fe4(NBut)S3 core. Attempts at chemical reduction of [Fe4(NBut)S3(SPh)4]2− lead to the formation of free thiolate and a complex paramagnetic NMR signal set; oxidation of this material, however, leads to recovery of the starting z = 2− cluster, suggesting that the Fe4(NBut)S3 core remains intact following the initial reduction. Using an alternative strategy adapted from recent Fe4S4 chemistry (Section 3.1.1), the reduced [Fe4(NBut)S3]1+ core can nevertheless be stabilized and isolated via terminal cyanide ligation. The bridging core imide is reactive and can be selectively replaced by less basic arylimides via transamination with arylamine to form 54. This behavior follows known reactivity in certain iron-imide clusters,123,124 but equivalent chemistry has not been demonstrated in iron-sulfur systems.
Figure 14.

Top: Superposition of the [Fe4NS3] core of the mono-core-heteroligated cluster [Fe4(NBut)S3Cl4]2− (47; solid blue, with three terminal chloride ligands also shown) and the corresponding [Fe4S3X] subunit of FeMo-co (dashed red). Bottom: Reactions of 47 illustrating core retention under terminal ligand substitution, oxidation and reduction, and replacement of the core imide by transamination with an arylamine to afford 54.
The mixed Fe-NR-S cubane cores are achieved via terminal reactions that range from the treatment of a symmetric dinuclear diimide complex 50 with an exogenous sulfide source (and implicit reductant) to more complicated transformations involving the combination of different polynuclear components (51-53, [FenSnCl4]2− (n = 2,4; Figure 13). All routes can be classified descriptively as fragment condensations according to the introduced reactants, and hypothetical balanced equations can be formulated based on experimental stoichiometries. Aspects of the precursor structures can be discerned in the final products, and the reactions themselves are moderately to highly selective for their respective core heteroligand compositions. These observations suggest that the empirical descriptions of fragment condensation in these systems might also apply to underlying mechanisms. To explore this possibility, selenide analogues of 53 and [Fe4S4Cl4]2− were prepared and deployed in an attempt to track the fate of the chalcogenide according to reactions [17]–[19].122 The results of these labelling experiments were mechanistically uninterpretable: congeners spanning all possible sulfide-selenide compositions were produced in reactions [17] and [18] whereas an all-selenide product dominated in reaction [19]. The cluster assembly pathways for these systems remain unknown at present.
| [17] |
| [18] |
| [19] |
5.2. Oxide-Sulfide Cubanes
The first oxide-sulfide cubane core was obtained in the form of the heterometallic cluster [(Cl4cat)2Mo2Fe2OS3(PEt3)3Cl]1+ and preceded shortly the discovery of the interstitial X ligand of FeMo-co. As shown in Figure 15, cluster 55 was synthesized by a fragment condensation reaction involving a heterodinuclear precursor and a mononuclear iron reactant, with the oxide core ligand apparently derived from a terminal Mo=O moiety in the dinuclear species.125
Figure 15.

Preparation of the oxide core cluster 55 and intermediates 56 and 57 leading to the oxide cluster 58 ([Fe8(μ3-S)6(μ4-O)(μ2-OCPh3)(μ2-SDmp)2(SDmp)2]) and the interstitial sulfide cluster 59 ([Fe8(μ3-S)6(μ6-S)(μ2-OCPh3)(μ2-SDmp)2(SDmp)2]).
More recently, the oxide-containing octanuclear cluster 58 has been synthesized by treatment of a diferrous thiolate alkoxide precursor 57 (obtained from the curious mononuclear complex 56) with a mixture of water and elemental sulfur126 (Figure 15). A second, structurally-related octanuclear product 59 is also formed. The two clusters were isolated as a mixture by HPLC, with crystallization yielding oxide-containing 58 with variable levels of 59 as a co-crystallized contaminant. Cluster 58 displays a disordered, asymmetric structure from which Fe4OS3 cubane and Fe4S3 cuboidal subunits can be identified; the oxide cubane vertex further coordinates to the Fe4S3 subunit to form a μ4 trigonal pyramidal bridge geometry. The iron enviroments are either explicitly tetrahedral (five sites) or implicitly tetrahedral (two sites) and trigonal bipyramidal (one site) if more distant iron-arene interactions are included in the metal coordination spheres. Cluster 59 presents a known topology marked by an interstitial μ6-S atom within a distorted Fe6 prism that, together with three additional bridging ligands, joins two Fe4S3 subclusters. The topology resembles the FeMo-co connectivity and is discussed with other examples in Section 6.3. The similarities between the two reaction products suggest that both 58 and 59 arise from the same or closely related cluster assembly pathways, with hydrolysis from introduced water leading to the oxide-containing core.
5.3. Selenide-Sulfide Cores: Selenide as a Surrogate for Sulfide
5.3.1. Core Selenide Incorporation
Selenide is an effective structural and electronic analogue of sulfide in iron-sulfur chemistry, and recent progress has demonstrated its selective incorporation into M-Fe-S clusters. One procedure utilizes the trisulfido complexes 60 in the self-assembly redox system [20] in Figure 16.127 The But3tach ligand supports the trigonal pyramidal MVIS3 group (M = Mo, W) in the formation of clusters 61 whose core oxidation state is dependent on M and the mol ratio of reactants. In this way, clusters with heteroligated [MFe3S3Q]2+,3+ cores can be prepared. The apparent stoichiometry leading to a 3+ cluster in system [20] (Q = S, Se) is indicated; that for a 2+ cluster is readily formulated. 57Fe isomer shifts support the designations M3+Fe2+2Fe3+ and M3+Fe2+Fe3+2 for the 2+ and 3+ cores, respectively. As is common with MFe3S4 clusters prepared by self-assembly, both Fe2+ and an additional reactant, usually thiolate, function as reductants leading to mixed-valence products.
Figure 16.
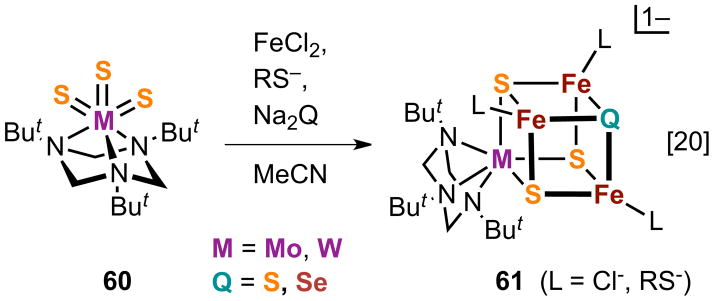
Formation of the cluster 61 with the heterocore [MFe3S3Q]2+ (M = Mo, W; Q = S, Se) from the MVIS3 complex 60 in assembly reaction [20].
| [20] |
In comparing the various Fe4S3Q and MFe3S3Q cores encountered thus far, perhaps the most impressive aspect is the range of Fe-Q distances permitted by the cubane-type geometry, albeit distorted by Q atom radius differences. In 47, 58, and 61, for example, mean Fe-Q bond lengths (Å) are approximately 1.95 (Q = N), 1.91-2-19 (Q = O, four-coordinate), 2.30 (Q = S), and 2.40 (Q = Se).122,126,127 Extensive metric data have been compiled for Fe4S4-n(NR)n clusters.122
5.3.2. Selenide as a Reaction Marker
Core conversion reactions [14] and [15] (Figure 11) can be minimally described by reaction [21] in which hydrosulfide is an external nucleophile. Because multiple core bonds are made and broken, the reaction pathway is not evident. However, an issue necessary to any ultimate mechanism that can be addressed is the locus of the attacking nucleophile in the product cluster. The approach, set out in Figure 17 (Q = S, Se), is one of chemical surrogacy. It utilizes the pronounced chemical similarity between sulfur and selenium in the same oxidation state and their ready distinction by crystallography.111 When the reaction is conducted with three equiv of hydroselenide, selenium is incorporated in the core of product 62. The mass spectrum of [(Tp)2Mo2Fe6S8Se(SEt)2]3− (derived from 62) discloses one selenide per cluster that, from X-ray structure analyis, is disordered over the two doubly bridging positions. No selenium was detected at the μ3- or μ6-positions, i.e., selenium incorporation is site-specific. It was further shown that selenium incorporation does not occur by Se/S exchange of all-sulfide 41b and hydroselenide. Consequently, we conclude that “the probable structural fate of the attacking sulfide nucleophile is as a μ2-S bridging atom in the PN-type topology.”111
Figure 17.

Core rearrangement of EBDC 41b to the PN-type cluster 62 by reaction with the external nucleophiles HQ− (Q = S, Se). The product cluster contains one μ2-Q bridge atom.
| [21] |
The structural fate of sulfide or selenide, both as an external and internal (bridging) nucleophile, has been further examined in the formation of four cluster types in Figure 18.128 For clarity, terminal ligands are omitted but are specified in the figure legend. The reactions are based on [(Tp*)WVIS3]1− 129 (63) as the initial precursor. This species presents three terminal sulfide nucleophiles as a template for self-assembly, here leading to single cubanes and hexanuclear cluster cores W2Fe6S9 of a new structural type. The left-hand column demonstrates analogous reactions of molybdenum and tungsten clusters leading to the PN-types 64a and 64b. Reduction of monoselenide cluster 65, formed by a reaction similar to [20] (Figure 16), affords the diselenide EBDC 66 with both selenium atoms in μ4-positions. Reaction with hydrosulfide induces core conversion to PN-type cluster 67 with a μ2-S bridge from the external nucleophile, and μ2-Se and μ6-Se bridges from two μ4-Se atoms that function as internal nucleophiles. With 66 and hydroselenide, the PN-type core 68 with three selenide bridges between the cubanes is produced.
Figure 18.

Simplified scheme showing the positions of incorporation of bridging selenide atoms in four different types of cluster cores as determined by X-ray structure analysis (adapted from ref. 128). Terminal ligands omitted for clarity are the following: W-Tp*, all clusters; Fe-Cl, single cubanes; Fe-PEt3, EBDCs; Fe-QH, PN-type. See ref. 128 for reaction stoichiometries and a detailed discussion of the scheme.
Under a different stoichiometry, the cluster [(Tp*)2W2Fe4S9]1− with the core [W2Fe4(μ2-S)6(μ3-S)2(μ4-S)]1+ (69) is assembled and is chemically reducible to the neutral cluster.128 These species are described as double-cuboidal clusters because of two WFe2S4 units (similar to 5, Figure 1) joined by a common μ4-S vertex and supported by two μ2-S bridges. The core connectivity, while not unprecedented, has been previously observed only in [(edt)2Mo2Fe4S9]3−/4− 130 and [Fe6S9(SR)2]4−.131,132 With selenide as the external nucleophile, cores 70a and 70b are formed in which three bridging positions are occupied by selenide. The collective results reveal three core oxidation states [W2Fe6Q9]0,1+,2+, all of which have been isolated and structurally characterized.
The research summarized in Figure 18 has two goals: (i) demonstration of the concept of template-assisted cluster synthesis with complex 63 as the template; (ii) determination of structural positions of sulfide and selenide introduced as external nucleophiles or present as internal nucleophiles in reactant clusters. The formation of single cubanes together with the assembly of double-cuboidal clusters is ample evidence of the efficacy of the WS3 group of 63 in template-assisted synthesis. Note that in all products selenium is bound only to iron, indicating that the WS3 group remains intact. Selenide positions in the synthetic progression SC (65) → EBDC (66) → PN-type (67, 68) provide the best available evidence addressing the function of external and internal sulfide nucleophiles. The first conversion proceeds with retention of the selenium position in the SC, and the second conversion introduces a μ2-Q bridge from the external nucleophile (consistent with Figure 17) and promotes the rearrangements 2 μ4-Se → μ2-Se + μ6-Se of internal nucleophiles.
The results of Figures 17 and 18 are insufficient for deduction of molecular pathways in cluster synthesis. Nonetheless, if the premise holds that selenide is a true surrogate of sulfide in molecules with equal numbers but differing populations of chalcogenides, the behavior of sulfide in sulfur-only reaction systems is revealed. One caveat is that the ca. 0.12 Å difference in covalent radii may allow sulfur to reside in sites too small for selenium. However, the well-documented structural analogy between Fe-S and Fe-Se clusters, including single cubanes,11,48,127,128 renders this situation unlikely. No such structural effect has yet been observed. The many similarities in the molecular chemistry of molybdenum and tungsten make it likely that the results obtained here would apply to Mo-Fe-S clusters as well. As yet, the corresponding potential template [(Tp*)MoVIS3]1− has not been prepared. We note finally the difference between the site-selective selenide incorporation manifested in these studies compared to the complex outcomes from the selenide-labeled reactions in the formation of the Fe-NR-S/Se clusters (Section 5.1). Although these two cases involve very different reaction types and are therefore not strictly comparable, the strength of heavy metal-ligand (i.e., W-S) bonds and the substitutional inertness of heavy metal centers may be significant factors in promoting site specificity in heterometal clusters. Selenide, whether introduced as an exogenous reactant or occurring as an internal ligand prior to core rearrangement, never disrupts a pre-existing W-S interaction.
5.4. Doubly-Bridging Heteroligands
The remaining occurrences of heteroligated cores involve doubly-bridging heteroligands μ2-Q (Q ≠ S) and span a range of cluster geometries. A number of examples appear elsewhere, including the alkoxide- and thiolate-bridged Fe8S7 clusters (Sections 5.2 and 6) and various W-Fe-S-Se clusters (Section 5.3). Bridges currently include monoanionic S-, O-, and N-donors (thiolate, alkoxide, and amide, respectively) and dianionic chalcogenides (oxide and selenide). (In the present context, thiolate, in contradistinction to sulfide, is a heteroligand.) Recent cluster types (post-2003) containing these bridging ligands are depicted, together with synthetic summaries, in Figure 19. Prior to 2003, very few Fe-S or M-Fe-S clusters with μ2-Q bridges between subcluster units were known and these were confined to thiolates for iron-ligated Q. Several types of recent heteroligand-bridged clusters are recognized: the PN M2Fe6S9 topology with one or two mono- or dianionic μ2-Q bridges in place of sulfide (71),105,111,128 double-cuboidal W2Fe4S9 congeners with μ2-Se in place of sulfide (72, Figure 18),128 and neutral Fe8S7 clusters (73, 74) and the related Fe8S6O cluster (58, Figure 15; see also Figure 20) containing two or three monanionic μ2-Q heteroligands.79,126,133–137
Figure 19.

Summary of three types of M-Fe-S clusters (M = Mo, W) containing cores with μ2-Q heteroatom bridges and compact frameworks (71-74), and two examples of bridged assemblies involving two or three recognizable fragments (75, 76).
Figure 20.

Schematic structures of synthetic Fe8S7 clusters: PN-type clusters 77a ([Fe8(μ3-S)6(μ6-S)(μ2-{N{SiMe3}2)2(SC(NMe2)2)2{N(SiMe3)2}2]) and 77b, and clusters with interstitial sulfide atoms 78 (Fe8(μ3-S)6(μ6-S)(μ2-STip)( μ2-SDmp)2(SDmp)2] and the minor byproduct 79 ([Fe8S7(μ2-SDmp)2(μ2-{N(SiMe3)2)})(STip)2]. Also shown is the Fe8 portion of 78 as an idealized bicapped trigonal prism with the mean Fe-S bond distance and mean length of Fe-Fe edges in trigonal faces; lengths of other Fe-Fe edges are indicated (adapted from ref. 134).
The foregoing clusters may be distinguished from others such as the diclusters 75104,105,111,112,128 and the tricluster 76.138 whose subclusters are bridged at formerly terminal positions by μ2-Q ligands. For this class, bridging results in more open or flexible structures that are better described as bridged assemblies, a concept previously introduced.139 These are constructs containing two or more recognizable fragments linked by covalent bridges. Cores 32 and 33 (Figure 10) are further examples of homo- and heteroligated assemblies, respectively. While there is no absolute demarcation between clusters and bridged assemblies, especially in the case of structurally complex, high-nuclearity structures, the former are typically distinguished by intimately bridged, compact frameworks whereas the latter possess obvious (i.e., chemically and physically significant) substructures.
6. Fe8S7 Clusters by Non-Polar Assembly
6.1. Background
Nearly all homo- and heterometal clusters described in the preceding sections, as well as others related to biological metal-sulfur clusters, are prepared by the reaction of an FeII,III and/or other metal compound, a core bridging ligand or its precursor (sulfur, sulfide, or a nitrogen anion salt), and a suitable terminal ligand such as thiolate or (pseudo)halide. These self-assembly systems are conventionally performed in methanol or nonprotic polar solvents, mainly acetonitrile, dichloromethane, DMF, or Me2SO. Product clusters are nearly always obtained as cations or anions and isolated as BF4−, BPh4−, R4N+, or Ph4P+ salts. There are of course some exceptions, including among others [Fe4S4(STip)2{SC(NMe2)2}2] prepared in toluene140 and, more recently, [Fe4S4{N(SiMe3)2}4] (13, Figure 3) also prepared in toluene. Octanuclear [Fe8S12(RNC)12] clusters have been synthesized in benzene from the fragment coupling reaction of [Fe4S4(SEt)2(RNC)6] and (PhCH2S)2S.141 Certain neutral clusters such as [Fe6S6(PEt3)4X2]116,142 (X = halide, PhS−), [Fe7S6(PEt3)4Cl3],143 and [Fe8S8(Pri2NHCMe2)6 (12, Figure 3) are pr143epared in THF whose dielectric constant (ε 7.52) is somewhat higher than that of toluene (ε 2.38).
Not surprisingly, the success of early assembly reactions (1972–1979) such as [22],144,145 [23],146 and [24]147,148 has stimulated development of other successful cluster syntheses with ionic reactants in polar solvents. Note that in these systems, thiolate acts as a reductant of FeIII, MoVI, and elemental sulfur, a frequent feature of assembly reactions containing these reactants. Such systems in general are useful in producing charged clusters of varying nuclearity.28
| [22] |
| [23] |
| [24] |
As shown in the following sections, departure from the foregoing ionic/polar to essentially nonpolar conditions by Tatsumi and coworkers79,126,133–135,137 affords entry to significant new cluster types. In this work, toluene is the solvent, [Fe{N(SiMe3)2}2]149,150 is a soluble source of FeII, elemental sulfur provides sulfide by reduction, (Me3Si)2N− serves as a ligand (both bridging and terminal) and a strong base for thiol deprotonation (pKa ≈ 26 for NH(SiMe3)2), and the sterically encumbered thiolates [2,6-(mesityl)2C6H3S]− (DmpS) and [2,4,6-Pri3C6H2S]− (TipS) function as reductants and ligands. Such systems provide three conditions favorable to the formation of large metastable clusters:134 a nonpolar solvent, suitably soluble precursors in appropriate mol ratios, and bulky thiolate ligands that solubilize and probably protect by steric size the product cluster from external nucleophiles. Schematic structures of product clusters 77-79 are provided in Figure 20 and bear comparison with the nitrogenase clusters 44-46 (Figure 12).
6.2. PN-Type Clusters
The assembly system 8 [Fe{N(SiMe3)2}2]/7 S/3 SC(NMe2)2/12 TipSH affords the cluster [Fe8S7{N(SiMe3)2}4(SC(NMe2)2)2] (77a) in 28% yield.133 Subsequent refinement of reaction and workup conditions increased the yield to 82%.135 An alternative procedure, the reaction of the previously known trinuclear cluster [Fe3(μ2-STip)4{N{SiMe3)2}2]151 with sulfur, HSTip, and tetramethylthiourea in toluene leads to 77a in 73% yield. A second assembly system in which Et3PS replaces SC(NMe2)2 affords [Fe8S7{N(SiMe3)2}4(SPEt3)2] (77b, 70%).79 Another synthetic protocol for this cluster is the reaction of [Fe4S4{N(SiMe3)2}4] with Et3P in toluene (29%). Sulfide appears to be removed as S0 from the initial cluster core, a rare reaction for any iron-sulfur cluster that is likely facilitated in this instance by the all-ferric oxidation state of the precursor. The net reaction couples two precursor clusters by means of sulfur removal and is a creative application of the fragment condensation concept.
The [Fe8S7]4+ portion of the foregoing clusters is indicative of the mixed-valence formulation Fe2+6Fe3+2; magnetic data reveal a diamagnetic ground state. Under the stiochiometry employed, probable events in the assembly systems include deprotonation of thiol by {(Me3Si)2N}−, and reduction of sulfur by FeII and thiolate. The amide provides bridging and terminal ligation and the thiourea or phosphine sulfide is terminally coordinated. Note that thiolate does not appear in either product. The structures of 77a and 77b clearly manifest the PN topology of 46, including the obtuse Fe-(μ6-S)-Fe angle (143.6° in 77a) but differ from the protein cluster in bridging and terminal ligands.
Clusters 77a and 77b are isoelectronic with the POX state 45 (sometimes symbolized as P2+) which is two electrons more oxidized than the all-ferrous PN state 46.152 Cluster 77a sustains replacement of the thiourea ligands by substituted benzenethiolate anions in fluorobenzene to afford the clusters [Fe8S7{N(SiMe3)2}4(SAr)2]2−.135 One of these displays two reduction steps in THF solution, thereby linking the oxidation states Fe2+6Fe3+2, Fe2+7Fe3+, and Fe2+8 in a three-membered electron transfer series. Other than noting that the POX cluster has access to additional coordinating protein ligands, it is unclear why this cluster and 77a and 77b have very different core structures.
6.3. Interstitial Sulfide Clusters
Treatment of the preformed binuclear FeII complex [Fe2(μ2-SDmp)2(STip)2] with sulfur in toluene results in the octanuclear cluster [Fe8S7(STip)(SDmp)4]] (78, 28%) and the minor byproduct 79. The most remarkable aspect of these species is the presence of an interstitial sulfur atom contained within a severely distorted Fe6 trigonal prism (Figure 20). This feature associates 78 and 79 with alkoxide-bridged cluster 59 (Figure 15). In 78, the two Fe-Fe edges of the prism bridged by SDmp are much longer (3.62, 3.71 Å) than that bridged by STip (2.91 Å), which is longer than the mean of the Fe-Fe distances in the triangular Fe3 faces (2.76 Å, Figure 20). The structure of 79 is very similar but with a μ2-amide bridge and two terminal STip ligands. The [Fe8S7]5+ cluster cores correspond to the oxidation state Fe2+5Fe3+3 and an apparent S = ½ ground state.
As recognized by Ohki et al.,134 cleavage of one Fe-(μ2-SR)-Fe bridge in 72 with thiolate could result in a cluster of general type [Fe8S7(μ2-SR)2(SR)4]1−. A cluster of this composition with two thiolate bridges would closely simulate the native PN cluster 46 with two cysteinate bridges and corresponding terminal ligation. Any such process would require reduction inasmuch as the cluster is one electron and three electrons more oxidized than POX (Fe2+6Fe3+2) and PN (Fe2+8), respectively.
The synthetic PN-type and interstitial clusters forge a “topological link”134 that connects the native PN cluster to FeMo-co (Figure 12) and the related all-iron FeFe-co, which is presumed to share the same core structure as the heterometallic FeMo-co. The shape of the Fe6 trigonal prism of the cofactor is more regular, all Fe-Fe edge lengths being in the 2.58–2.69 Å interval, and is too small to accommodate a sulfide ion. The cofactor has been shown to incorporate an interstitial carbide (Section 5). The only example of a synthetic Fe6 molecular cluster containing carbide is [Fe6C(CO)16]2−.153 Mean values of Fe-Fe separations (2.67 Å) and Fe-Ccarbide bond lengths (1.89 Å) are comparable with those in FeMo-co (2.59, 2.63 Å; 2.00 Å). However, the host polyhedron is octahedral rather than trigonal prismatic, and the iron oxidation state is much reduced compared to the cofactor for which various assessments have led to a mean oxidation state in the range Fe72.4+ to Fe72.7+.154
Nonpolar conditions are not a necessity for the formation of higher nuclearity iron-sulfur clusters,28 as illustrated by the preparation of the prismanes [Fe6S6X6]2−,3−,155,156 [Fe6S8(PEt3)6]0,..,4−,157, [Fe6S9(SR)2]4− (Section 5.3), and [Fe8S6I8]3−,4−,158,159 in solvents such as methanol, acetonitrile, and dichloromethane. In nonpolar solvents like toluene, the formation of charged products is suppressed. With suitable empirically-determined reaction stoichiometries, uncharged clusters can form under these nonpolar conditions, although predictive powers are so limited that the nuclearity of the products and other structural features must be left to experiment. In the synthesis of 77–79, stepwise cluster buildup must occur, but the steps are unknown. The authors of this work do not propose balanced equations for cluster formation, perhaps because of competing reactions and lower yields in some cases. While much remains to be learned, the synthesis of Fe8S7 clusters is a noteworthy achievement in the biomimetic chemistry of iron-sulfur clusters.
7. Conspectus
In bioinorganic enzymology, all aspects of catalytic sites--from biosynthesis to reaction mechanisms--are subjects of contemporary inquiry. This account is concerned with synthesis, and by possible implication, the biosynthesis of certain types of metalloclusters. The foregoing sections describe the current state of biomimetic metal chalcogenide and imide cluster chemistry implicating species of nuclearities four and eight. Clusters with cubane-type [Fe4S4]z cores are emphasized because these structures are ubiquitous in biology and enjoy an import comparable to other prosthetic groups such as hemes and flavins. Throughout, emphasis has been placed on original methods of synthesis because this is the most challenging part of the synthetic analogue approach to biological metal sites.160 This work is intended to contribute to the broad and developing area of metallocenter biosynthesis,161 particularly that of iron-sulfur clusters162–166 and FeMo-co,167,168 by showing which synthetic routes are actually feasible for cluster construction in the absence, and possibly in the presence, of proteins.
The synthesis of biologically relevant metalloclusters addresses two primary goals. The first is to determine in as much detail as possible how a given cluster is formed. In a biological system, this requires the elucidation of the overall reaction pathway, which describes the order of events and the proteins and other reactants necessary at each stage of cluster assembly. Thereafter, the chemical mechanism which includes atom-by-atom, even electron-by-electron, events is sought. While the sequence of events in the biosynthesis of any metallocenter is presumably controlled by proteins, it seems probable that the step-by-step formation of the final cluster implicates reactivity properties intrinsic to the reactants that form the core structure. In this context, we repeat an earlier statement: “The often empirical nature of cluster synthesis should not be a deterrent to experimentation. As Pasteur observed, ‘serendipity…appears to be most generous to those positioned to detect and exploit the accidental.’”23 It should be borne in mind that, despite much progress in defining biosynthetic pathways, the complete biosynthetic scenario for a polynuclear metal center in biology has not been elucidated at a truly mechanistic level.
A second goal is the exposition from a credible representation of a biological cluster of intrinsic structural, electronic, and reactivity properties. Differences may then be a consequence of protein environment. While the mechanism of any biological reaction must be extracted from the protein itself, reactivity properties of a synthetic cluster may, at the least, show what is possible.
Lastly, any ultimate description of cluster biosynthesis must rely on demonstrated inorganic chemical reactivity. For example, virtually any scheme for biological iron-sulfur cluster formation subsumes one or more of the proven reactions [25]–[27] (R = Cys) or their equivalent. These include cluster assembly with an FeII salt and a persulfide ([25]), fragment condensation by reductive core dimerization ([26]), and ligand substitution to bind the core to the protein ([27]). The intention of much of the work described here is to extend this understanding of fundamental cluster reactivity. Without such research, there is no way to know what is possible. Presently, the sequential pathways and component proteins involved in the biosynthesis of simple iron-sulfur clusters and FeMo-co are emerging, but the mechanisms of biological cluster assembly remain largely unknown at the molecular level.
| [25] |
| [26] |
| [27] |
Acknowledgments
Research on metal clusters at the University of Waterloo was supported by NSERC, CFI, and ORF, and at Harvard University by NIH Grant GM-28856.
ABBREVIATIONS
- ACN
acenaphthylenide(1−)
- Av
Azotobacter vinelandii
- Bu3tach
1,3,5-tri-tert-butyl-1,3,5-triazacyclohexane
- CD
cyclodextrin
- Cl4cat
tetrachlorocatcholate(2−)
- co
cofactor
- EBDC
edge-bridged double cubane
- edt
ethane-1,2-dithiolate(2−)
- Fd
ferredoxin
- HP
high-potential
- HSDmp
2,6-bis(mesityl)benzene-1-thiol
- L/M
generalized ligand/metal
- LS3
1,3,5-tris(4,6-dimethyl-3-mercaptophenylthio)-2,4,6-tris(p-tolylthio)- benzenate(3−)
- Pri2NHCMe2
1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene
- R*
chiral alkyl substituent
- Q
S, Se, RN (dianions)
- SC
single cubane
- SCE
standard calomel electrode
- SMes
mesitylthiolate(1−)
- STip
2,4,6-tris(isopropyl)benzene-1-thiolate(1−)
- Tp
tris(pyrazolyl)hydroborate(1−)
- Tp*
tris(3,5-dimethylpyrazolyl)hydroborate(1−)
Biographies
 Sonny C. Lee was born in Ping-Tung, Taiwan and moved to the United States at an early age. He received his chemical education at Caltech (B.S. and postdoctoral study with Professor H. B. Gray) and Harvard University (Ph.D. with Professor R. H. Holm), and is currently a faculty member at the University of Waterloo. His research interests emphasize aspects of synthetic inorganic chemistry, particularly the preparation and study of clusters containing weak-field iron centers and nitrogen anions as biomimetic analogues for the cluster chemistry of biological nitrogen fixation.
Sonny C. Lee was born in Ping-Tung, Taiwan and moved to the United States at an early age. He received his chemical education at Caltech (B.S. and postdoctoral study with Professor H. B. Gray) and Harvard University (Ph.D. with Professor R. H. Holm), and is currently a faculty member at the University of Waterloo. His research interests emphasize aspects of synthetic inorganic chemistry, particularly the preparation and study of clusters containing weak-field iron centers and nitrogen anions as biomimetic analogues for the cluster chemistry of biological nitrogen fixation.
 Wayne Lo was born in Taipei, Taiwan. He graduated from National Chung-Hsing University (B.S.), University of Massachusetts-Boston (M.Sc.), and Boston College (Ph.D., research advisor Professor W. H. Armstrong). He worked as postdoctoral associate with Professor R. H. Holm at Harvard University (2008–2012). His research interests are in inorganic and bioinorganic chemistry, with emphasis on metalloenzymes with iron-sulfur and manganese clusters and the design of biologically related metallocompounds.
Wayne Lo was born in Taipei, Taiwan. He graduated from National Chung-Hsing University (B.S.), University of Massachusetts-Boston (M.Sc.), and Boston College (Ph.D., research advisor Professor W. H. Armstrong). He worked as postdoctoral associate with Professor R. H. Holm at Harvard University (2008–2012). His research interests are in inorganic and bioinorganic chemistry, with emphasis on metalloenzymes with iron-sulfur and manganese clusters and the design of biologically related metallocompounds.
 Richard H. Holm was born in Boston, Massachusetts. He spent his early years on Nantucket Island and in Falmouth, Massachusetts where he received his secondary school education. He is a graduate of the University of Massachusetts (B.S.) and Massachusetts Institute of Technology (Ph.D. in chemistry). His graduate advisor was Professor F. A. Cotton. He has served on the faculties of the University of Wisconsin, the Massachusetts Institute of Technology, and Stanford University. Since 1980, he has been at Harvard University, where he has been Chair of the Department of Chemistry and, from 1983, Higgins Professor of Chemistry. As of 2006, he has been Higgins Research Professor of Chemistry. His research interests are centered in inorganic and bioinorganic chemistry, with particular reference to the synthesis and properties of molecules whose structures and reactivity are relevant to biological processes. With Professor Edward I. Solomon, he has been co-editor of three thematic issues in Chemical Reviews: Bioinorganic Enzymology I (1996), Biomimetic Inorganic Chemistry (2004), and Bioinorganic Enzymology II (this issue).
Richard H. Holm was born in Boston, Massachusetts. He spent his early years on Nantucket Island and in Falmouth, Massachusetts where he received his secondary school education. He is a graduate of the University of Massachusetts (B.S.) and Massachusetts Institute of Technology (Ph.D. in chemistry). His graduate advisor was Professor F. A. Cotton. He has served on the faculties of the University of Wisconsin, the Massachusetts Institute of Technology, and Stanford University. Since 1980, he has been at Harvard University, where he has been Chair of the Department of Chemistry and, from 1983, Higgins Professor of Chemistry. As of 2006, he has been Higgins Research Professor of Chemistry. His research interests are centered in inorganic and bioinorganic chemistry, with particular reference to the synthesis and properties of molecules whose structures and reactivity are relevant to biological processes. With Professor Edward I. Solomon, he has been co-editor of three thematic issues in Chemical Reviews: Bioinorganic Enzymology I (1996), Biomimetic Inorganic Chemistry (2004), and Bioinorganic Enzymology II (this issue).
Footnotes
The authors declare no competing financial interest.
References
- 1.Holm RH, Solomon EI, editors. Biomimetic Inorganic Chemistry; Chem Rev. 2004. p. 104. [DOI] [PubMed] [Google Scholar]
- 2.Cotton FA. Quart Rev. 1966;20:389. [Google Scholar]
- 3.Wu AJ, Penner-Hahn JE, Pecoraro VL. Chem Rev. 2004;104:903. doi: 10.1021/cr020627v. [DOI] [PubMed] [Google Scholar]
- 4.Mukhopadhyay S, Mandal SK, Bhaduri S, Armstrong WH. Chem Rev. 2004;104:3981. doi: 10.1021/cr0206014. [DOI] [PubMed] [Google Scholar]
- 5.Mullins CS, Pecoraro VL. Coord Chem Rev. 2008;252:416. doi: 10.1016/j.ccr.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Umena Y, Kawakami K, Shen JR, Kamiya N. Nature. 2011;473:55. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 7.Que L, Jr, Tolman WB. Angew Chem Int Ed. 2002;41:1114. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Lee D, Lippard SJ. In: Bio-coordination Chemistry (Comprehensive Coordination Chemistry II) Que L, Tolman WB, editors. Chapter 8.13. Oxford: 2004. [Google Scholar]
- 9.Andreini C, Bertini I, Cavallaro G, Najmanovich RJ, Thornton JM. J Mol Biol. 2009;388:356. doi: 10.1016/j.jmb.2009.02.052. [DOI] [PubMed] [Google Scholar]
- 10.Cotruvo JA, Jr, Stubbe J. Annu Rev Biochem. 2011;80:733. doi: 10.1146/annurev-biochem-061408-095817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rao PV, Holm RH. Chem Rev. 2004;104:527. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 12.Johnson DC, Dean DR, Smith AD, Johnson MK. Annu Rev Biochem. 2005;74:247. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 13.Koay MS, Antonkine ML, Gartner W, Lubitz W. Chem Biodiversity. 2008;5:1571. doi: 10.1002/cbdv.200890145. [DOI] [PubMed] [Google Scholar]
- 14.Seefeldt LS, Hoffman BM, Dean DR. Annu Rev Biochem. 2009;78:701. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ciurli S. Met Ions Life Sci. 2007;2:241. [Google Scholar]
- 16.van der Vlugt JI, Meyer F. Met Ions Life Sci. 2007;2:181. [Google Scholar]
- 17.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem Rev. 2007;107:4273. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 18.Lubitz W, van Gastel M, Gaertner W. Met Ions Life Sci. 2007;2:279. [Google Scholar]
- 19.Tard C, Pickett CJ. Chem Rev. 2009;109:2245. doi: 10.1021/cr800542q. [DOI] [PubMed] [Google Scholar]
- 20.Kratz H-B, Metzler-Nolte N. Concepts and Models in Bioinorganic Chemistry. Wiley-VCH; New York: 2006. [Google Scholar]
- 21.Lu Y. Angew Chem Int Ed. 2006;45:5588. doi: 10.1002/anie.200600168. [DOI] [PubMed] [Google Scholar]
- 22.Groysman S, Holm RH. Biochemistry. 2009;48:2310. doi: 10.1021/bi900044e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SC, Holm RH. Chem Rev. 2004;104:1135. doi: 10.1021/cr0206216. [DOI] [PubMed] [Google Scholar]
- 24.Steed JW, Atwood JL. Supramolecular Chemistry. Wiley; New York: 2009. [Google Scholar]
- 25.Lee SC, Holm RH. Angew Chem Int Ed Engl. 1990;29:840. [Google Scholar]
- 26.Yaghi OM, Scott MJ, Holm RH. Inorg Chem. 1992;31:4778. [Google Scholar]
- 27.Magliocchi CM, Xie X, Hughbanks T. Inorg Chem. 2004;43:1902. doi: 10.1021/ic035050u. [DOI] [PubMed] [Google Scholar]
- 28.Holm RH. In: Bio-coordination Chemistry (Comprehensive Coordinaton Chemistry II) Que L, Tolman WB, editors. Chapter 8.2. Oxford: 2004. [Google Scholar]
- 29.Yoo SJ, Angove HC, Burgess BK, Hendrich MP, Munck E. J Am Chem Soc. 1999;121:2534. [Google Scholar]
- 30.Hans M, Buckel W, Bill E. J Biol Inorg Chem. 2008;13:568. doi: 10.1007/s00775-008-0345-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou C, Raebiger JW, Segal BM, Holm RH. Inorg Chim Acta. 2000;300–302:892. [Google Scholar]
- 32.Scott TA, Zhou HC. Angew Chem Int Ed. 2004;43:5628. doi: 10.1002/anie.200460879. [DOI] [PubMed] [Google Scholar]
- 33.Scott TA, Berlinguette CP, Holm RH, Zhou HC. Proc Natl Acad Sci USA. 2005;102:9741. doi: 10.1073/pnas.0504258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng L, Holm RH. J Am Chem Soc. 2008;130:9878. doi: 10.1021/ja802111w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chakrabarti M, Deng L, Holm RH, Munck E, Bominaar EL. Inorg Chem. 2009;48:2735. doi: 10.1021/ic802192w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chakrabarti M, Deng L, Holm RH, Munck E, Bominaar E. Inorg Chem. 2010;49:1647. doi: 10.1021/ic902050k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohki Y, Sunada Y, Tatsumi K. Chem Lett. 2005;34:172. [Google Scholar]
- 38.Sharp CR, Duncan JS, Lee SC. Inorg Chem. 2010;49:6697. doi: 10.1021/ic100742c. [DOI] [PubMed] [Google Scholar]
- 39.Henderson RA. Chem Rev. 2005;105:2365. doi: 10.1021/cr030706m. [DOI] [PubMed] [Google Scholar]
- 40.Job RC, Bruice TC. Proc Natl Acad Sci USA. 1975;72:2478. doi: 10.1073/pnas.72.7.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hill CL, Renaud J, Holm RH, Mortenson LE. J Am Chem Soc. 1977;99:2549. doi: 10.1021/ja00450a024. [DOI] [PubMed] [Google Scholar]
- 42.Henderson RA, Sykes AG. Inorg Chem. 1980;19:3103. [Google Scholar]
- 43.Ye H, Rouault TA. Biochemistry. 2010;49:4945. doi: 10.1021/bi1004798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lo W, Scott TA, Zhang P, Ling CC, Holm RH. J Inorg Biochem. 2011;105:497. doi: 10.1016/j.jinorgbio.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lo W, Zhang P, Ling CC, Huang S, Holm RH. Inorg Chem. 2012;51:9883. doi: 10.1021/ic301324u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuroda Y, Sasaki Y, Shiroiwa Y, Tabushi I. J Am Chem Soc. 1988;110:4049. [Google Scholar]
- 47.Sharma AK, Kim N, Cameron CS, Lyndon M, Gorman CB. Inorg Chem. 2010;49:5072. doi: 10.1021/ic1002447. [DOI] [PubMed] [Google Scholar]
- 48.Tan LL, Holm RH, Lee SC. Polyhedron. 2013;58:206. doi: 10.1016/j.poly.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grzyb J, Xu F, Weiner L, Reijerse EJ, Lubitz W, Nanda V, Noy D. Biochim Biophys Acta. 2010;1797:406. doi: 10.1016/j.bbabio.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 50.Davies SC, Evans DJ, Henderson RA, Hughes DL, Longhurst S. J Chem Soc, Dalton Trans. 2002:3470. [Google Scholar]
- 51.Lo W, Huang S, Zheng SL, Holm RH. Inorg Chem. 2011;50:11082. doi: 10.1021/ic2016269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goh C, Segal BM, Huang J, Long JR, Holm RH. J Am Chem Soc. 1996;118:11844. [Google Scholar]
- 53.Zhou HC, Holm RH. Inorg Chem. 2003;42:11. doi: 10.1021/ic020464t. [DOI] [PubMed] [Google Scholar]
- 54.Beinert H, Kennedy MC, Stout CD. Chem Rev. 1996;96:2335. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 55.Lloyd SJ, Lauble H, Prasad GS, Stout CD. Protein Science. 1999;8:2655. doi: 10.1110/ps.8.12.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Volbeda A, Charon MC, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. Nature. 1995;373:580. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 57.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Science. 2004;303:76. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vey JL, Drennan CL. Chem Rev. 2011;111:2487. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. Science. 1998;282:1853. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 60.Crane BR, Siegel LM, Getzoff ED. Science. 1995;270:59. doi: 10.1126/science.270.5233.59. [DOI] [PubMed] [Google Scholar]
- 61.Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC. Nature Struct Biol. 2003;10:271. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 62.Svetlitchnyi V, Dobbek H, Meyer-Klaucke W, Meins T, Thiele B, Romer P, Huber R, Meyer O. Proc Natl Acad Sci USA. 2004;101:446. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stack TDP, Holm RH. J Am Chem Soc. 1988;110:2484. [Google Scholar]
- 64.Daley CJA, Holm RH. J Inorg Biochem. 2003;97:287. doi: 10.1016/s0162-0134(03)00280-0. [DOI] [PubMed] [Google Scholar]
- 65.Terada T, Wakimoto T, Nakamura T, Hirabayashi K, Tanaka K, Li J, Matsumoto T, Tatsumi K. Chem Asian J. 2012:920. doi: 10.1002/asia.201200039. [DOI] [PubMed] [Google Scholar]
- 66.Ohki Y, Tanifuji K, Yamada N, Imada M, Tajima T, Tatsumi K. Proc Natl Acad Sci USA. 2011;108:12635. doi: 10.1073/pnas.1106472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Sullivan T, Millar MM. J Am Chem Soc. 1985;107:4096. [Google Scholar]
- 68.Deng L, Majumdar A, Lo W, Holm RH. Inorg Chem. 2010;49:11118. doi: 10.1021/ic101702b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cai L, Holm RH. J Am Chem Soc. 1994;116:7177. [Google Scholar]
- 70.Zhou C, Cai L, Holm RH. Inorg Chem. 1996;35:2767. doi: 10.1021/ic960216v. [DOI] [PubMed] [Google Scholar]
- 71.Tard C, Liu X, Ibrahim SK, Bruschi M, De Giola L, Davies SC, Yang X, Wang LS, Sawers G, Pickett CJ. Nature. 2005;433:610. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]
- 72.Fritsch J, Scheerer P, Frielingsdorf S, Kroschinsky S, Friedrich B, Lenz O, Spahn CMT. Nature. 2011;479:249. doi: 10.1038/nature10505. [DOI] [PubMed] [Google Scholar]
- 73.Shomura Y, Yoon KS, Nishihara H, Higuchi Y. Nature. 2011;479:253. doi: 10.1038/nature10504. [DOI] [PubMed] [Google Scholar]
- 74.Mouesca JM, Fontecilla-Camps JC, Amara P. Angew Chem Int Ed. 2013;52:2002. doi: 10.1002/anie.201209063. [DOI] [PubMed] [Google Scholar]
- 75.Goris T, Wait AF, Saggu M, Fritsch J, Heidary N, Stein M, Zebger I, Lendzian F, Armstrong FA, Friedrich B, Lenz O. Nature Chem Biol. 2011;7:310. doi: 10.1038/nchembio.555. [DOI] [PubMed] [Google Scholar]
- 76.Scott MJ, Holm RH. Angew Chem Int Ed Engl. 1993;32:564. [Google Scholar]
- 77.Goh C, Holm RH. Inorg Chim Acta. 1998;270:46. [Google Scholar]
- 78.D’Addario S, Demartin F, Grossi L, Iapalucci MC, Laschi F, Longoni G, Zanello P. Inorg Chem. 1993;32:1153. doi: 10.1021/ic950482t. [DOI] [PubMed] [Google Scholar]
- 79.Ohki Y, Tanifuji K, Yamada N, Cramer RE, Tatsumi K. Chem Asian J. 2012;7:2222. doi: 10.1002/asia.201200568. [DOI] [PubMed] [Google Scholar]
- 80.Rao PV, Bhaduri S, Jiang J, Hong D, Holm RH. J Am Chem Soc. 2005;127:1933. doi: 10.1021/ja040222n. [DOI] [PubMed] [Google Scholar]
- 81.Kure B, Ogo S, Inoki D, Nakai H, Isobe K, Fukuzumi S. J Am Chem Soc. 2005;127:14366. doi: 10.1021/ja051748q. [DOI] [PubMed] [Google Scholar]
- 82.Cramer SP, Hodgson KO, Gillum WO, Mortenson LE. J Am Chem Soc. 1978;100:3398. [Google Scholar]
- 83.Beinert H, Thomson AJ. Arch Biochem Biophys. 1983:333. doi: 10.1016/0003-9861(83)90531-3. [DOI] [PubMed] [Google Scholar]
- 84.Kissinger CR, Adman ET, Sieker LC, Jensen LH. J Am Chem S oc. 1988;110:8721. [Google Scholar]
- 85.Johnson MK, Duderstadt RE, Duin EC. Adv Inorg Chem. 1999;47:1. [Google Scholar]
- 86.Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O. Science. 2001;293:1281. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]
- 87.Drennan CL, Heo J, Sintchak MD, Schreiter E, Ludden PW. Proc Natl Acad Sci USA. 2001;98:11973. doi: 10.1073/pnas.211429998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dobbek H, Svetlitchnyi V, Liss J, Meyer O. J Am Chem Soc. 2004;126:5382. doi: 10.1021/ja037776v. [DOI] [PubMed] [Google Scholar]
- 89.Holm RH. Adv Inorg Chem. 1992;38:1. [Google Scholar]
- 90.Zhou J, Scott MJ, Hu Z, Peng G, Munck E, Holm RH. J Am Chem Soc. 1992;114:10843. [Google Scholar]
- 91.Cen W, Lee SC, Li J, MacDonnell FM, Holm RH. J Am Chem Soc. 1993;115:9515. [Google Scholar]
- 92.Cen W, MacDonnell FM, Scott MJ, Holm RH. Inorg Chem. 1994;33:5809. [Google Scholar]
- 93.Zhou J, Raebiger JW, Crawford CA, Holm RH. J Am Chem Soc. 1997;119:6242. [Google Scholar]
- 94.Zhou J, Scott MJ, Hu Z, Peng G, Munck E, Holm RH. J Am Chem Soc. 1992;114:10843. [Google Scholar]
- 95.Zhou J, Hu Z, Munck E, Holm RH. J Am Chem Soc. 1996;118:1966. [Google Scholar]
- 96.Holm RH, Simhon ED. Molybdenum Enzymes. Chapter 1 Wiley; New York: 1985. [Google Scholar]
- 97.Malinak SM, Coucouvanis D. Prog Inorg Chem. 2001;49:599. [Google Scholar]
- 98.Lee SC, Holm RH. Proc Natl Acad Sci USA. 2003;100:3595. doi: 10.1073/pnas.0630028100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coucouvanis D, Al-Ahmad SA, Salifoglou A, Papaefthymiou V, Kostikas A, Simopoulos A. J Am Chem Soc. 1992;114:2472. [Google Scholar]
- 100.Jensen KP, Ooi BL, Christensen HEM. J Phys Chem A. 2008;112:12829. doi: 10.1021/jp8014782. [DOI] [PubMed] [Google Scholar]
- 101.Hauser C, Bill E, Holm RH. Inorg Chem. 2002;41:1615. doi: 10.1021/ic011011b. [DOI] [PubMed] [Google Scholar]
- 102.Fomitchev DV, McLauchlan CC, Holm RH. Inorg Chem. 2002;41:958. doi: 10.1021/ic011106d. [DOI] [PubMed] [Google Scholar]
- 103.Zuo J-L, Zhou H-C, Holm RH. Inorg Chem. 2003:4624. doi: 10.1021/ic0301369. [DOI] [PubMed] [Google Scholar]
- 104.Zhang Y, Holm RH. J Am Chem Soc. 2003;125:3910. doi: 10.1021/ja0214633. [DOI] [PubMed] [Google Scholar]
- 105.Zhang Y, Holm RH. Inorg Chem. 2004;43:674. doi: 10.1021/ic030259t. [DOI] [PubMed] [Google Scholar]
- 106.Berlinguette CP, Miyaji T, Zhang Y, Holm RH. Inorg Chem. 2006;45:1997. doi: 10.1021/ic051770k. [DOI] [PubMed] [Google Scholar]
- 107.Pesavento RP, Berlinguette CP, Holm RH. Inorg Chem. 2007;46:510. doi: 10.1021/ic061704y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scott TA, Holm RH. Inorg Chem. 2008;47:3426. doi: 10.1021/ic702372f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Demadis KD, Campana CF, Coucouvanis D. J Am Chem Soc. 1995;117:7832. [Google Scholar]
- 110.Malinak SM, Demadis KD, Coucouvanis D. J Am Chem Soc. 1995;117:3126. [Google Scholar]
- 111.Berlinguette CP, Holm RH. J Am Chem Soc. 2006;128:11993. doi: 10.1021/ja063604x. [DOI] [PubMed] [Google Scholar]
- 112.Hlavinka ML, Miyaji T, Staples RJ, Holm RH. Inorg Chem. 2007;46:9192. doi: 10.1021/ic701070w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Peters JW, Stowell MHB, Soltis SM, Finnegan MG, Johnson MK, Rees DC. Biochemistry. 1997;36:1181. doi: 10.1021/bi9626665. [DOI] [PubMed] [Google Scholar]
- 114.Mayer SM, Lawson DM, Gormal CA, Roe SM, Smith BE. J Mol Biol. 1999;292:871. doi: 10.1006/jmbi.1999.3107. [DOI] [PubMed] [Google Scholar]
- 115.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 116.Snyder BS, Holm RH. Inorg Chem. 1988;27:2339. [Google Scholar]
- 117.Reynolds JG, Holm RH. Inorg Chem. 1981;20:1873. [Google Scholar]
- 118.Spatzal T, Aksoyoglu M, Zhang L, Andrade SLA, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergman U, DeBeer S. Science. 2011;334:974. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wiig JA, Hu Y, Lee CC, Ribbe MW. Science. 2012;337:1672. doi: 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen XD, Duncan JS, Verma AK, Lee SC. J Am Chem Soc. 2010;132:15884. doi: 10.1021/ja106478k. [DOI] [PubMed] [Google Scholar]
- 122.(a) Chen XD, Zhang W, Duncan JS, Lee SC. Inorg Chem. 2012;51:12891. doi: 10.1021/ic301868m. [DOI] [PubMed] [Google Scholar]; (b) Tan LL, Devgan MK, Lee SC. unpublished results. [Google Scholar]
- 123.Duncan JS, Zdilla MJ, Lee SC. Inorg Chem. 2007;46:1071. doi: 10.1021/ic061133+. [DOI] [PubMed] [Google Scholar]
- 124.Verma AK, Lee SC. J Am Chem Soc. 1999;121:10838. [Google Scholar]
- 125.Han J, Koutmos M, Al-Ahmad S, Coucouvanis D. Inorg Chem. 2001;40:5985. doi: 10.1021/ic0104914. [DOI] [PubMed] [Google Scholar]
- 126.Ohta S, Ohki Y, Hashimoto T, Cramer RE, Tatsumi K. Inorg Chem. 2012;51:11217. doi: 10.1021/ic301348f. [DOI] [PubMed] [Google Scholar]
- 127.Majumdar A, Holm RH. Inorg Chem. 2011;50:11242. doi: 10.1021/ic2018117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zheng B, Chen XD, Zhang SL, Holm RH. J Am Chem Soc. 2012;134:6479. doi: 10.1021/ja3010539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Seino H, Arai Y, Iwata N, Nagao S, Mizobe Y, Hidai M. Inorg Chem. 2001;40:1677. doi: 10.1021/ic0008823. [DOI] [PubMed] [Google Scholar]
- 130.Zhou HC, Su W, Achim C, Rao PV, Holm RH. Inorg Chem. 2002;41:3191. doi: 10.1021/ic0201250. [DOI] [PubMed] [Google Scholar]
- 131.Christou G, Sabat M, Ibers JA, Holm RH. Inorg Chem. 1982;21:3518. [Google Scholar]
- 132.Strasdeit H, Krebs B, Henkel G. Inorg Chem. 1984;23:1816. [Google Scholar]
- 133.Ohki Y, Sunada Y, Honda M, Katsada M, Tatsumi K. J Am Chem Soc. 2003;125:4052. doi: 10.1021/ja029383m. [DOI] [PubMed] [Google Scholar]
- 134.Ohki Y, Ikagawa Y, Tatsumi K. J Am Chem Soc. 2007;129:10457. doi: 10.1021/ja072256b. [DOI] [PubMed] [Google Scholar]
- 135.Ohki Y, Imada M, Murata A, Sunada Y, Ohta S, Honda M, Sasamori T, Katada M, Tatsumi K. J Am Chem Soc. 2009;131:13168. doi: 10.1021/ja9055036. [DOI] [PubMed] [Google Scholar]
- 136.Hashimoto T, Ohki Y, Tatsumi K. Inorg Chem. 2010;49:6102. doi: 10.1021/ic100692v. [DOI] [PubMed] [Google Scholar]
- 137.Ohta S, Yokozawa S, Ohki Y, Tatsumi K. Inorg Chem. 2012;51:2645. doi: 10.1021/ic2025928. [DOI] [PubMed] [Google Scholar]
- 138.Koutmos M, Coucouvanis D. Angew Chem Int Ed. 2004;43:5023. doi: 10.1002/anie.200460154. [DOI] [PubMed] [Google Scholar]
- 139.Holm RH. Pure Appl Chem. 1995;67:217. [Google Scholar]
- 140.Bierbach U, Saak W, Haase D, Pohl S. Z Naturforsch. 1991;46b:1629. [Google Scholar]
- 141.Goh C, Novorozhkin A, Yoo SJ, Bominaar EL, Munck E, Holm RH. Inorg Chem. 1998;37:2926. [Google Scholar]
- 142.Reynolds MS, Holm RH. Inorg Chem. 1988;27:4494. [Google Scholar]
- 143.Noda I, Snyder BS, Holm RH. Inorg Chem. 1986;25:3851. [Google Scholar]
- 144.Herskovitz T, Averill BA, Holm RH, Ibers JA, Phillips WD, Weiher JF. Proc Natl Acad Sci USA. 1972;69:2437. doi: 10.1073/pnas.69.9.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Averill BA, Herskovitz T, Holm RH, Ibers JA. J Am Chem Soc. 1973;95:3523. doi: 10.1021/ja00792a013. [DOI] [PubMed] [Google Scholar]
- 146.Christou G, Garner CD. J Chem Soc, Dalton Trans. 1979:1093. [Google Scholar]
- 147.Wolff TE, Berg JM, Warrick C, Hodgson KO, Holm RH, Frankel RB. J Am Chem Soc. 1978;100:4630. [Google Scholar]
- 148.Wolff TE, Berg JM, Hodgson KO, Frankel RB, Holm RH. J Am Chem Soc. 1979;101:4140. [Google Scholar]
- 149.Andersen RA, Faegri K, Jr, Green JC, Haaland A, Lappert MF, Leung W-P, Rypda K. Inorg Chem. 1988;27:1782. [Google Scholar]
- 150.Olmstead MM, Power PP, Shoner SC. Inorg Chem. 1991;30:2547–2551. [Google Scholar]
- 151.MacDonnell FM, Ruhlandt-Senge K, Ellison JJ, Holm RH, Power PP. Inorg Chem. 1995;34:1815. [Google Scholar]
- 152.Rupnik K, Hu Y, Lee CC, Wiig JA, Ribbe MW, Hales BJ. J Am Chem Soc. 2012;134:13749. doi: 10.1021/ja304077h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Churchill MR, Wormald J. J Chem Soc, Dalton Trans. 1974:2410. [Google Scholar]
- 154.Harris TV, Szilagyi R. Inorg Chem. 2011;50:4811. doi: 10.1021/ic102446n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kanatzidis MG, Hagen WR, Dunham WR, Lester RK, Coucouvanis D. J Am Chem Soc. 1985;107:953. [Google Scholar]
- 156.Kanatzidis MG, Salifoglou A, Coucouvanis D. Inorg Chem. 1986;25:2460. [Google Scholar]
- 157.Goddard CA, Long JR, Holm RH. Inorg Chem. 1996;35:4347. doi: 10.1021/ic960052i. [DOI] [PubMed] [Google Scholar]
- 158.Pohl S, Saak W. Angew Chem Int Ed Engl. 1984;23:907. [Google Scholar]
- 159.Pohl S, Opitz U. Angew Chem Int Ed Engl. 1993;32:863. [Google Scholar]
- 160.Holm RH, Solomon EI. Chem Rev. 2004;104:347. doi: 10.1021/cr0206364. [DOI] [PubMed] [Google Scholar]
- 161.Kukchar J, Hausinger RP. Chem Rev. 2004;104:509. doi: 10.1021/cr020613p. [DOI] [PubMed] [Google Scholar]
- 162.Lill R. Nature. 2009;460:831. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 163.Bandyopadhyay S, Chandramouli K, Johnson MK. Biochem Soc Trans. 2008;36:1112. doi: 10.1042/BST0361112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Raulfs EC, O’Carroll IP, Dos Santos PC, Unciuleac M, Dean DR. Proc Natl Acad Sci USA. 2008;105:8591. doi: 10.1073/pnas.0803173105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fontecave M, Ollagnier-de Choudens S. Arch Biochem Biophys. 2008;474:226. doi: 10.1016/j.abb.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 166.Shepard EM, Boyd ES, Broderick JB, Peters JW. Curr Opinion Chem Biol. 2011;15:319. doi: 10.1016/j.cbpa.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 167.Hu Y, Ribbe MW. Coord Chem Rev. 2011;255:1218. doi: 10.1016/j.ccr.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hu Y, Ribbe MW. J Biol Chem. 2013;288:13173. doi: 10.1074/jbc.R113.454041. [DOI] [PMC free article] [PubMed] [Google Scholar]