

Abstract
Inhibition of Golgi α-mannosidase II (GMII), which acts late in the N-glycan processing pathway, provides a route to blocking cancer-induced changes in cell surface oligosaccharide structures. To probe the substrate requirements of GMII, oligosaccharides were synthesized that contained an α(1,3)- or α(1,6)-linked 1-thiomannoside. Surprisingly, these oligosaccharides were not observed in X-ray crystal structures of native Drosophila GMII (dGMII). However, a mutant enzyme in which the catalytic nucleophilic aspartate was changed to alanine (D204A) allowed visualization of soaked oligosaccharides and led to the identification of the binding site for the α(1,3)-linked mannoside of the natural substrate. These studies also indicate that the conformational change of the bound mannoside to a high-energy B2,5 conformation is facilitated by steric hindrance from, and the formation of strong hydrogen bonds to, Asp204. The observation that 1-thio-linked mannosides are not well tolerated by the catalytic site of dGMII led to the synthesis of a pentasaccharide containing the α(1,6)-linked Man of the natural substrate and the β(1,2)-linked GlcNAc moiety proposed to be accommodated by the extended binding site of the enzyme. A cocrystal structure of this compound with the D204A enzyme revealed the molecular interactions with the β(1,2)-linked GlcNAc. The structure is consistent with the ~80-fold preference of dGMII for the cleavage of substrates containing a nonreducing β(1,2)-linked GlcNAc. By contrast, the lysosomal mannosidase lacks an equivalent GlcNAc binding site and kinetic analysis indicates oligomannoside substrates without non-reducing-terminal GlcNAc modifications are preferred, suggesting that selective inhibitors for GMII could exploit the additional binding specificity of the GlcNAc binding site.
Introduction
Aberrant glycosylation of glycoproteins and glycolipids is a molecular change typical of malignant transformations.1–3 For example, human cancers of the breast, colon, and melanomas often overexpress the glycosyltransferase N-acetylglucosaminyltransferase V (GlcNAc-TV).4–11 This enzyme introduces a specific branching N-acetylglucosamine moiety at the N-linked oligosaccharides of glycoproteins, thereby forming the biosynthetic precursor for polylactosamine chains.12 It has been proposed that these polysaccharides on integrins and cadherins or adhesion receptors facilitate focal adhesion turnover, cell migration, and tumor metastasis.13
There is evidence that inhibition of GlcNAc-TV may be useful for the treatment of malignancies. However, it has been difficult to design and synthesize potent and cell-permeable inhibitors of glycosyltransferases.14–16 Therefore, efforts to block the biosynthesis of polylactosamine chains of N-linked glycoproteins have focused on inhibitors of glycosidases that act earlier in the biosynthesis of N-glycans to prevent the formation of the biosynthetic pathway primer of GlcNAc-TV. Most efforts in the field have focused on Golgi α-mannosidase II (GMII),17–19 which trims two mannosyl residues from GlcNAcMan5GlcNAc2 to form the core GlcNAcMan3GlcNAc2 moiety.20
The alkaloids swainsonine and mannostatin are potent inhibitors of Golgi α-mannosidase II.18 However, these compounds also inhibit the GH38 lysosomal α-mannosidases (LM) with potencies equal to those of the GMII and therefore induce symptoms similar to those of the lysosomal storage disease α-mannosidosis.18,21–23 Attempts to develop more selective inhibitors of GMII by chemically modifying swainsonine or mannostatin have been unsuccessful to date.17–19,24,25 Under standing the molecular basis for the substrate specificities of these enzymes may provide unique opportunities for the design of more selective inhibitors.
GMII is a retaining glycosylhydrolase of family GH38. Retaining glycosyl hydrolases employ a two-step mechanism involving the formation of a covalent glycosyl enzyme complex. Two carboxylic acids positioned within the active site act in concert: one as a catalytic nucleophile and the other as a general acid/base catalyst.26–28 Studies with 5-fluoro pseudosubstrates and deuterium-labeled substrates have shown that the transition states on either side of the covalent intermediate have a marked oxacarbenium ion character, involving electron delocalization across the C-1–O-5 bond. This demands planarity of C-2, C-1, O-5, and C-5 at or near the transition state, which for the pyranose ring can be accomplished by several boat and half-chair conformations, namely, 2,5B, B2,5, 4H3, and 3H4.29–31 X-ray crystal structures of wild-type and mutant (D341N) Drosophila melanogaster GMII (in which the D204 side chain is present) with fluorinated mannose analogues have revealed that the covalent mannosyl-enzyme intermediate adopts a distorted 1S5 skew boat conformation.32 In this conformation, the leaving group is placed antiperiplanar to the lone pair of the ring oxygen, which is required for the departure of the leaving group according to Deslongchamp’s antiperiplanar lone-pair hypothesis. Furthermore, steric clashes between the syn-hydrogens at C-3 and C-5 and the attacking water molecule are minimal in the 1S5 skew boat conformation. There are only a limited number of pseudorotational itineraries that glycosidase can take,29–31 and hence it has been suggested that retaining α-mannosidases follow a 1S5 → B2,5 → 0S2 itinerary.32 Interestingly, it has been shown that the Michaelis complex of a retaining endo-β-mannanase adopts a 1S5 and its covalent intermediate an 0S2 conformation, strongly implicating a B2,5 transition state for this enzyme.33 Thus, it appears that these two disparate enzymes follow similar pseudorotational itineraries but in opposite directions.
The presence of an extended binding site is common for glycosidases and may be important for substrate specificity. For example, GMII is dependent upon the β(1,2)-GlcNAc moiety of the natural substrate GlcNAcMan5GlcNAc2 for efficient catalysis,34 and it has been suggested that this saccharide moiety makes important interactions with the enzyme.
To probe the substrate requirements of GMII, we report here the chemical synthesis of a number of oligosaccharides (1–5) that have been employed in cocrystallographic studies with wild-type and mutant (D204A) Drosophila GMII (dGMII). These studies uncovered the molecular interactions of the enzyme with the α(1,3)-linked mannoside and β(1,2)-linked GlcNAc moiety of the natural substrate. The importance of the GlcNAc binding site is consistent with a ~80-fold preference of dGMII for cleavage of substrates containing the β(1,2)GlcNAc residue.
Results
Oligosaccharide Synthesis
Oligosaccharides 1–5, derived from the natural substrate of GMII, were synthesized for cocrystallization studies. Compounds 1–4 contain an α(1,6)-or α(1,3)-linked 1-thio-α-mannoside. Such compounds are resistant toward hydrolysis by mannosidases and were expected to allow cocrystallographic studies with wild-type dGMII.35–37 Furthermore, the C–S bond is significantly longer than a C–O linkage, which was expected to make the compounds better ligands because the longer C–S bond mimics lengthening of the exocyclic C–O bond in the transition state of the cleaved α-mannoside. Thus, compound 1 is an α(1,3)-linked dimannoside in which the exocyclic oxygen of the nonreducing mannoside is replaced by sulfur (Figure 1). Compound 2 is derived from 1 but contains an additional α(1,6)-mannoside. Thus, this derivative contains both mannosides that can be cleaved by GMII and was initially designed to determine whether the 1-thio-linked mannoside would be preferentially recognized by GMII. However, studies described in the next section established that thio-linked mannosides are not well tolerated by the catalytic site of dGMII. On the other hand, a crystal structure of compound 2 with a mutant enzyme revealed the binding site for the α(1,3)-linked mannoside of the natural substrates. Compounds 3 and 4 are more complex oligosaccharides and contain an α(1,6)-linked 1-thiomannoside. Finally, the observation that the 1-thio-linked mannosides are not well tolerated by the catalytic site of wild-type dGMII led to the synthesis of pentasaccharide 5, which is derived from the natural substrate of GMII and contains the α(1,6)-linked Man that is cleaved by the enzyme and the β(1,2)-linked GlcNAc moiety, which is proposed to be accommodated by the extended binding site of the enzyme.
Figure 1.
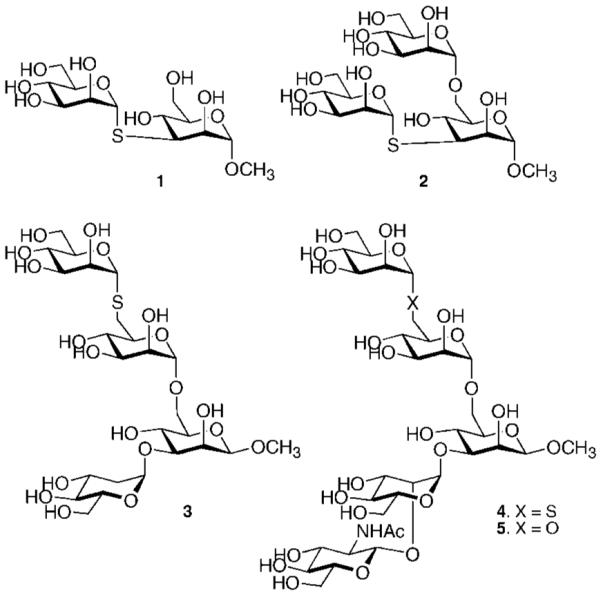
Structures of synthetic targets.
Compounds 4 and 5 were prepared by a convergent 3 + 2 glycosylation strategy using glycosyl acceptor 12 and glycosyl donors 18 and 22, respectively (Schemes 1 and 2). The trisaccharide acceptor 12 could easily be prepared from known monosaccharides 6, 7, and 8.38–40 The thio-linked mannosyl donor 22 was obtained by a nucleophilic displacement of triflate 19 with a thioaldose prepared by in situ deacetylation of 2036,41 followed by a two-step conversion of the trimethylsilylethyl (SE) glycoside of the resulting compound into a trichloroacetimidate.
Scheme 1a.
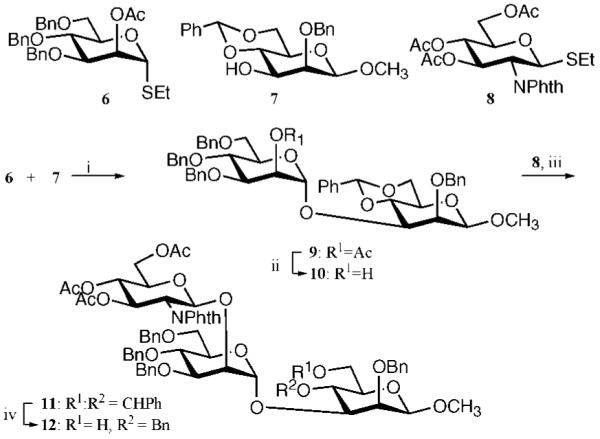
a Reagents and conditions: (i) NIS, TfOH, DCM, 0 °C (80%); (ii) NaOMe, MeOH (89%); (iii) NIS, TfOH, DCM, 0 °C (76%); (iv) BH3 in THF, Bu2BOTf in DCM, 0 °C (67%).
Scheme 2a.

a Reagents and conditions: (i) TrCl, pyridine, 80 °C, and then Ac2O, pyridine (96%); (ii) FeCl3 · 6H2O, DCM (82%); (iii) Tf2O, 2,6-lutidine, DCM, −40 °C; (iv) NIS, TfOH, DCM, 0 °C (76% for 17, 86% for 22); (v) diethylamine, DMF, 0 °C (73%); (vi) TFA, DCM, and then trichloroacetonitrile, DBU, DCM; (vii) TMSOTf, DCM, (80% for 23, 83% for 25); (viii) H2NNH2 · H2O, EtOH, 90 °C, and then Ac2O, pyridine; (ix) NaOMe, MeOH, and then Na (s), NH3 (l), THF, −78 °C (86% for 4, 87% for 5).
Thus, NIS/TfOH-mediated coupling of thioglycoside 640 with glycosyl acceptor 739 (Scheme 1) gave disaccharide 9 in an excellent yield of 80%. Neighboring group participation by the C-2 acetyl ester of 6 ensured exclusive formation of the α-anomer. Next, the acetyl ester of 9 was removed by treatment with NaOMe in methanol to give glycosyl acceptor 10, which was employed in a NIS/TfOH-mediated coupling with thioglycosyl donor 838 to give trisaccharide 11in a yield of 76%. The bulky C-2 phthalidene of 8 ensured that only the β-anomer was formed. The benzylidene ring of 11 was selectively opened by treatment with borane in THF and Bu2BOTf in DCM,42 resulting in the formation of glycosyl acceptor 12 having a C-4 benzyl ether and a C-6 hydroxyl.
Next, attention was focused on the preparation of glycosyl donors 18 and 22 (Scheme 2). Thus, glycosyl acceptor 15 was obtained by regioselective tritylation of the C-6 hydroxyl of mannoside 1343 with trityl chloride and pyridine, followed by acetylation with acetic anhydride and pyridine and removal of the trityl ether by employing FeCl3 · 6H2O in DCM. Glycosylation of 15 with 1644 with NIS/TfOH as the promoter45 gave disaccharide 17 in an excellent yield of 76% as only the α-anomer. The 1-thio-α-mannoside 21 was prepared by treatment of 15 with triflic anhydride in the presence of 2,6-lutidine in DCM, followed by displacement of the triflate of the resulting compound 19 with 2,3,4,6-tetra-O-acetyl-1-thio-α-d-mannose, which was prepared by in situ S-deacetylation of 2046 by diethylamine in DMF.
The requisite trichloroacetimidates 18 and 22 were obtained by hydrolysis of the trimethylsilyl ethyl glycosides of 17 and 21, respectively, by use of trifluoracetic acid in DCM followed by treatment of the resulting lactols with trichloroacetonitrile in the presence of DBU.47,48
A TMSOTf-promoted glycosylation of glycosyl acceptor 12 with glycosyl donors 18 and 22 in DCM gave pentasaccharides 23 and 25, respectively, in excellent yields. Cleavage of the phthalimido group of 23 and 25 was accomplished by treatment with hydrazine in ethanol, which was followed by N-acetylation with acetic anhydride in methanol to afford compounds 24 and 26, respectively. Finally, deprotection of 24 and 26 to give target compounds 4 and 5, respectively, was accomplished by a two-step procedure entailing treatment with NaOMe in methanol to hydrolyze the acetyl esters and Birch reduction to remove the benzyl ethers.
The chemical synthesis of trimannoside 2 was more challenging due to the presence of a 1-thiomannoside linked to a secondary C-3 hydroxyl. Thus, the configuration of the C-3 hydroxyl of 27 was inverted by a two-step procedure involving Swern oxidation to give a ketone, which was immediately reduced with NaBH4 in a mixture of dichloromethane to give taloside 28 as the only diastereoisomer in good yield (Scheme 3). Triflation of the C-3 hydroxyl of 28 with triflic anhydride in the presence of 2,6-lutidine in DCM followed by displacement of the resulting compound with 2,3,4,6-tetra-O-acetyl-1-thio-α-d-mannose, which was prepared by in situ S-deacetylation of 2046 by use of diethylamine in DMF, gave 1-thio-linked 29. The benzylidene ring of 29 was selectively opened by treatment with borane in THF and Bu2BOTf in DCM,42 and the C-6 hydroxyl of the resulting compound 30 was glycosylated with thioglycoside 16 by use of NIS/TfOH as the promoter system to yield trisaccharide 31. Deprotection of 31 to give target compound 2 was easily accomplished by the standard two-step procedure. Compound 1 was easily obtained by a similar deprotection of 29. Finally, tetrasaccharide 3 was prepared by regioselective opening of the benzylidene acetal of 9 followed by glycosylation with trichloroacetimidate 22 and deprotection by standard procedures (for details see Supporting Information).
Scheme 3a.
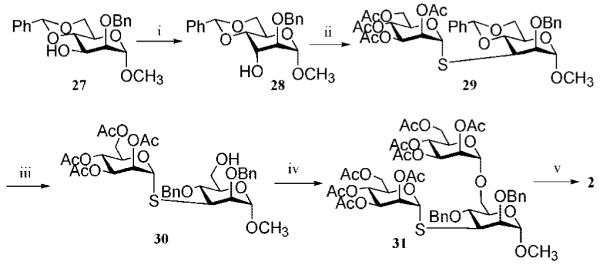
a Reagents and conditions: (i) DMSO, 1:2 Ac2O/DMSO, and then NaBH4, 1:1 DCM/MeOH (75%); (ii) Tf2O, 1:2 pyridine/DCM, and then 20, DMF, diethylamine, 0 °C (61%); (iii) BH3 in THF, Bu2BOTf (63%); (iv) 16, NIS, TfOH, DCM, 0 °C (77%); (v) NaOMe, MeOH, and then Na/NH3(l), −78 °C (70%).
Crystallography
The crystallographic results of 10 complexes of Drosophila GMII are reported here to resolutions between 2.03 and 1.10 Å, with Rfree between 20% and 15.5%. The statistics for the refinement and PDB codes are listed in the Supporting Information.
Initially, we attempted to determine the structure of noncleavable 1-thiomannosides 1–4 bound in the active side of native dGMII. X-ray crystallographic studies were carried out on crystals grown in the presence of the analogues (cocrystals) or soaked into phosphate-washed crystals. The washing step removes bound Tris from the active site49 and previously led to improvements in the visualization of weakly binding compounds.49,50 However, after analysis of electron densities from more than 10 different crystals prepared under various conditions, no evidence was found of an oligosaccharide in complex with the native enzyme.
Preparation and Crystallographic Analysis of D204A Mutant
The catalytic nucleophile of Drosophila GMII was previously identified by trapping of a covalently linked intermediate at aspartate 204 (in the residue numbering of the Drosophila construct used for crystal studies).32 Therefore, a mutant in the catalytic nucleophile Asp204 in which the aspartate was changed to an alanine (D204A) was prepared as an alternative tool for visualizing bound substrates. The D204A protein could be purified in an identical manner to the wild-type enzyme, in similar yields and with nearly identical crystallization properties. The crystals formed by D204A gave the same space group (P212121) and crystal cell dimensions as the native dGMII, yielding comparable high-resolution diffraction (to 1.1 Å) (Supporting Information, Table 1S).
The absence of the catalytic aspartate changed the environment of the active site. One of the principal changes is that the active-site zinc, which normally coordinates with four amino acid side chains (Asp204, His90, Asp92, and His471), now only coordinates with three side chains and shifts approximately 0.3 Å toward Asp92. Second, Arg228, which normally interacts with Asp204, shows more freedom of movement and a new alternate conformation becomes visible. Third, Trp95, which forms the cap of the cleavage pocket, moves upward, generating a slightly more open pocket. The arrangement of solvent waters within the empty active-site pocket differs between the native and mutant GMII, and Tris also binds differently in the active site of the mutant enzyme (Supporting Information).
Crystallography of dGMII Mutants with Mannose
Initially, the D204A nucleophile mutant as well as the previously prepared acid–base catalyst mutant D341N32 was employed to trap the artificial substrate 2,4-dinitrophenyl α-d-mannopyranoside. There is a slight amount of residual mannosidase activity in these mutants, which was evident as the crystals turned yellow, indicating substrate cleavage. As a result the uncleaved substrate could not be visualized; however, the electron density for mannose was visible in the active site (−1 site) of each mutant (Figure 2).
Figure 2.
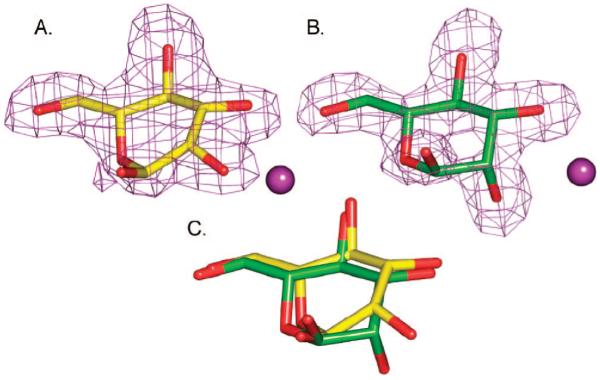
Binding of mannose in the active site of (A) D341A dGMII [PDB code 3BUP] or (B) D204A dGMII [PDB code 3BUQ]. Fo – Fc omit electron density maps are shown contoured at 3σ (0.2 e/Å3). A magenta ball represents the active-site zinc. (C) Superposition of the bound mannosides. Superposition was based on the protein atoms, and the mannose was extracted from the fit structures. Mannose in the active site of D341N (yellow) is in a high-energy boat conformation, while in D204A (green) it is in a 4C1 chair conformation.
In the acid–base mutant (D341N) containing natural Asp204, the ring was found in what is best modeled as a distorted high-energy B2,5 conformation (Figure 2A), which is necessary for it to fit in the confined space of the binding pocket. It should be noted, however, that the fitting is not unambiguous, and a distorted 0S2 skew-boat or 0H5 half-boat could also be accommodated reasonably well. The high-energy 1,4B boat conformation with equatorial hydroxyls has been previously noted for inhibitors such as kifunensine,49 ghavamiol,50 or mannonoeuromycin51 bound to the wild-type enzyme, while the 1S5 conformation was observed in the trapped covalent intemediate.32 All these complexes lie on the same conformational interconversion itinerary from 4C1 (i.e., 4C1 ↔ 0H5 ↔ 0S2 ↔ B2,5 ↔ 1S5 ↔ 1,4B).
Interestingly, mannose complexed in the nucleophile mutant (D204A) adopted the low-energy 4C1 conformation (Figure 2B), previously unobserved for dGMII binding. The change in conformation of zinc-bound mannose is a reflection of the difference in the environment around the zinc in the D204A mutant. Superposing the two structures and extracting the bound mannose indicates that the position of the hydroxyls other than O2 also differ slightly. (Figure 2C). While D204A can bind some compounds in its active site (albeit in a nonnative conformation), we have failed to find evidence of binding of a number of inhibitors, including the nanomolar inhibitor swainsonine.
Crystallography of D204A dGMII with Thioglycoside-Containing Oligosaccharides
Successful visualization of the oligosaccharides in the active-site pocket required extended soaking (16-24 h) of the synthetic oligosaccharides with phosphate or MOPS-washed D204A crystals. As will be discussed below, the thio-linked mannosyl moiety of compounds 1–4 was not well tolerated by the active site of the mannosidase, and when possible the binding mode was altered such that this moiety was complexed in another subsite of the enzyme. The latter feature is best illustrated in the complex of dGMII with compound 2.
The D204A/2 crystal diffracted to high resolution (1.12 Å with R and Rfree of 11.4% and 15.5%, respectively) and the density was exceptionally clear for the complete trisaccharide (Figure 3A). The α(1,6)-O-linked mannosyl residue of compounds 2 was complexed in the active site (−1) and the α(1,3)-S-linked mannosyl moiety was bound in a newly identified pocket, which is designated as the +2′ site (to distinguish it from the +2, +3, and +4 sites, which bind with the other branch of the natural substrate). The B-factors for the trisaccharide were comparable along its length (average of 9.9 for the zinc-associated −1 mannoside and 16.4 and 15.7 for the mannosides in the +1 and +2′ sites, respectively). Furthermore, the sugar rings of 2 adopted low-energy 4C1 conformations; however, there was a slight twist in the C2, C3, C5, O5 plane of the mannoside in the −1 site.
Figure 3.

Binding of 2 and 4 to D204A: stereoviews (divergent) of (A) 2 bound in Fo – Fc omit map contoured at 4σ (0.38 e/Å3) or (B) 4 contoured at 1.6σ (0.15 e/Å3) to visualize density in the +1 and +2 positions. The MPD displaced by GlcNAc binding is shown in panel A. The orientation in both panels is the same to illustrate the differences in position of each compound. (C, D) Interactions less than 3.2 Å are shown for (C) 2 or (D) 3. Distances are given in angstroms. Interacting waters are shown as orange spheres. Zinc is represented as a magenta sphere.
The trisaccharide 2 makes extensive water contacts (a total of 10; Figure 3C). The +2′ site is formed primarily by Arg343, Asp340, and Asp341, which make hydrogen bonds with the O3 and O4 of the α(1,3)-S-linked mannoside of 2. Furthermore, the O3 is at the apex of an interaction triangle consisting of the Nε of Arg343, the carbonyl oxygen of Asp341, and a water molecule (Wat2343 in PDB structure 3BVU). Water 2343 in turn interacts with the terminal amino group of Lys288. The O4 lies equidistant between the NH2 nitrogen of Arg343 and the Oδ2 oxygen of Asp340 (2.8 Å). The O6 of the sugar bound in the +2′ site is found in two almost equally populated conformations, making contacts in either conformation with two water molecules. One of the A-conformation interacting waters makes contact in turn with the Ser926 backbone amine.
Apart from a hydrogen bond between the O4 and Asp341 Oδ2 (2.6 Å), the interactions with the central mannoside (+1 site or swivel position) are primarily with loosely bound (high B-factors) water molecules, which are not directly attached to the amino acid backbone. Only O2 of the swivel residue makes a hydrogen bond to a well-defined water molecule (Wat2012), which in turn hydrogen-bonds to the Oδ1 of Asp340.
The complex with disaccharide 1 (Supporting Information) demonstrates that a thio-linked mannoside can be complexed in the active site (−1) of the enzyme. In this case, the position of the S-linked mannoside complexed to zinc in the −1 site was clearly defined. In contrast, the poor quality of the density for the mannoside in the +1 site and its higher B-factors (average of 27.7 versus 10.9 for the mannoside in the −1 site) indicates that it is loosely bound and probably somewhat mobile.
The cocrystal structures with oligosaccharides 3 and 4 further support the notion that 1-thiomannosides are not well tolerated by the catalytic site of dGMII. The structural details of these complexes will be discussed in the next section because they also provide evidence of the importance of the GlcNAc moiety of compound 5 and the natural substrate as an anchoring residue.
Structure of Cocomplex of D204A with Pentasaccharide 5 and Comparison with the Mode of Binding of Thio-Linked Oligosaccharides 3 and 4
In view of the difficulties encountered in visualizing the thio-linked oligosaccharides in the active site of D204A, the oxygen-containing derivative 5 was synthesized. The high-resolution structure (1.10 Å) of crystals soaked with 5 showed density for the complete pentasaccharide, with the density for the swivel residue (+1 site) being somewhat less defined but assignable. The density is shown in Figure 3B, and a schematic illustration of the protein and water contacts is shown in Figure 3D. The interactions made by the mannoside in the −1 site are almost identical to those observed in the other structures and involve primarily amino acid contacts and tight complexation to the zinc. The three central mannosyl residues of 5 in the +1, +2, and +3 sites make only a few direct hydrogen bonds with the protein, and most interactions are by water contact. Thus, the O4 of the mannoside in the +1 site interacts with Asp341 Oδ2 and the O4 of the mannoside in the +2 site interacts with Tyr267 OH. Furthermore, three amino acids (Asp340, Arg410, and Glu875) form water-mediated hydrogen bonds with the mannosides in the +1, +2, and +3 sites.
The GlcNAc residue of 5 is accommodated in an important binding site (+4), and as expected for its role as an anchor residue, it makes extensive interactions with the protein. There is a direct hydrogen bond between His273 and O3 of the GlcNAc moiety as well as three single water molecule-mediated hydrogen bonds to the protein. Furthermore, an important stacking interaction is present between the Tyr267 ring and GlcNAc ring.
The GlcNAc binding site has never been observed to be unoccupied,24,32,49,53 containing either 2-methylpentane-2,4-diol (MPD, Figure 3A) or glycerol depending on the cryocondition used. The binding of free N-acetylglucosamine to this site has also been observed in crystals of dGMII soaked with this compound (D. Kuntz, unpublished observation). Previously, we proposed that the non-reducing-terminal GlcNAc of the natural substrate GlcNAcMan5GlcNAc2-Asn would bind in this location, and the data presented here support this proposal.52
The crystal structures of compounds 3 and 4 with D204A provide further support of the importance of the GlcNAc moiety as an anchoring residue. Thus, the 1-thiomannosyl and GlcNAc residues of 4 are accommodated in the −1 and +4 sites, respectively, and superimpose with similar residues of 5 (Supporting Information). The middle mannosides, however, diverge and the density for the mannoside in the +1 site was ill-defined despite complete redundant data and the highly refined nature of the model. This lack of density most likely results from the unfavorable nature of complexation of a thiomannoside and the very few interactions made by this residue with the protein. The latter is a reflection of the role of the mannoside in the +1 site as a swivel residue.
The tetrasaccharide 3 has a similar structure as 4 except that it does not contain a GlcNAc moiety (Figure 1). Interestingly, the lack of the latter residue resulted in a completely different mode of complexation (Figure 4). Thus, the electron density of the soaked crystal structure does not show the thio-linked mannoside and only a trimannoside moiety could be visualized. Although the density of the trimannoside is well-defined (Supporting Information), the modeling was consistent with an occupancy of 70%. In a second crystallization attempt, under slightly different conditions, the occupancy was less than 10%, and thus the affinity of 3 for D204A appears quite low. The α(1,3)-O-linked and α(1,6)-O-linked mannosyl residues of 3 are complexed in the active site (−1) and the newly identified +2′ site, respectively. A superimposition of 2 with 3 (Figure 5) demonstrates that the mannosides in the −1 and +2′ subsites are in almost identical positions and make similar types of interactions. However, the central swivel mannosides (+1), which are α(1,3)-linked mannoside in 3 and α(1,6)-linked mannoside in 2, are less clearly resolved and located in different positions. In particular, the interactions made between the protein and the mannoside of 3 in the +1 site differ from those of the complexes with 1 and 2: O4 interacts with the presumptive acid–base catalyst Asp341 Oδ2 (2.3 Å), while a close contact (2.9 Å) is formed between the exocyclic oxygen of the α(1,3)-glycosidic linkage and Oδ2 of Asp341. The O2 interaction of the residue in the +1 site to a tightly bound water seen in the complex with 2 is no longer present. Instead, it interacts with the Arg876 backbone carbonyl, which in turn hydrogen-bonds with the O6 of the mannoside in the −1 site.
Figure 4.
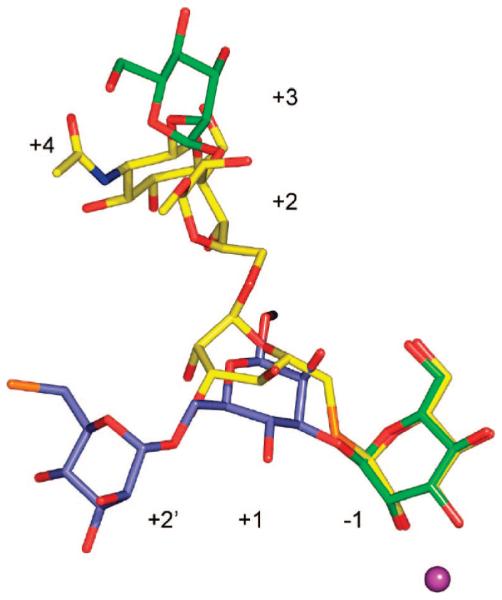
The presence of GlcNAc causes a radical rearrangement of oligosaccharides bound to Golgi mannosidase II. Shown is the binding of 3 (blue, from PDB structure 3BVV) and GlcNAc-modified 4 (yellow, from PDB structure 3BVW). For orientation purposes, one of the mannoside residues is colored green. This mannoside binds in the −1 site of D204A/3 complex and in the +3 site of the D204A/4 complex. The active-site zinc is colored magenta. Although 3 is a tetrasaccharide, only three sugars could be assigned to the electron density.
Figure 5.
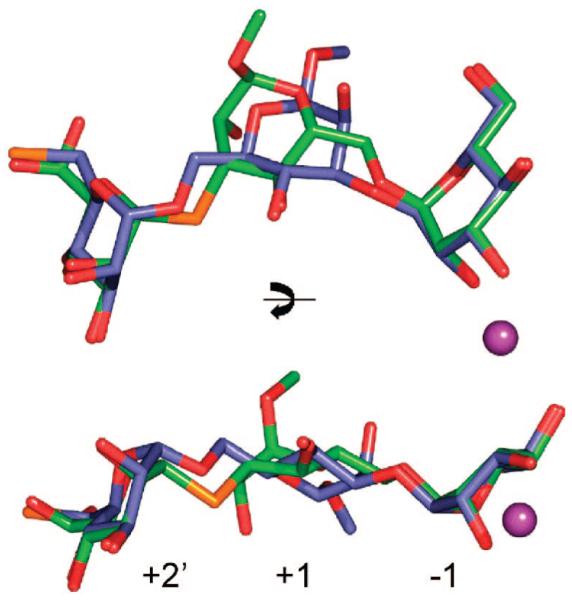
Comparison of binding of 2 (green) and 3 (slate) to D204A. Compound 2 has an α(1,6)-linked mannoside while 3 has an α(1,3)-linked mannoside bound in the −1 site. Compound 3 is a tetramannoside but only three mannosides are visible in the electron density; the terminal thio-linked mannose cannot be assigned. The thio bonds are colored orange, and zinc in the active site is represented by a magenta ball.
Enzymology
To establish the importance of the GlcNAc binding subsite for hydrolysis, time course studies were first performed with pyridylamine- (PA-) tagged derivatives of the natural substrates GlcNAcMan5GlcNAc2-PA and Man5GlcNAc2-PA. In addition, comparative assays were performed with human lysosomal mannosidase (hLM), a GH38 enzyme with sequence and structural similarity to dGMII.
To determine relative activities of both enzymes, experiments were carried out with 4-methylumbelliferyl α-mannoside (4MU-α-Man) as substrate. This information was employed in time course studies, which demonstrated rapid cleavage of GlcNAc-Man5GlcNAc2-PA by dGMII through a GlcNAcMan4GlcNAc2-PA intermediate to produce GlcNAcMan3GlcNAc2-PA, similar to the established substrate specificity for the equivalent mammalian enzyme. In sharp contrast, dGMII cleaved Man5GlcNAc2-PA >80-fold more slowly (Figure 6).
Figure 6.
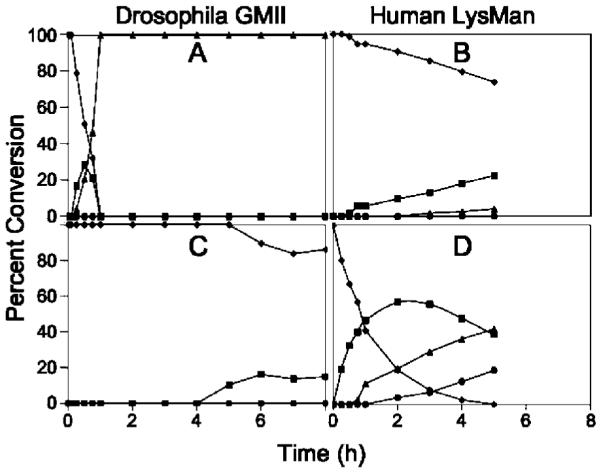
In Vitro digestion time course of Man5GlcNAc2-PA and GlcNAcMan5GlcNAc2-PA by dGMII and hLM. Purified recombinant dGMII (A, C) and hLM (B, D) were used in digestion time course studies with GlcNAcMan5GlcNAc2-PA (A, B) or Man5GlcNAc2-PA (C, D) as substrates. Cleavage of the substrates (GlcNAcMan5GlcNAc2-PA or Man5GlcNAc2-PA, ◆) to smaller glycan structures (GlcNAcMan4GlcNAc2-PA or Man4GlcNAc3-PA, ■; GlcNAcMan3GlcNAc2-PA or Man3GlcNAc3-PA, ▴; GlcNAcMan2GlcNAc2-PA or Man2GlcNAc3-PA, ●) were quantitated by HPLC. dGMII cleaved GlcNAcMan5GlcNAc2-PA ~80-fold faster than Man5GlcNAc2-PA at equivalent enzyme concentrations. Minimal digestion of Man5GlcNAc2-PA or GlcNAcMan5GlcNAc2-PA by hLM was detected when equivalent enzyme activity units (based on 4MU-α-Man activity) of hLM and dGMII were employed (not shown). Increasing the enzyme concentration of hLM in the in vitro assays by 100-fold (B, D) resulted in detectable cleavage of Man5GlcNAc2-PA, but cleavage of GlcNAcMan5GlcNAc2-PA remained ~16-fold slower.
Insufficient quantities of the natural substrates prevented detailed Km analyses, but the time course data clearly indicate that the non-reducing-terminal GlcNAc residue confers additional binding affinity and substrate preference for the GlcNAcMan5GlcNAc2-Asn glycan processing intermediate. hLM cleaved Man5GlcNAc2-PA at a similar rate (based on equivalent 4MU-α-Man hydrolytic activity) as dGMII, while cleavage of GlcNAcMan5GlcNAc2-PA was undetectable at equivalent enzyme concentrations (data not shown). When the concentration of hLM was increased 100-fold, cleavage of Man5GlcNAc2-PA to smaller structures was clearly evident, but cleavage of GlcNAcMan5GlcNAc2-PA by hLM was ~16-fold slower (Figure 6). These results indicate that the non-reducing-terminal GlcNAc in the GlcNAcMan5GlcNAc2-PA substrate enhances the rate of glycan cleavage by dGMII, whereas the same residue is inhibitory for glycan cleavage by hLM.
The X-ray crystallographic studies indicate that the active site of dGMII poorly recognized thio-linked mannosides. To support this observation, the rate of hydrolysis of different concentrations of 4MU-α-Man alone and in the presence of different concentrations of compounds 3 or 4 was measured fluorometrically, and Ki values and IC50 values were estimated to be larger than 5 mM, confirming that the thio derivatives are poorly recognized by dGMII.
Discussion
The crystal structures of various synthetic oligosaccharides complexed to Golgi α-mannosidase II containing a mutation of the active-site nucleophile Asp204 define two previously unidentified carbohydrate-binding subsites and provide a model for natural substrate recognition. The binding cleft of Golgi mannosidase II consists of a long solvent-accessible groove with a buried catalytic pocket, the −1 site. A defining feature of the −1 site is a tightly bound zinc, which interacts with a mannoside with the strongest coordination through the C-2 hydroxyl. There is an unusual architecture of the protein backbone around the −1 pocket, as Trp95, which can be considered a flap residue, and the 204 residue, whether catalytic aspartate or mutant alanine, always appears as outliers in Ramachandran plot analyses. Access to the −1 site is somewhat restricted as the pocket is covered by Trp95. However, Trp95 demonstrates a degree of mobility in the various structures and can reposition itself to maintain stacking interactions with bound carbohydrates or inhibitors. The incoming substrate’s approach path to the zinc is further restricted by the surrounding amino acid side chains. Previous structures with a number of inhibitors,24,32,50,51 as well as the structure of mannose bound to D341N presented here, indicate that binding typically occurs in a high-energy distorted B2,5 conformation.
In the case of the D204A nucleophile mutant studied here, changing the aspartate to the smaller-sized alanine allows more free space in the cleavage pocket and a more unrestricted approach to the zinc. As a result, the sugars bound in the −1 pocket of D204A retained a low-energy 4C1 conformation. An overlay of the bound structure of compound 5 onto the native enzyme indicates that a spatial clash between Asp204 Oδ2 and O2 would occur if the −1 mannoside was bound in a 4C1 conformation (Figure 7). In native structures of dGMII, Asp204 does not show positional flexibility because Oδ1 is tightly associated with the zinc (2.1–2.3 Å in various native structures) and Oδ2 makes close contacts with the N∊1 and NH2 of Arg228. Thus, the data presented here indicate that sugar distortion is being imposed by the spatial restraints of the cleavage pocket. This distortion is further assisted by the formation of strong hydrogen bonds to Asp204 when mannose is bound in the high-energy conformation. Conformational distortion driven by steric constraints has been demonstrated previously. For example, an X-ray crystal structure of an inverting endoglucanase with a nonhydrolyzable substrate showed the glucosyl unit in the catalytic binding site adopting a 2S0 conformation.35 It appears that steric hindrance by Tyr73 forces the glucoside into the distorted conformation, as mutation of Tyr73 to a less bulky serine gives an enzyme to which the glucoside binds in a low-energy 4C1 conformation. Thus, steric hindrance imposed by the side chain of a catalytic residue or other amino acid in the binding pocket of a glycosidase, in conjunction with favorable interactions elsewhere, can drive a conformational change required for glycosidic bond cleavage.
Figure 7.
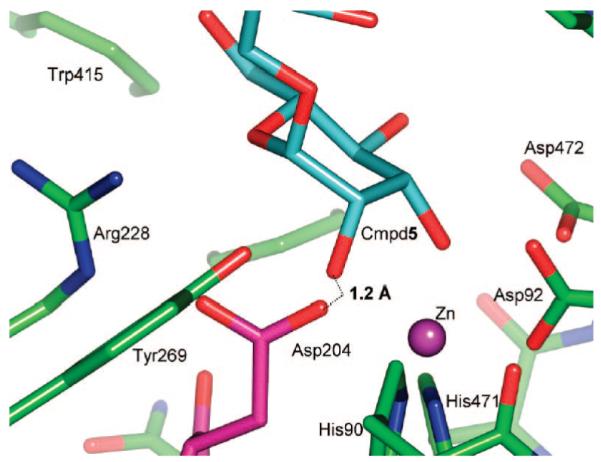
Spatial clash between Asp204 and sugar bound in the low-energy 4C1 conformation. Coordinates for the D204A/5 complex (PDB code 3BVX) were superimposed with those of the unliganded native enzyme (PDB code 3BVT). If the pentasaccharide bound to the native enzyme in an identical manner to which it binds to D204A, the distance between the Asp204 Oδ2 and O2 of 5 would be only 1.2 Å. The pentasaccharide is colored in cyan, while amino acid side chains of the native protein are shown in green, except for Asp204, which is highlighted by coloring it magenta.
The present study focused on the identification of additional sugar binding subsites within the binding cleft of dGMII in support of the unique substrate specificity for this enzyme. The mode of binding of the synthetic oligosaccharides confirmed the presence of two previously suspected sugar-binding subsites and provide a model for substrate cleavage and product release. On the basis of the presence of bound 2-methylpentane-2,4-diol or glycerol in the crystal structure of dGMII, we previously proposed a GlcNAc binding site at one end of the binding groove, which would anchor the natural substrate GlcNAcMan5GlcNAc2-Asn into the groove.52 The intact substrate can be thought of as a Y, where each arm of the Y is cleaved off in successive reactions. The arms of the Y are made up of respectively the α(1,6)-linked (initially bound in the −1 site) and α(1,3)-linked (initially bound in the +2′ site) non-reducing-end mannosides. The intervening mannoside residue at the base of the Y (+1 site) swivels to reposition the arms between the two cleavage steps. To fulfill its function as a swivel residue, the mannoside in the +1 site must be loosely associated with the enzyme. The oligosaccharide chain would remain tethered in the binding site during the rotational event due to the presence of the GlcNAc anchor, but the product would be released, as binding sites are lost due to the cleavage events.
The complexes of D204A with compounds 2 and 5 best define the +2′ and +4 carbohydrate binding sites, respectively, while the other complexes provide support for the findings as well as providing a clear indication of the positional mobility of the swivel mannoside (Figure 8). Binding at the +2′ site is primarily facilitated by direct contacts to Arg343, Asp340, and Asp341, while Lys288 and Ser296 are also involved through intermediary waters. The GlcNAc (+4) binding site involves a direct hydrogen-bond interaction with His273, and a tightly bound water-mediated contact with the Val61–Gln64 main chain. The tight binding of the GlcNAc is primarily due to a ring-stacking interaction between the GlcNAc ring and Tyr267. The importance of the GlcNAc binding site for anchoring the full-length GMII substrate is clearly shown in the complexes with compounds 3 and 4, which adopt completely different binding positions (e.g., the sugar formerly in the −1 site of the D204A/3 complex is now bound in the +3 site of the D204A/4 complex, the +1 mannose is in the +2 site, the +2′ sugar is now in the +1 site, and the previously undefined thio-linked sugar is in the −1 site) (Figure 4).
Figure 8.
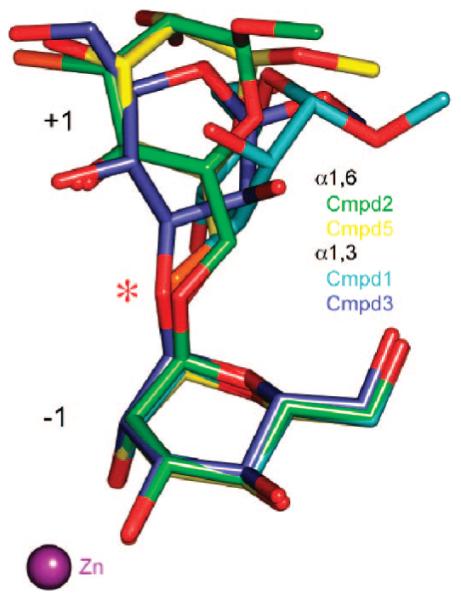
The swivel sugar is observed in a large number of positions. The two mannosides closest to the active-site zinc are shown for the α(1,6)-linked compounds 2 (green) and 5 (yellow) and for the α(1,3)-linked compounds 1 (cyan) and 3 (slate). In all cases, the position of the zinc-bound −1 mannoside is almost invariant, whereas the mannoside in the +1 site, which is described as the swivel residue, is highly variable. The oxygen at the cleavage site, indicated by an asterisk, is in almost the same position in all complexes.
Studies on mammalian GMII have demonstrated the requirement for the prior action of N-acetylglucosaminyltransferase I (GnT I) to add a non-reducing-terminal GlcNAc residue to the core α(1,3)mannosyl residue prior to cleavage of the non-reducing-terminal α(1,3)- and α(1,6)-mannosyl residues by GMII.34 These results are extended here for the homologous Drosophila GMII, which demonstrates a ~80-fold preference for cleavage of GlcNAcMan5GlcNAc2 over structures lacking the terminal GlcNAc residue. The structural basis for this specificity, first suggested through the observed interaction of cryoprotectant molecules, is confirmed in the present studies with the description of the anchor site. Recently, the structure of dGMII D204A with the natural substrate has confirmed that the observations reported here on synthetic compounds are relevant to the true substrate as well (N. Shah, D. A. Kuntz, and D. R. Rose, unpublished results).
Time course studies presented here, examining the cleavage of glycan substrates by the catabolic GH38 hydrolase hLM (an enzyme with sequence and structural similarity to GMII) indicate that hLM prefers oligomannoside substrates lacking the terminal GlcNAc residue, a finding that is consistent with the absence of the extended GlcNAc binding subsite in bovine LM.53
The observation that the wild-type dGMII does not accommodate thio-substituted glycosides remains an important but unresolved question. Evidently, there are some chemical characteristics of the catalytic site, the thioglycosidic linkage, or both that preclude binding. Possibly the distortion of the substrate that occurs on binding, mediated by the nucleophile Asp204, is not achievable in the substrate analogues, because of either increased rigidity or a greater repulsion of the S-substituent in the binding region.
The data presented here extend the focus for selective GMII inhibitor design through the identification of a GlcNAc binding site that is open, accessible, and amenable to virtual and experimental inhibitor screening as a potential route to identifying selective antimetastatic agents without the secondary complications from lysosomal storage disorders.
Supplementary Material
Acknowledgment
Funding was provided by National Institutes of Health Grants 5P41RR05351-18 (G.-J.B. and K.W.M.) and GM47533 (K.W.M.) and by the Canadian Institutes of Health Research (D.A.K. and D.R.R.). We thank Ruth-Ann Stewart and Tara Signorelli for help with mutagenesis and preparation of D204A, and Tamika Hamlet and Kayla Shea for crystal screening. This work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation under Award DMR-0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by Award RR-01646 from the National Institutes of Health, through its National Center for Research Resources.
Footnotes
Supporting Information Available: Experimental procedures and analytical data for all new compounds; crystallization conditions, data collection and refinement statistics, and refinement protocols for Drosophila Golgi mannosidase II complexes; and detailed structural information for dGMII/water, D204A/water, D204A/Tris, D204A/1, D204A/2, and D204A/4 complexes. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Hakomori S. Cancer Res. 1996;56:5309–5318. [PubMed] [Google Scholar]
- (2).Livingston PO. Immunol. Rev. 1995;145:147–166. doi: 10.1111/j.1600-065x.1995.tb00080.x. [DOI] [PubMed] [Google Scholar]
- (3).Yu LG. Glycoconjugate J. 2007;24:411–420. doi: 10.1007/s10719-007-9034-3. [DOI] [PubMed] [Google Scholar]
- (4).Demetriou M, Nabi IR, Coppolino M, Dedhar S, Dennis JW. J. Cell Biol. 1995;130:383–392. doi: 10.1083/jcb.130.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dennis JW, Laferte S, Waghorne C, Breitman ML, Kerbel RS. Science. 1987;236:582–585. doi: 10.1126/science.2953071. [DOI] [PubMed] [Google Scholar]
- (6).Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Nat. Med. 2000;6:306–312. doi: 10.1038/73163. [DOI] [PubMed] [Google Scholar]
- (7).Lu Y, Pelling JC, Chaney WG. Clin. Exp. Metastasis. 1994;12:47–54. doi: 10.1007/BF01784333. [DOI] [PubMed] [Google Scholar]
- (8).Pierce M, Arango J. J. Biol. Chem. 1986;261:772–777. [PubMed] [Google Scholar]
- (9).Seberger PJ, Chaney WG. Glycobiology. 1999;9:235–241. doi: 10.1093/glycob/9.3.235. [DOI] [PubMed] [Google Scholar]
- (10).Seberger PJ, Scholar EM, Kelsey L, Chaney WG, Talmadge JE. Clin. Exp. Metastasis. 1999;17:437–444. doi: 10.1023/a:1006629119030. [DOI] [PubMed] [Google Scholar]
- (11).Yamashita K, Tachibana Y, Ohkura T, Kobata AJ. Biol. Chem. 1985;260:3963–3969. [PubMed] [Google Scholar]
- (12).Cummings RD, Trowbridge IS, Kornfeld S. J. Biol. Chem. 1982;257:3421–3427. [PubMed] [Google Scholar]
- (13).Dennis JW, Pawling J, Cheung P, Partridge E, Demetriou M. Biochim. Biophys. Acta. 2002;1573:414–422. doi: 10.1016/s0304-4165(02)00411-7. [DOI] [PubMed] [Google Scholar]
- (14).Compain P, Martin OR. Curr. Topics Med. Chem. 2003;3:541–560. doi: 10.2174/1568026033452474. [DOI] [PubMed] [Google Scholar]
- (15).Walker-Nasir E, Ahmad I, Saleem M, Hoessli DC. Curr. Org. Chem. 2007;11:591–607. [Google Scholar]
- (16).Zou W. Curr. Top. Med. Chem. 2005;5:1363–1391. doi: 10.2174/156802605774642999. [DOI] [PubMed] [Google Scholar]
- (17).Asano N, Nash RJ, Molyneux RJ, Fleet GW. J. Tetrahedron: Asymmetry. 2000;11:1645–1680. [Google Scholar]
- (18).Goss PE, Baker MA, Carver JP, Dennis JW. Clin. Cancer Res. 1995;1:935–944. [PubMed] [Google Scholar]
- (19).Michael JP. Nat. Prod. Rep. 2007;24:191–222. doi: 10.1039/b509525p. [DOI] [PubMed] [Google Scholar]
- (20).Moremen KW. Biochim. Biophys. Acta. 2002;1573:225–235. doi: 10.1016/s0304-4165(02)00388-4. [DOI] [PubMed] [Google Scholar]
- (21).Bowen D, Adir J, White SL, Bowen CD, Matsumoto K, Olden K. Anticancer Res. 1993;13:841–844. [PubMed] [Google Scholar]
- (22).Elbein AD, Solf R, Dorling PR, Vosbeck K. Proc. Natl. Acad. Sci. U.S.A. 1981;78:7393–7397. doi: 10.1073/pnas.78.12.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Goss PE, Baptiste J, Fernandes B, Baker M, Dennis JW. Cancer Res. 1994;54:1450–1457. [PubMed] [Google Scholar]
- (24).Kawatkar SP, Kuntz DA, Woods RJ, Rose DR, Boons GJ. J. Am. Chem. Soc. 2006;128:8310–8319. doi: 10.1021/ja061216p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li B, Kawatkar SP, George S, Strachan H, Woods RJ, Siriwardena A, Moremen KW, Boons GJ. ChemBioChem. 2004;5:1220–1227. doi: 10.1002/cbic.200300842. [DOI] [PubMed] [Google Scholar]
- (26).Vasella A, Davies GJ, Bohm M. Curr. Opin. Chem. Biol. 2002;6:619–629. doi: 10.1016/s1367-5931(02)00380-0. [DOI] [PubMed] [Google Scholar]
- (27).Zechel DL, Withers SG. Curr. Opin. Chem. Biol. 2001;5:643–649. doi: 10.1016/s1367-5931(01)00260-5. [DOI] [PubMed] [Google Scholar]
- (28).Heightman TD, Vasella AT. Angew. Chem., Int. Ed. 1999;38:750–770. doi: 10.1002/(SICI)1521-3773(19990315)38:6<750::AID-ANIE750>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- (29).Sinnott ML. In: Enzyme Mechanisms. Page MI, Williams A, editors. Royal Society of Chemistry; London: 1987. pp. 259–297. [Google Scholar]
- (30).Davies GJ, Sinnott ML, Withers SG. In: ComprehensiVe Biological Catalysis. Sinnott ML, editor. Vol. 1. Academic Press; London: 1997. pp. 119–209. [Google Scholar]
- (31).Davies GJ, Ducros VMA, Varrot A, Zechel DL. Biochem. Soc. Trans. 2003;31:523–527. doi: 10.1042/bst0310523. [DOI] [PubMed] [Google Scholar]
- (32).Numao S, Kuntz DA, Withers SG, Rose DR. J. Biol. Chem. 2003;278:48074–48083. doi: 10.1074/jbc.M309249200. [DOI] [PubMed] [Google Scholar]
- (33).Ducros VMA, Zechel DL, Murshudov GN, Gilbert HJ, Szabo L, Stoll D, Withers SG, Davies GJ. Angew. Chem., Int. Ed. 2002;41:2824–2827. doi: 10.1002/1521-3773(20020802)41:15<2824::AID-ANIE2824>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- (34).Harpaz N, Schachter H. J. Biol. Chem. 1980;255:4894–4902. [PubMed] [Google Scholar]
- (35).Larsson AM, Bergfors T, Dultz E, Irwin DC, Roos A, Driguez H, Wilson DB, Jones TA. Biochemistry. 2005;44:12915–12922. doi: 10.1021/bi0506730. [DOI] [PubMed] [Google Scholar]
- (36).Pachamuthu K, Schmidt RR. Chem. Rev. 2006;106:160–187. doi: 10.1021/cr040660c. [DOI] [PubMed] [Google Scholar]
- (37).Witczak ZJ. Curr. Med. Chem. 1999;6:165–178. [PubMed] [Google Scholar]
- (38).Cirla A, McHale AR, Mann J. Tetrahedron. 2004;60:4019–4029. [Google Scholar]
- (39).Misra AK, Roy N. Carbohydr. Res. 1995;278:103–111. doi: 10.1016/0008-6215(95)00231-x. [DOI] [PubMed] [Google Scholar]
- (40).Peters T. Liebigs Ann. Chem. 1991:135–141. [Google Scholar]
- (41).Yu HN, Ling CC, Bundle DR. J. Chem. Soc., Perkin Trans. 2001;1:832–837. [Google Scholar]
- (42).Jiang L, Chan TH. Tetrahedron Lett. 1998;39:355–358. [Google Scholar]
- (43).Paulsen H, Pries M, Lorentzen JP. Liebigs Ann. Chem. 1994:389–397. [Google Scholar]
- (44).Weingart R, Schmidt RR. Tetrahedron Lett. 2000;41:8753–8758. [Google Scholar]
- (45).Veeneman GH, van Leeuwen SH, van Boom JH. Tetrahedron Lett. 1990;31:1331–1334. [Google Scholar]
- (46).Hamachi I, Nagase T, Shinkai S. J. Am. Chem. Soc. 2000;122:12065–12066. [Google Scholar]
- (47).Schmidt RR. Angew. Chem., Int. Ed. 1986;25:212–235. [Google Scholar]
- (48).Schmidt RR, Kinzy W. AdV. Carbohydr. Chem. Biochem. 1994;50:21–123. doi: 10.1016/s0065-2318(08)60150-x. [DOI] [PubMed] [Google Scholar]
- (49).Shah N, Kuntz DA, Rose DR. Biochemistry. 2003;42:13812–13816. doi: 10.1021/bi034742r. [DOI] [PubMed] [Google Scholar]
- (50).Kuntz DA, Ghavami A, Johnston BD, Pinto BM, Rose DR. Tetrahedron: Asymmetry. 2005;16:25–32. [Google Scholar]
- (51).Kuntz DA, Liu H, Bols M, Rose DR. Biocatal. Biotransform. 2006;24:55–61. [Google Scholar]
- (52).van den Elsen JMH, Kuntz DA, Rose DR. EMBO J. 2001;20:3008–3017. doi: 10.1093/emboj/20.12.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Heikinheimo P, Helland R, Leiros HKS, Leiros I, Karlsen S, Evjen G, Ravelli R, Schoehn G, Ruigrok R, Tollersrud OK, McSweeney S, Hough E. J. Mol. Biol. 2003;327:631–644. doi: 10.1016/s0022-2836(03)00172-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.