

Abstract
Panitumumab is a fully human monoclonal antibody approved for the treatment of epidermal growth factor receptor (EGFR) positive colorectal cancer. Recently, panitumumab has been radiolabeled with 89Zr and evaluated for its potential to be used as immuno-positron emission tomography (PET) probe for EGFR positive cancers. Interesting preclinical results published by several groups of researchers have prompted us to develop a robust procedure for producing clinical-grade 89Zr-panitumumab as an immuno-PET probe to evaluate EGFR-targeted therapy. In this process, clinical-grade panitumumab is bio-conjugated with desferrioxamine chelate and subsequently radiolabeled with 89Zr resulting in high radiochemical yield (>70%, n=3) and purity (>98%, n=3). All quality control (QC) tests were performed according to United States Pharmacopeia specifications. QC tests showed that 89Zr-panitumumab met all specifications for human injection. Herein, we describe a step-by-step method for the facile synthesis and QC tests of 89Zr-panitumumab for medical use. The entire process of bioconjugation, radiolabeling, and all QC tests will take about 5h. Because the synthesis is fully manual, two rapid, in-process QC tests have been introduced to make the procedure robust and error free.
Keywords: 89Zr-immuno-PET, Clinical production, Panitumumab, PET imaging
Introduction
Epidermal growth factor receptor (EGFR, erb1, HER1) is a glycoprotein belonging to the subclass I of the tyrosine kinase (TK) receptor super family.1 This receptor is dysregulated in a variety of cancers, including lung, colorectal, head and neck, prostate, breast, glioma, pancreatic, and ovarian.2 Overexpression of this receptor is associated with disease progression and treatment resistance. Therefore, EGFR is widely recognized as a promising target for the development of new oncology drugs. Although the large amount of clinical data accumulated over the last few years in EGFR positive cancers dramatically improved understanding of the EGFR's role in solid tumors, the treatment prediction is still an open question in clinical practices.3 The questions are as follows: (i) how do we identify patients who will most likely benefit from treatment with the available targeted drugs and (ii) which of these targeted therapies is most likely to succeed in a given patient? It can be argued that the most efficient approach should avoid traditional large-scale randomized clinical trials and instead adopt the concept of ‘personalized medicine’ by choosing targeted drugs based on an individual's receptor expression levels.4 In order to do so, a suitable method is needed to assess the EGFR status of each patient before the treatment and monitor the effect during the treatment process. At present, invasive methods such as tissue biopsy are used to identify EGFR expression in the primary tumor or metastatic sites in cancer patients. Although these methods are useful for tumor characterizations, repeated biopsy for therapy monitoring and disease management is impractical and unreliable. Moreover, EGFR assessment in ex vivo tumor specimens is still controversial from both the methodological and biological points of view.5 Other techniques for EGFR assessment, such as immunohistochemistry, enzyme-linked immunosorbent assay or Western blotting for protein level quantification, and flourescent in situ hybridization for gene amplification, still need to be validated.6 Essentially, there are no reliable methods available for everyday practice for use in monitoring EGFR status.3A non-invasive, in-vivo identification and quantitative evaluation of EGFR expression may be a novel and powerful tool for assessing this receptor. Functional imaging of tumors via nuclear medicine modalities (positron emission tomography (PET) or single-photon emission computed tomography (SPECT)) allows for non-invasive in vivo visualization of receptor levels with high sensitivity, in the subnanomolar concentration range, as opposed to the millimolar concentration range for other imaging modalities.7 Thus, PET or SPECT along with suitable surrogate radioligands may provide a complementary, noninvasive option for obtaining real-time information and facilitating the selection of patients for EGFR-targeted therapy assessment of response to therapeutic drugs.
On the basis of recent literature,4,8 it is quite challenging to develop EGFR-targeted radiotracers. Many small molecule EGFR TK inhibitors (both reversible and irreversible) including Food and Drug Administration (FDA)-approved therapeutic drugs (erlotinib, gefitinib, and lapatinib) have been radiolabeled in the last few years and preclinically evaluated to determine their suitability for imaging EGFRs. Most of these tracers showed very promising in vitro results for imaging; however, subsequent in vivo studies have been unfavorable. To date, almost all the radiotracers that target EGFR TK (intracellular domain) have shown very low to moderate specific uptake, causing insufficient signal to noise for clinical imaging. In many cases, significant non-specific binding has been observed.8 Imaging EGFRs with radiolabeled anti-EGFR mAbs that bind to the extracellular domain of EGFR have been more successful than small molecular probes (e.g., TK inhibitors), and this imaging is the subject of intense investigation.
The anti-HER1 mAb panitumumab (Vectibix) is a fully human mAb approved by the FDA for the treatment of EGFR-expressing colorectal cancers.9,10 Currently, it is being evaluated in patients with other types of EGFR-expressing cancers, such as breast, lung, head and neck, renal, and ovarian. 89Zr has emerged as a promising positron emitting radionuclide for diagnostic immuno-PET imaging because of its longer half-life (78.4 h), which provides a close match to the biological half-life of an intact mAb.11–14 89Zr can be labeled with mAbs via desferrioxamine (DFO) B chelate (Figure 1) resulting in high radiochemical yield (RCY) and purity.11–18 Recent preclinical studies showed that 89Zr-labeled panitumumab is a promising quantitative PET biomarker of EGFR expression.17,18 89Zr-panitumumab microPET/CT showed very high uptake to EGFR-expressing tumor and correlated strongly with EGFR expression (Figure 2).18 Dosimetry estimates predict a 0.578 mSv/MBq of whole body effective radiation dose, which would allow human imaging with only a low tracer dose of 89Zr-panitumumab.18 Because of its high tumor uptake and high sensitivity of PET technique, the use of 89Zr-panitumumab at low tracer dose is expected to be clinically feasible. 89Zr-immuno-PET may be useful in patient selection and monitoring of EGFR-targeted therapies.
Figure 1.
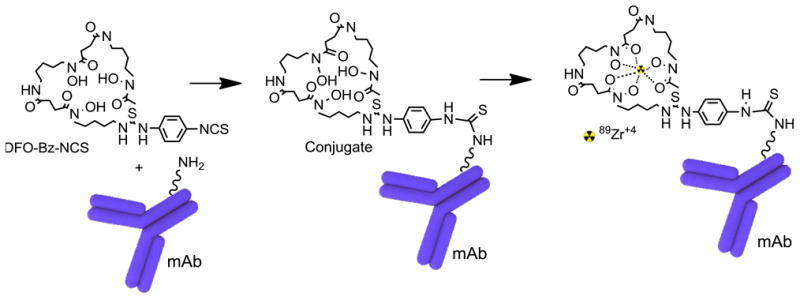
Schematic representation of desferrioxamine (DFO) panitumumab (mAb) conjugation reaction and radiolabeling of DFO-panitumumab conjugate with 89Zr. At basic pH, isothiocynate group of DFO reacts with lysine-NH2 of mAb to form a stable thiourea linkage. 89Zr-oxalate is used to make stable 89Zr-DFO-panitumumab at room temperature.
Figure 2.
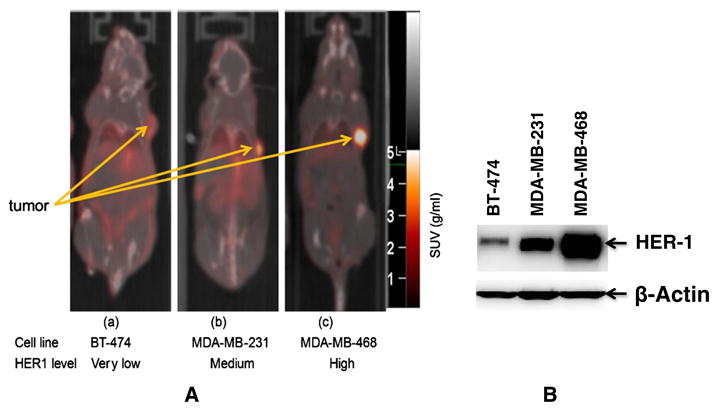
(A) Tumor uptake of 89Zr-panitumumab in various subcutaneous athymic nude female xenograft models. 10.18 ± 1.24 MBq of 89Zr-panitumumab were administered intravenously via tail-vein, and a 5-min CT scan followed by a 30-min static PET scan were performed at 96-h post-injection; (B) Comparative epidermal growth factor receptor (EGFR)-expression level of BT-474, MDA-MB-231, and MDA-MB-468 cell lines by western blot analysis. Cartesian molecular imaging software v5.0.2.30 and data are presented as EGFR/Actin ratio. The images are reprinted with the permission from Elsevier Inc. Bhattacharyya et al., Nucl. Med. Biol. 2013, 40, 451–457.
In our newly established United States Pharmacopeia laboratory (USP, general chapter <823>) at Frederick National Laboratory for Cancer Research, we have developed clinical-grade 89Zr-panitumumab as a surrogate biomarker to support a clinical trial on the evaluation of EGFR-targeted therapy in colorectal cancer patients.19 PET scans using 89Zr-panitumumab before (baseline) and after treatment can predict the binding efficiency of the therapeutic drug to the target.
Recently, the synthesis of 89Zr-panitumumab has been reported by few research groups including ours.17,18,20 However, synthesis and QC procedures described in all of this literature were only for preclinical studies, not human injection. Developing radiometal-based immuno-PET agents that can meet all the criteria specified by the FDA for human injection is quite challenging. Herein, we report a stepwise ∼5-h production procedure of the clinical-grade 89Zr-panitumumab, starting from clinical-grade panitumumab, which resulted in high RCY and purity. QC data from three qualification runs are provided to show that this is indeed a clinical-grade product for human administration. Unless otherwise stated, the whole process is carried out following the instructions of USP general chapter <823>for PET radiopharmaceuticals.
Materials and methods
Reagents, supplies, and equipment
Each reagent or supply has a unique internal specification code of three characters and a unique sequential number, for each lot for each specification code. When an item is ordered, it is given a unique internal tracking number for the reagent or supply. The specification sheets list all of the reagent and supply manufacturers addresses and contact information, including any pertinent certificate of analysis or certificate of quality criteria that these reagents/supplies must meet prior to their release for further use. Any additional testing beyond what may be listed on the certificate of analysis or certificate of quality is described and documented properly.
Clinical-grade panitumumab vials (100 mg/5 mL) manufactured by Amgen, Inc., Thousand Oaks, CA, USA were purchased from a pharmacy wholesaler. Isothiocyanatobenzyl derivative of desferrioxamine (DFO-Bz-NCS) was obtained from Macrocyclics, Inc, TX, USA. The purity of DFO-Bz-NCS was checked with HPLC before use. 89Zr was produced at the National Institutes of Health's (Bethesda, MD, USA) cyclotron facility by proton irradiation (beam energy: 14 MeV; current: 20 μA) (p,n) reaction (2–5 h) on yttrium-89 metal mesh (200mg, 4 N purity, American Elements), using a 16.5 MeV proton cyclotron (PETtrace, General Electric, Fairfield, CT, USA). 89Zr was separated as [89Zr]Zr-oxalate from irradiated 89Y-metal mesh.21 The specific concentration was ∼740 MBq/mL of 1.0 M oxalic acid, and amount of 88Zr present as impurity was <0.2%. Caution! 89Zr emits very high energy gamma photons. Adequate shielding is essential to reduce exposure during manipulation. Na2CO3, NaN3, dimethyl sulfoxide (DMSO), gentisic acid, oxalic acid, citric acid,1.0 M HEPES buffer, NaCl, and 10M phosphate buffered saline (PBS) were obtained from Sigma-Aldrich, St. Louis, MO, USA. Other reagents are sterile pyrogen-free 0.9% saline (APP Pharmaceuticals, IL, USA); chelex Resin (Bio-rad, Hercules, CA, USA); water for injection (Hospira, Inc. Lakeforest, IL, USA); HPLC grade water (Sigma-Aldrich, St. Louis, MO, USA or VWR Scientific, West Chester, PA, USA); pH buffers (VWR Scientific); Limulus Amebocyte Lysate reagent water (Charles River, Charleston, SC, USA); Calibrated standard pH buffers (VWR Scientific, West Chester, PA, USA). All other chemicals, unless otherwise stated, were purchased from Sigma-Aldrich (St. Louis, MO, USA).
MDA-MB-468 human breast cancer cells were obtained from the Development Therapeutics Program/DCTD/NCI/NIH Tumor Repository (Frederick National Laboratory for Cancer Research, Frederick, MD, USA).
Supplies used in this process are PD10 size exclusion chromatography column (GE Healthcare, Piscataway, NJ, USA); 0.22 μm sterile syringe filter (Pall Corp. Ann Arbor, MI, USA, pore size 0.22 μm, dia. 25mm, hydrophilic polyethersulfone as filtering media, Cat. No. PN4612); empty sterile vials 10 mL (Hollister-Stier Laboratories, Spokane, WA, USA); sterile metal-free pipette tips (Bio-rad, Hercules, CA, USA); sterile disposable syringes of different sizes (BD, Franklin Lake, NJ, USA); sterile disposable needles (BD, Franklin Lake, NJ, USA); sterile vent filter (Millipore Corp., Billerica, MA, USA); HPLC column (Superdex TM 200 10/300GL, GE Healthcare); radio-thin layer chromatography (RTLC) plate (Auburn Biostrips, Auburn, CA, USA); and pH test strips ( VWR Scientific, West Chester, PA, USA).
All instruments were maintained and calibrated properly as per routine quality control (QC) methods.22 For accurate quantification of 89Zr-activities, the samples were counted for 1 min on a gamma counter (Wallac Wizard 1480 3″, Perkin Elmer, Waltham, MA, USA) using an energy window of 800–1000 keV for 89Zr (909 keV emission). 89Zr radiolabeling yield and purity were checked using thin layer chromatography (radio-ITLC or radio-TLC) paper and analyzed on the gamma counter (Wallac Wizard 1480 3″). A Phosphor-Imager (FLA 5100R, Fujifilm, Stamford, CT, USA) is used as an alternative to a gamma counter. Radio-TLC plates were imaged, and analysis was performed with an image reader. RCY/purity was calculated from the intensities obtained from the top and bottom spots of the TLC-image. This technique was used mainly in the developmental stage to cross-validate the general radio-TLC technique (counting in a gamma counter). All activity measurements were performed in a dose calibrator (CRC-15R Capintec, Inc., Ramsey, NJ, USA), which was appropriately calibrated with a calibration factor of 465 for 89Zr. A Shimadzu HPLC system equipped with an auto injector, a variable UV detector preset to 280 nm, and a BioScan radioactive detector was used for QC test. HPLC column (Superdex 200 10/300 GL) was equilibrated with HPLC eluent for 30 min at the flow rate of 0.8 mL/min. For the QC test, injected amount was 20 μL of the protein sample, and the flow rate was 0.8 mL/min. The HPLC retention time (tR) of the product was15-18 min.
High-performance liquid chomtography eluent
About 0.65 g of NaN3 was transferred to a 1 L bottle filled with 500 mL of HPLC grade water. A total of 5 mL of 10 M PBS and 30 mL of 5 M NaCl were transferred to the 1 L bottle. Bottle was then filled with HPLC-grade water up to 1 L mark. Solution pH was ∼7.0.
Gentisic acid solution
A bout 0.25g of gentisic acid was dissolved in 100mL of 0.9% NaCl (USP). A total of 0.4mL of 2.0M Na2CO3 was added. Solution was homogenized until no more CO2 was formed. The pH of the solution was ∼5. This solution was used in 89Zr-labeling reaction and was also used as mobile phase in 89Zr-panitumumab purification (PD10) step.
Authentic standard
The DFO-panitumumab conjugate is used as the authentic standard, which has been prepared following the procedure documented in the literature.17,18,20 The purity was checked using HPLC to compare its retention time with pure authentic panitumumab (Figure 3). The conjugate concentration is determined with the Lowry assay,23 and the DFO to mAb molar ratio is determined with the radiometal-binding assay using 25mM non-radioactive ZrCl4 mixed with trace amount of 89Zr-oxalate.24 This freshly prepared conjugate is then used to develop a standard calibration curve (Figure 4) with HPLC (21–210 μg/mL of DFO-panitumumab) to determine the DFO-protein concentration in every batch. The amount of mAb per dose and the specific activity (SA) of the final product can also be measured with this calibration curve. Curve will be validated at least once in a month using standard sample with known concentration. New calibration curve will be made if the validation result deviates ≥10%.
Figure 3.

HPLC chromatograms at 280 nm: (a) pure panitumumab and (b) desferrioxamine-panitumumab conjugate.
Figure 4.

(a) Standard calibration curve using desferrioxamine (DFO)-panitumumab conjugate as an authentic standard. X-axis represents the concentration in milligrams/millililiters (0.021–0.21 mg/mL) and Y-axis represents the absorbance (peak area). (b) HPLC chromatogram for a 20-μL injection of a 0.021 mg/mL DFO-panitumumab conjugate showing conjugate's UV sensitivity at 280 nm.
Probe synthesis
This protocol is for preparing small scale (for one to two patients), clinical-grade 89Zr-panitumumab. Conjugation and radiolabeling are performed in same day. No conjugate is stored overnight before radiolabeling. The overall process flow is shown in Figure 5. Sterile and metal-free pipette tips have been used all the time for liquid transfer during the synthesis.
Figure 5.

Process flow of 89Zr-panitumumab synthesis for medical use.
Bioconjugation and purification
Approximately 0.5mL (∼ 10 mg) of panitumumab from purchased panitumumab vial (100 mg/5 mL) was transferred to a 1.5mL microcentrifuge tube (reaction vial). The pH was adjusted to ∼9.0 by administering 0.1M Na2CO3 (∼0.2 mL) with a micropipette and pH paper. Threefold molar excess of DFO-Bz-NCS (10 μL of 20 mM DFO stock solution in DMSO) over the molar amount of panitumumab was added to the reaction vial. Reaction mixture was incubated for 30 to 60 min at 37°C using a Thermomixer. Reaction mixture was then transferred to a previously equilibrated (20 mL of 0.9% NaCl solution) PD10 column and flow-through was discarded. About 1.5mL of 0.9% NaCl was transferred onto the column and flow-through was discarded. About 2mL more of 0.9% NaCl was pipetted onto the PD-10 column, and the DFO-protein was collected in 0.5 mL fractions. Fractions 2–4 were combined to obtain DFO-conjugated protein in a 4 mL glass vial.
Concentration and purity of the conjugate
A total of 20 μL of the purified conjugate was transferred to a microcetrifuge tube containing 0.5mL of saline (diluted to 500 times). About 20 μL of this diluted sample was injected to the HPLC instrument. HPLC chromatogram was integrated to obtain the UV peak (280 nm) area. Protein concentration (mg/mL) was calculated using the standard calibration curve (Figure 4). The volume needed for 1mg protein was also calculated for radiolabeling step.
Radiolabeling with 89Zr
The required volume (150–180 MBq, ∼250 μL) of 89Zr oxalic acid solution was pipetted into a reaction vial. Amount of activity in millicurie or MBq was confirmed by calibrating the reaction vial in a dose calibrator. Approximately ∼100 μL of 2 M Na2CO3 was added, and the reaction vial was incubated for 3min. The pH of the solution was adjusted to 7.5 by further addition of 2 M Na2CO3 (∼100 μL). While gently shaking, the reaction mixtures were added: (i) 0.5mL 0.5M HEPES (pH7.2) buffer; (ii) adequate volume of conjugate to obtain 1–1.2mg mass of the conjugate; and (iii) 0.2 mL gentisic acid solution. Reaction was incubated for 1h at room temperature with occasional gentle shaking.
Radiochemical yield of crude product
Approximately 2 μL of crude product was diluted to 500 times with 0.9% NaCl. ITLC experiment was performed by pipetting ∼1 μL of diluted sample to the ITLC plate (Auburn Biostrip, Auburn, CA, USA) and was developed in the developing chamber using 20 mM of citric acid solution as the mobile phase. The plate was cut in the middle and counted both the bottom and top parts separately for radioactivity in a gamma well counter. RCY was calculated using the equation RCY(%)=(counts of the bottom/counts of (top+ bottom))× 100. RCY should be ≥50%.
Purification of 89Zr-panitumumab
Reaction mixture was transferred onto PD10 column previously equilibrated with 20mL of PD10 mobile phase (gentisic acid solution), and the flow-through was discarded. About 1.5mL of the mobile phase was pipetted onto the column, and the flow-through was discarded. About 2 mL more of mobile phase was transferred onto the column, and 89Zr-panitumumab was collected in 0.5 mL fractions in small glass vials. Amount of activity in each vial was determined in a dose calibrator. Radiochemical purity (RCP) of fractions two and three (usually highest activity fractions) were checked by ITLC. If the RCY is >90%, the two fractions are combined in a 10 mL product vial. The total amount of radioactivity of the product vial was measured in a dose calibrator.
Sterile filtration of the product
About 5 mL of 0.9% saline was added to the product vial. Product solution was drawn in a 10-mL syringe. Needle of the syringe was replaced by a sterile syringe filter. The filter needle was inserted through the septum of the 10mL sealed sterile vial with a venting needle on it. The product solution was passed through the sterile filter slowly by pressing the plunger of the syringe. Product vial was then detached from the syringe. This whole process was performed inside an aseptic isolator hood (ISO class 5 environment). Filter integrity test of the sterile filter was performed following the standard procedure (vide infra).
Post-synthesis quality control tests
All the QC tests, except the sterility test of 89Zr-panitumumab, were carried out according to the USP <823 >recommendations. After successfully meeting all release criteria, doses are released to physicians for human administration. A post-release sterility test for every batch must be completed and recorded.25
Sampling for quality assurance
The product vial was assayed for total radioactivity in a dose calibrator. It was inspected visually for particulates and color. The final drug product in the vial should be clear and colorless, without any visible particulates as per USP chapters <823>and <631>on color and achromaticity.
Filter integrity test of the sterilizing filter was performed by placing the filter on a gas line with a pressure gage and the outlet of the filter under water. The gas pressure was increased slowly on the inlet to the filter until a steady stream of bubbles was observed at the filter outlet. The pressure at which the bubble stream begins was recorded and compared with the manufacturer's pressure rating (≥46 psi) for the sterile syringe filter (Acrodisc, Pall corp., PN4612, Ann Arbor, MI, USA).
After a successful filter integrity test, 300 μL of the product was withdrawn for further QC and sterility testing.
Chemical and radiochemical purity/identity
The purity of the drug product can be determined by HPLC analysis. A blank injection must be completed before the sample was injected to make sure there were not any UV and/or radiation impurities from the column or the instrument. About 20 μL of 89Zr-panitumumab (auto-injection preferable) was injected. All of the UV responses were integrated to obtain the peak areas for the [89Zr]-panitumumab. The reverse correlation (UV peaks) was performed from the linear equation of DFO-panitumumab to determine the concentration of [89Zr]-panitumumab in the final solution. Typically, the final concentration of [89Zr]-panitumumab in the [89Zr]-panitumumab product to be tested will be <0.2 mg/mL. Any other 280-nm UV absorbing chromatographic peaks that elute after more than 3.0 min up to 40 min post-injection should be integrated individually and shown as a sum of less than ‘10%’ in the final product. It was assumed that the molar UV absorption coefficients for the unknown contaminants were the same as those for [89Zr]-panitumumab, and that the [89Zr]-panitumumab standard curve was applicable to the unknown contaminants.
All of the radioactive peaks were integrated in the chromatogram, and the percent of the [89Zr]-panitumumab radioactivity in the final product was calculated. The radioactivity of [89Zr]-panitumumab in the final product should be ≥90%.
Radiochemical purity by radio-thin layer chromatography
Approximately 0.5mL of the RTLC eluent (20 mM of citric acid solution) was transferred to a 15mL centrifuge tube (developing chamber). Using a micropipette, ∼1μL of the sample was spotted at the origin of the RTLC strip. The spot was allowed to dry prior to placing it in developing chamber. RTLC plate was developed by carefully positioning it in the developing chamber. The solvent front was allowed to migrate at least 90% of the length of the RTLC plate. RTLC plate was cut into the middle and place each part in separate counting tubes. Parts were counted for radioactivity with an appropriate counter (Perkin Elmer gamma counter). The RCP was calculated using this equation: RCP (%) =(counts of the bottom/counts of (top+ bottom))×100. The acceptable RCP should be ≥90%.
Determination of pH
Because the product volume was small and the product was radioactive, pH test strips were used instead of a pH meter. The pH test strips were checked by pipetting pH5 and pH7 calibrated commercial pH standards onto individual strips. The color on the strips must match the pH 5 and pH7 on the color key supplied with the test strips. Then, the 89Zr-panitumumab was pipetted onto another test strip, and the color was checked against the color key. The measured pH must be between pH 6 and pH 8.
Bacterial endotoxin test
The levels of bacterial endotoxin in 89Zr-panitumumab were tested and qualified by portable testing system (PTS) limulus amebocyte lysate test using FDA approved Endosafe®-PTS from Charles River Laboratories, Wilmington, MA, USA. All of the bacterial endotoxin levels were <175 EU per batch for the initial qualification syntheses. This testing required around 10–15 min.
Radionuclidic identity
Half-life determination is utilized for verifying the radionuclidic identity of the radionuclide in the product. The USP radioactivity general chapter, 821, states that the half-life can be ‘readily determined by successive counting of a given source of a radionuclide over a period of time that is long compared to its half-life’. The variation used here is to count for only a fraction of the half-life of 78 h. To count a sample for more than 78 h would reduce the radiopharmaceutical dose.
For the test, an aliquot of the 89Zr-panitumumab or 89Zr-oxalate (used in this preparation) was counted in an ion chamber or gamma counter at least three times at a particular time point. The half-life of the radioactivity was determined using the following equation.
The half-life test result for 89Zr must be in between 74 h and 82 h.
Sterility test
Sterility testing can be performed using the direct inoculation method, following USP <71>, after releasing the product for human administration. This test requires ∼14 days.
Dose preparation time
| Bioconjugation and purity checking: | 1 h and 40 min. |
| 89Zr-labeling and purification: | 2 h and 00 min. |
| QC testing (except sterility): | 1 h and 20 min. |
Further evaluation of the product
89Zr-panitumumab prepared in three qualification runs following the aforementioned protocol was further tested to determine the stability of the product and its target specificity toward EGFR-expressing cell lines.
Specificity of the 89Zr-Panitumumab
In vitro binding and target specificity of 89Zr-panitumumab were assessed using methanol-fixed EGFR-expressing MDA-MB-468 cells. 89Zr-panitumumab was diluted in 20% BSA in PBS (∼100 000 counts in 50μL). Cells were fixed in six small test tubes (∼1 million cells per tube). Cells in three test tubes were pretreated with a solution of pure (non-radioactive) panitumumab (20 μg in 50μL of 20% BSA in 1 X PBS) and incubated for 1h at 37°C in order to block the EGF receptors. Cells were then washed three times with PBS. All six tubes were then treated with 50 μL of 89Zr-panitumumab. After 1 h of incubation, the cells were washed with PBS, and washings were collected for gamma counting. After washing with PBS, each tube was counted in a gamma counter to determine the amount of activity bound to the cells. The found radioactivity was used to calculate the percentage of antibody bound to the cells using following equation:
Stability and expiration dating
The final drug product was left at 4°C for up to 72 h. At periodic times during the 72 h, the product was measured for RCP using analytical HPLC and radio-TLC. In addition, the 89Zr-panitumumab product has been examined for changes in UV absorbance of the product peak over time.
Results and discussion
Bioconjugation and radiolabeling
This method is developed on the basis of the modified literature procedures.16-18,20 The bioconjugation and radiolabeling of 89Zr-DFO-panitumumab (i.e., 89Zr-panitumumab) are shown schematically in Figure 1. This is a two-step manual synthesis process starting from the clinical drug product panitumumab, purchased as a drug product, and chelate p-isothiocyanatobenzyl-desferrioxamine, commercially available with a certificate of analysis.
Panitumumab is used directly from the purchased drug vial for bioconjugation without any solvent exchange or dilution. Chelate p-isothiocyanatobenzyl-desferrioxamine can form stable thiourea linkage with the lysine residue of panitumumab at basic pH levels (8–9) within 30 min (Figure 1). The conjugate is separated from free chelate by size exclusion chromatography on a PD10 column, with 0.9% saline elution. Both literature reports and our radiometal-binding assay of the purified conjugate have demonstrated that three equivalents of chelate are sufficient to keep the chelate-to-protein labeling ratio within the desirable range (1.6 ± 0.2, n = 6). An increase in the amount of chelate increased the number of chelate per antibody (Figure 6). During clinical production, the purified conjugate is analyzed by HPLC before radiolabeling to determine its concentration and purity. A calibration curve (Figure 4) developed by using different concentrations (0.021-0.21 mg/mL) of DFO-panitumumab conjugate has been used to determine the concentration of purified protein. Isolated yield and purity (HPLC) of the conjugate are typically >75% and >98%, respectively.
Figure 6.
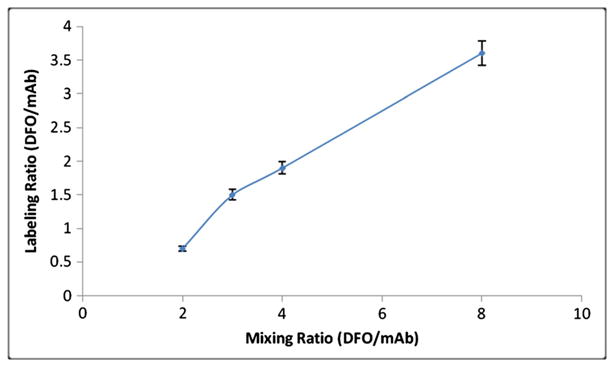
Chelate (desferrioxamine (DFO)) to mAb (panitumumab) molar labeling ratio was measured at various chelates to mAb mixing ratios. Reaction was repeated three times for each data point.
If the HPLC purity of the conjugate >90%, approximately 1–1.2 mg of the conjugate are added to the reaction vial containing ∼180.0 MBq of 89Zr-oxalate, which was previously neutralized (∼pH7.5) with 2.0M Na2CO3. The reaction mixture is incubated for 1h at room temperature in the presence of 0.2mL of gentisic acid and 0.5mL of 0.5M HEPES buffer. Maintaining the pH range (7.0–8.0) of the radiolabeling reaction is very important. At a lower pH, RCY decreases dramatically. HEPES buffer is used to maintain this pH range. RCY of the crude product is checked quickly by RTLC before purification. The typical RCY of the crude product is 80 ± 5% (n = 3). If RCY is >50%, the 89Zr-panitumumab crude mixture is purified and formulated (dilution and filtration) to make the final dose, as described in the experimental section. The volume of the purified product is typically 1–2 mL, and the volume of the formulated 55MBq dose is ∼5.0 mL. Process flow of the whole synthesis is shown in Figure 5.
Dose amount and specific activity
Table 1 shows the composition of the final product. 89Zr-panitumumab is the only active ingredient in the final product. The drug is intravenously injected into the subject in a solution of <10mL preservative-free 0.9% saline. The drug product solution is stored at 2–8 °C in a gray, butyl septum-sealed, sterile, pyrogen-free glass vial with an expiration time of 48h. The injectable dose of 89Zr-panitumumab for the initial clinical study will be <55MBq with an SA >55MBq/mg (>8100MBq/μmol) at the end of the synthesis. The participant will be injected with <1 mg of panitumumab drug, which is less than 1/420th of the initial therapeutic dose and less than 7nmol of panitumumab, well under the microdose guideline for an exploratory IND of 30 nmol of a protein drug. In our clinical production, the amount of panitumumab per dose was 400 ± 100 μg (n= 3).
Table 1. 89Zr-panitumumab final product components.
| Components | Amount in injectate |
|---|---|
| 89Zr-panitumumab | <55MBq |
| DFO-panitumumab | <1mg |
| Gentistic acid | < 5mg |
| Saline for injection | Remainder |
Gentisic acid (2,5-dihydroxybenzoic acid), an active metabolite of salicylic acid degradation, is used both in radiolabeling and in the final product purification mobile phase to protect the protein from radiolysis.26 It is a component of the approved kit for TechneScan HDP.
Quality control
In-process quality control tests
Because the whole clinical production process is fully manual, two in-process QC tests are performed to make sure synthesis contain no manual error in the crucial steps and to ensure that the quality of the intermediates is good enough to produce clinical-grade end product.
The first in-process QC is performed after the purification (PD10) of the conjugate. The purity and concentration of the conjugate are determined by analytical HPLC. The conjugate is used for the next radiolabeling step if its HPLC purity is >90%. The product is identified by its matching retention time (tR) with clinical-grade panitumumab (Figure 3). The typical yield and purity of the in-process intermediate conjugate is >75% and >98%, respectively. An appropriate quantity of this purified conjugate (∼1.0mg, ∼200μL) is immediately used for 89Zr labeling.
The second in-process QC is performed just after radiolabeling. The crude radiolabeled product is analyzed by radio-TLC. In radio-TLC, unbound 89Zr-oxalate and free DFO-bound 89Zr move with the solvent front (Rf > 0.5), and only the 89Zr bound with protein stays near the origin (Rf < 0.5), Figure 7. The product is processed further if the RCY is >50%. Radiolabeling is highly pH sensitive. Any error in pH adjustment will decrease the RCY, that is, the quality of the product.
Figure 7.
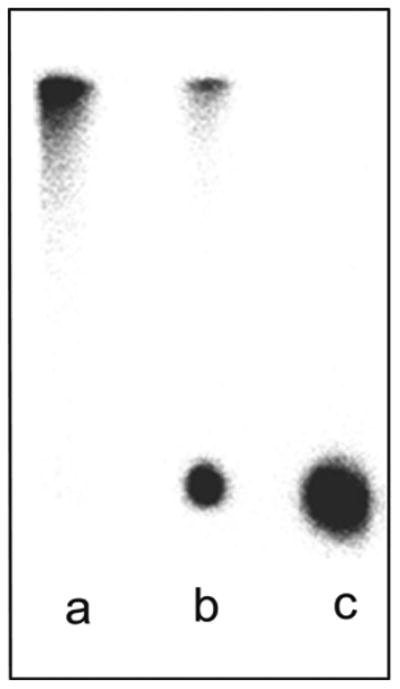
Representative phosphor images of radio-TLC strips: (a) 89Zr-oxalate (control); (b) 89Zr-panitumumab (crude product) before purification; and (c) purified 89Zr-panitumumab.
Quality control tests of the product
The drug product is assayed for total radioactivity and is examined for particulates. The integrity of the sterilizing filter is tested. Samples are removed for QC tests to determine pH, SA, radiochemical and chemical purities, radionuclidic purity, apyrogenicity, and sterility of the product. At least 0.1 mL of the sample is retained for further testing, if necessary. The product dose is drawn, labeled, and once all but the sterility tests have been passed, the product dose is released for injection. A PTS chromogenic endotoxin test must be completed before the product is released. The sample for sterility testing is inoculated within 48h. QC results from three of the completed qualification 89Zr-panitumumab radiosyntheses are listed in Table 2.
Table 2. Quality control results of the three batches (ZR-PAN-MBR-01, ZR-PAN-MBR-02, and ZR-PAN-MBR-03) of 89Zr-panitumumab.
| Quality control test | Description | Acceptance criteria | ZR-PAN-MBR-01 | ZR-PAN-MBR-02 | ZR-PAN-MBR-03 |
|---|---|---|---|---|---|
| Particulates and color | Visual inspection for color and particulates | Clear and colorless | Clear and colorless | Clear and colorless | Clear and colorless |
| Filter Integrity | Bubble point test | ≥46 PSI | 58 | 57 | 60 |
| pH | pHas per USP<791> pH | pH must be between 6 and 8 | 7.0 | 7.0 | 7.0 |
| Radiochemical ID and purity(%) | Radio-TLC | Rf <0.5; purity ≥90% | >98 | >98 | >98 |
| Radiochemical identity | Half-life test | 74-82 h | 76.4 | 75.8 | 76.3 |
| Radiochemical purity | HPLCa, consistent with guidelines of USP <621> | Radiochemical purity ≥90% | 97 | 95 | 96 |
| Chemical purity | HPLCa, consistent with guidelines of USP <621> | Chemical purity ≤1 mg of panitumumab per dose | 0.38 | 0.42 | 0.40 |
| Specific activity | Expressed as MBq/mg of panitumumab | ≥1.5 mCi (55.5 MBq)/mg of panitumumab | 3.8(140.6) | 3.4 (125.8) | 3.5(129.5) |
| Bacterial endotoxin levels | Limulus Amebocyte Lysate (LAL) by PTS | <175 EU per dose | <2 | <2 | <2 |
| *Immunoreactivity | In vitro cell-binding assay | ≥60% | >60% | >60% | >60% |
| *Sterility test (14 days) | USP sterility test (USP <71>) | No growth | No growth | No growth | No growth |
HPLC column (Superdex 200 10/300 GL) with mobile phase flow rate of 0.8 mL/min
not a product releasing criterion
Chemical and radiochemical purity
High-performance liquid chomtography analysis is used to determine purity for this drug product. The final RCP is proposed to be greater than or equal to 90%. As the preparation process can be completed under very mild conditions, non-radioactive UV impurities are not generated. In HPLC analyses, no new UV peaks are observed before and after the 89Zr-panitumumab peak compared with those of pure panitumumab. Both chemical and RCP are measured by UV absorbance at 280 nm, and the radiation peak of the 89Zr-panitumumab eluted from a Superdex 200 gel filtration column (GE Healthcare), Figure 8. The column flow rate is 0.8 mL/min and is kept at room temperature, 25–30 ° C. The typical retention of 89Zr-panitumumab is between 16min and 18min for the UV absorbance, and the radioactivity is ∼0.8 min further downstream from the UV detector. The standard concentrations must bracket the sample or bracket the minimum acceptable mass limit. All standards must be baseline resolved (resolution >1.5) for a valid analysis. A linear regression is determined for UV absorbance peak areas of the standards (DFO-protein, Figure 4). This constitutes the calibration curve. Then, the HPLC peak area of the 89Zr-panitumumab drug product is fitted on the calibration curve to determine the protein concentration in the drug product sample. Typically, the final concentration of the DFO-protein in the 89Zr-panitumumab product to be tested is <200 μg/mL and the protein mass is <1mg per dose. No low or high molecular weight UV impurities were found in the qualification runs.
Figure 8.
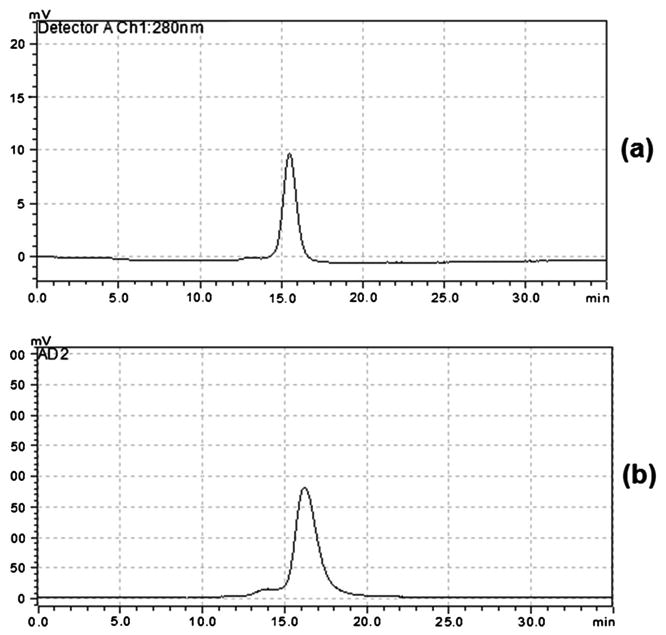
HPLC chromatograms of 89Zr-panitumumab: (a) UV chromatogram at 280 nm and (b) corresponding radiation chromatogram. Early small shoulder of radiation peak could be due to the 89Zr labeling to the trace amount of impurity (high molecular weight) present in the original protein.
The use of radio-TLC to determine the RCP and RCY was validated using citric acid solution as the mobile phase. The Rf values obtained from these studies were below 0.5 for the three qualification runs. The limit has been set at Rf < 0.5 for the final 89Zr-panitumumab because the TLC test is included only to separate the unbound 89Zr (solvent front) from the product (Figure 7). The specification for the purity is greater than or equal to 90% using this methodology. This is primarily a test for free 89Zr and 89Zr bound in free DFO. If present, both will move with the solvent front with the Rf value >0.5, so this Rf value is an adjunct test to the analytical HPLC.
Two PD-10 purification steps are associated with this preparation from GMP grade protein panitumumab. In the first purification step, the DFO-protein conjugate is separated from the excess DFO. The 89Zr-labeled DFO-protein is purified on a PD-10 column (second purification step) to separate 89Zr-panitumumab from excess 89Zr-oxalate. An ITLC test of the purified 89Zr-panitumumab showed >98% RCP (Rf <0.5). The >98% RCP by ITLC means that there is little or no DFO chelate impurity present in the final product. The product purity is further checked by HPLC. In HPLC chromatogram, no notable high-molecular or low-molecular weight radiation or UV peaks are observed, indicating that no fragmented or aggregated byproducts of the protein are generated during the manufacturing.
Between 7μL and10μL of DMSO solution of DFO is used in the bioconjugation step. After passing through two purification steps, the possibility of the presence of this solvent (DMSO) in the final product is very low. Moreover, as per FDA's ‘Guidance for Industry’, ICH Q3C, DMSO is a class 3 solvent that the FDA recommends be limited to less than 50mg/day in pharmaceuticals.27 Even if all the DMSO remained in the final product, it is still only a trace amount, which is well below the FDA's guidance. Therefore, the percentage of DMSO has not been determined in the final product.
Sterility and expiration dating
Conventional sterility testing requires 14 days. For this reason, the PET drug product (89Zr-panitumumab) was filtered through a 0.22 μm sterile syringe filter, and the filter integrity was tested to make sure the product was likely to be pyrogen-free. If the 0.22 μm filter used to filter the final product fails the integrity test, then the final product will be resterilized using a new sterile filter. After releasing the drug for human administration, a conventional sterility test must be completed for each batch.
The expiration time is 48h for the product stored at 4°C. The HPLC measurements suggest that there is little or almost no loss of radiolabeling during the 72h at 4°C, but TLC measurements after 48h show that the product starts to slowly lose the labeling. Even after 72 h, the purity (> 90%) of the product by radio-TLC is acceptable for human injection. For the HPLC, no other UV or radioactive peaks were detected.
In vitro binding and target specificity
89Zr-panitumumab produced using the method described earlier has been evaluated in vitro using an EGFR-expressing MDA-MB-468 cell line. Specific binding of 89Zr-panitumumab was 64–72% (n= 3) in cells without pretreatment with panitumumab. In blocking experiment when cells were pretreated with excess panitumumab, the 89Zr-panitumumab radioactivity bound to the cells was only 4–8% (n=3).
These results clearly demonstrate that binding was target-specific.
Conclusions
In this process, we have described a robust and reproducible protocol to prepare clinical-grade 89Zr-panitumumab with very high RCY (>70%, n=3) and purity (>98%, n=3). Introduction of two QC tests have dramatically improved the reproducibility and quality of the drug product. Starting from the approved panitumumab drug, 89Zr-panitumumab will be ready for medical use within 5h. 89Zr-panitumumab produced in this method is highly stable and can be stored at 4 °C for 48 h before use. Products passed post-release sterility testing performed in accordance with the USP specifications.
Quality control analysis of the product of the three qualification runs showed that the quality of the product consistently met all the specifications of a clinical-grade (USP general chapter <823>) PET-radiopharmaceutical. This protocol can be used to produce other 89Zr-mAb tracers for medical use without any significant modifications. 89Zr-panitumumab produced following this protocol will be used as a surrogate PET biomarker to evaluate EGFR-targeted therapy of cancer patients.19
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. The authors are grateful to Dr. Lawrence Szajek of cyclotron facility at NIH Bethesda for providing 89Zr-oxalate.
Footnotes
Conflict of Interest The authors did not report any conflict of interest.
References
- 1.Burgess AW. Growth Factors. 2008;26:263–274. doi: 10.1080/08977190802312844. [DOI] [PubMed] [Google Scholar]
- 2.Ciardiello F, Tortora G. Expert Opin Invest Drugs. 2002;11:755–768. doi: 10.1517/13543784.11.6.755. [DOI] [PubMed] [Google Scholar]
- 3.Pantaleo MA, et al. Ann Oncol. 2009;20:213–226. doi: 10.1093/annonc/mdn625. [DOI] [PubMed] [Google Scholar]
- 4.Slobbe P, Poot AJ, Windhorst AD, et al. Drug Discov Today. 2012;17:1174–1187. doi: 10.1016/j.drudis.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 5.Dei Tos AP. Int J Biol Biomarkers. 2007;22:S3–S9. [PubMed] [Google Scholar]
- 6.Dei Tos AP, Ellis I. Eur J Cancer. 2005;41:1383–1392. doi: 10.1016/j.ejca.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 7.Bhattacharyya S, Dixit M. Dalton Trans. 2011;50:6112–6128. doi: 10.1039/c1dt10379b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hicks JW, VanBrocklin HF, Wilson AA, et al. Molecules. 2010;15:8260–8278. doi: 10.3390/molecules15118260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu M, Rivkin A, Pham T. Clin Ther. 2008;30:14–30. doi: 10.1016/j.clinthera.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 10.2012 Sep 30; http://www.cancer.gov/cancertopics/druginfo/panitumumab.
- 11.Deri AM, et al. Nucl Med Biol. 2013;40:3–14. doi: 10.1016/j.nucmedbio.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nayak TK, Germastani K, Baidoo KE, Milenic DE, Brechbiel MW. J Nucl Med. 2010;51:942–950. doi: 10.2967/jnumed.109.071290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holland JP, Caldas-Lopes E, Divilov V, Longo VA, Taldone T, Zatorska D, Chiosis G, Lewis JS. PloS ONE. 2010;5:1–11. doi: 10.1371/journal.pone.0008859. e8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perk LR, Vosjan M, Visser GWM, Budde M, Jurek P, Keifer G, van Dongen GAMS. Eur J Nucl Med Mol Imaging. 2009;37:250–258. doi: 10.1007/s00259-009-1263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Börjesson PKE, Jauw YWS, et al. Clin Cancer Res. 2006;12:2133. doi: 10.1158/1078-0432.CCR-05-2137. [DOI] [PubMed] [Google Scholar]
- 16.Vosjan JWDM, et al. Nat Protocol. 2010;5:739–743. doi: 10.1038/nprot.2010.13. [DOI] [PubMed] [Google Scholar]
- 17.Nayak TK, Germestani K, Milenic DE, Brechbiel MW. J Nucl Med. 2012;53(1):113–120. doi: 10.2967/jnumed.111.094169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhattacharyya S, et al. Nucl Med Biol. 2013;40:451–457. doi: 10.1016/j.nucmedbio.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.2013 Apr 04; http://clinicaltrials.gov/show/NCT00326495.
- 20.Chang AJ, et al. Mol Imaging. 2013;12:17–27. [PMC free article] [PubMed] [Google Scholar]
- 21.Holland JP, Sheh Y, Lewis J. Nucl Med Biol. 2009;36:729–739. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanzonico P. J Nucl Med. 2009;49:1114–1131. doi: 10.2967/jnumed.107.050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 24.Meares CF, McCall MJ, Reardan DT, Goodwin DA, Diamanti CI, McTigue M. Anal Biochem. 1984;142:68–78. doi: 10.1016/0003-2697(84)90517-7. [DOI] [PubMed] [Google Scholar]
- 25.Dixit M, Shi J, Ling W, et al. Int J Mol Imaging. 2013;2013:1–10. 278607. [Google Scholar]
- 26.Liu S, Edward DS. Bioconj Chem. 2001;12(4):554–558. doi: 10.1021/bc000145v. [DOI] [PubMed] [Google Scholar]
- 27.FDA Guidance for Industry: ICH Q3C—Tables and List, Revision 2 (2/2012) 2012 Aug 13; http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatory-Information/Guidances/ucm073395.pdf.