

Abstract
Although the Hsp90 chaperone family, comprised in humans of four paralogs, Hsp90α, Hsp90β, Grp94 and Trap-1, has important roles in malignancy, the contribution of each paralog to the cancer phenotype is poorly understood. This is in large part because reagents to study paralog-specific functions in cancer cells have been unavailable. Here we combine compound library screening with structural and computational analyses to identify purine-based chemical tools that are specific for Hsp90 paralogs. We show that Grp94 selectivity is due to the insertion of these compounds into a new allosteric pocket. We use these tools to demonstrate that cancer cells use individual Hsp90 paralogs to regulate a client protein in a tumor-specific manner and in response to proteome alterations. Finally, we provide new mechanistic evidence explaining why selective Grp94 inhibition is particularly efficacious in certain breast cancers.
The Hsp90 family of molecular chaperones regulates and maintains cell homeostasis under proteotoxic stress and pathogenic pressure1. In humans, Hsp90α and Hsp90β in the cytoplasm, Grp94 in the endoplasmic reticulum and Trap-1 in the mitochondria are the four Hsp90 paralogs2,3. Hsp90s are characterized by a distinct ‘Bergerat fold’ in the N-terminal ATP-binding domain (NTD)4. Binding and release of the nucleotide drives the Hsp90 catalytic cycle and the refolding of client proteins. Occupancy of this pocket by small-molecule inhibitors inactivates Hsp90 chaperone function. Pan-Hsp90 inhibitors have demonstrated potent reversal of disease phenotypes when tested in models of cancer, neurodegeneration, infection and inflammatory disease5. Some of these compounds have moved to the clinic for the treatment of cancers6.
Despite considerable interest in the use of Hsp90 inhibitors for the treatment of disease, little is known about the contribution of each paralog to the observed therapeutic benefit. To date, published studies have used pan-Hsp90 inhibitors to inactivate all of the Hsp90s and the processes that depend on them, making it impossible to correlate the role of individual paralogs with the biological effects. This is unsatisfying as the chaperoning roles of these Hsp90s do not overlap2,3,7,8.
Much of our inability to study individual paralogs in cancer cells stems from a lack of suitable tools. Strategies that address the biology of Hsp90s and their individual paralogs in an endogenous cellular environment where the chaperones are limiting but not absent (that is, in unengineered cancer cell lines and in primary samples) are needed. Ideally, this gap would be filled by chemical tools that probe a protein’s function in a controlled manner. Such tools would complement traditional approaches by aiding the molecular characterization of biomolecules both in vitro and within their natural biological contexts.
The discovery of paralog-specific Hsp90 inhibitors is challenging because of the high degree of conservation in their ATP-binding cavities, the pocket to which the known synthetic ligands bind7,8. Most reported Hsp90 inhibitors bind equally well to the majority of these paralogs9,10. Crystal structures of apo-Hsp90α NTD or nucleotide- or inhibitor-bound Hsp90α and Hsp90β NTDs are essentially superimposable11,12. In addition, though different poses were observed for some inhibitors when bound to Hsp90 and Grp94, these have not yet resulted in appreciable selectivity and specific cellular activity through individual paralog inhibition7,13.
Paradoxically, despite the high degree of sequence conservation in their ATP-binding pockets, crystallographic and biochemical studies have shown that, when bound to nucleotides, Hsp90α, Hsp90β, Grp94 and Trap-1 adopt distinctly different conformations and hydrolyze ATP with notably different rates14–17. The overall structure and conformational flexibility of the paralogs thus have an important role in configuring their ATP-binding sites.
Here, we take advantage of the conformational distinctions between the Hsp90 paralogs and use the chemical diversity of the purine-scaffold class18,19 to identify Hsp90 paralog–specific ligands. We explain the source of paralog binding specificity using structural and modeling analyses. We then use several of the identified paralog-selective inhibitors to provide new insights into the tumor-specific chaperoning of a client protein by individual Hsp90s.
RESULTS
Screening identifies paralog-selective chemical spaces
To identify paralog-selective Hsp90 inhibitors, we combined library screening with structural and computational analysis. We screened an in-house library of over 130 purine-scaffold (PU) compounds (Fig. 1a,b) in a fluorescence polarization assay10,20 to test for binding to Hsp90α and Grp94. Select derivatives were also analyzed for binding to Hsp90β and Trap-1. Although most of the compounds had similar affinities for each of the paralogs, we also identified compounds with selectivity for Grp94, which we term Type 2 chemical space, (Fig. 1b and Supplementary Results, Supplementary Fig. 1a) and others with selectivity for Hsp90α, which we term Type 1 chemical space (Fig. 1b and Supplementary Fig. 2a).
Figure 1. Library screening identifies paralog specific chemical spaces.

(a) General structure of the purine-scaffold chemical library. Screening identified the Grp94-selective compounds PU-H54, PU-WS13 and PU-H39 (5) and the Hsp90-selective compounds PU-11 (6), PU-29F and PU-20F (7). All but PU-H54 have X3 attached at N9. (b) Top, graphical representation of screening data from the paralog-specific fluorescence polarization assay. Over 130 purine-scaffold compounds were screened. Each x-axis entry represents a different compound. Compounds were tested in triplicate. Bottom, 21 paralog-selective purine-scaffold compounds were identified. Several Grp94-selective compounds (Type 2) had greater than 100-fold preference for Grp94 over Hsp90α and Hsp90β and a 10- to 100-fold preference over Trap-1 (Supplementary Fig. 1b,c). In the Hsp90α-selective subtype (Type 1), we found ligands that showed a 5- to 15-fold and a 5- to 35-fold selectivity over Grp94 and Trap-1, respectively. Unexpectedly, in spite of the high homology between Hsp90α and Hsp90β, we also identified compounds with three- to fivefold selectivity for the Hsp90α paralog (Supplementary Fig. 2b,c). Detailed structures and binding data are presented in Supplementary Figures 1 and 2.
Crystal structures reveal a new binding site in Grp94
Modeling the Grp94-selective compounds into the ATP-binding pockets of existing structures of Grp94, Hsp90α and Hsp90β could not account for the observed binding specificity. Therefore, we determined the structure of a representative Grp94-specific ligand, PU-H54 (1), in complex with the NTDs of both Grp94 and Hsp90α (Fig. 2, Supplementary Fig. 3 and Supplementary Table 1). The structure of Hsp90N–PU-H54 is essentially identical to that of all of the other Hsp90N–PU complexes11,21 (Supplementary Fig. 3a,b). The lower affinity of PU-H54 for Hsp90α and Hsp90β can be explained by the lack of X2-Ar substituents that make tight interactions with the ATP-binding pocket.
Figure 2. Structural and computational analyses define differences in Hsp90 paralog pockets and paralog-selective chemical spaces.

(a) Overlay of Hsp90α- and Grp94-bound PU-H54 reveals an 80° torsional rotation about the sulfanyl linker (highlighted in red) when inserted into the Grp94-specific channel. (b) Interactions of PU-H54 bound to Grp94 showing the increased hydrophobic stabilization of the 8-aryl group (X2-Ar) when bound into Site 2. (c) Surface area (left) and conformation (right) of paralog-selective chemical spaces as generated by Macromodel/Molecular Surface. Figures were created by superimposing the favored docking pose of all of the Hsp90α (top, Type 1) and Grp94 (bottom, Type 2) inhibitors described in Supplementary Figures 1 and 2.
In the structure of the Grp94–PU-H54 complex, rearrangements occur in the lid region, resulting in a substantially altered ATP-binding pocket (Supplementary Fig. 3c–e). Although the purine moiety of PU-H54 maintains previously observed contacts with the ATP pocket (Supplementary Fig. 3f), the X2-Ar adopts a markedly different conformation compared to that of the Hsp90α-bound PU-H54. Overlaying the Hsp90α- and Grp94-bound PU-H54 ligands reveals an ~80° rotation of the 8-aryl group about the sulfanyl linker, with the Hsp90α-bound ligand adopting the ‘forward’ rotation and the Grp94-bound ligand adopting a previously uncharacterized ‘backwards’ rotation (Fig. 2a). Concurrent with this backwards pose of the ligand, Phe199 of Grp94 swings away from the binding pocket by 4 Å to expose a deep, almost completely hydrophobic cleft, which we term Site 2, lined by Leu104, Leu163, Phe199, Ala202, Phe203, Val211, Ile247 and Leu249 (Fig. 2b and Supplementary Fig. 3f). The hydrophobic X2-Ar of Grp94-bound PU-H54 is inserted into this newly revealed nonpolar Site 2 and makes stabilizing contacts with several cleft residues (Fig. 2b). A similar cleft composed of the equivalent, conserved residues is also present in Hsp90α and Hsp90β, but access to it is blocked by the side chain of Phe138, the equivalent of Phe199 in Grp94. Together, our structure uncovers a new site in Grp94 that is occupied by PU-H54 and potentially all of the Type 2 compounds, accounting for their paralog-selective binding.
Paralog pockets differ in their architecture and polarity
We used the structure of Grp94–PU-H54, other available Hsp90 paralog structures and a Trap-1 homology model and performed a SiteMap analysis to investigate structural differences between their ATP-binding pockets (Supplementary Fig. 4a). This study identified substantial differences in the position and orientation of amino acids that compose the NTDs of the four paralogs. Although the amino acids lining the ATP-binding site are similar among the paralogs (Supplementary Fig. 4b), their conformation and orientation are different, as evidenced by the hydrophobic and hydrophilic contours of the ATP-binding sites (Supplementary Fig. 4c). We found the Grp94 site to be the most hydrophobic, with a ratio of hydrophobic to hydrophilic character of 2.98 to 0.63 (fivefold), followed by Trap-1 (threefold), Hsp90β (twofold) and Hsp90α (1.5-fold) (Supplementary Table 2).
Selective ligands differ in conformation, volume and polarity
Docking individual compounds into the available Hsp90 paralog structures showed that all of the Type 2 ligands bound favorably into Site 2 of Grp94, as revealed by the Grp94–PU-H54 structure (GScores < −10), in agreement with the observed hydrophobicity of X2-Ar in these ligands. These compounds scored poorly when docked into the pockets of Hsp90α, Hsp90β, Trap-1 or Site 1 of Grp94 (GScore ≥ −4). Type 1 ligands, in contrast, positioned the X2-Ar moiety into Site 1 and scored favorably in both Hsp90α and Hsp90β (GScore ≤ −10), in line with the experimental binding data.
The preferred docked pose of ligands comprising the two chemical spaces differed (Fig. 2c). Type 1 ligands adopted the ‘forward bent’ conformation of the phenyl ring, situating the X2-Ar into Site 1. In contrast, all of the Type 2 ligands, which prefer Site 2 in Grp94, positioned this ring similar to the ‘backwards bent’ conformation observed in the Grp94–PU-H54 structure (Fig. 2a). The volume and polarity of the Ar-X2 rings and the X3 N9-N3 substitutions also differed. In the Type 1 chemical space, the Ar-X2 was balanced between hydrophobic and hydrophilic (Fig. 2c), and the X3 appendage occupied a limited space. The Type 2 ligands, in contrast, preferred a hydrophobic Ar-X2 (Fig. 2c), and a larger volume was described by the X3 appendage.
Ligand characteristics that confer Grp94 selectivity
The Grp94-selective inhibitors had Ar-X2– and X3-dependent subtypes (Supplementary Figs. 1a and 5a). The Ar-X2–dependent subtype compounds bound well to Grp94 (Supplementary Fig. 1b) and showed approximately 100-fold selectivity for Grp94 over Hsp90α, Hsp90β and Trap-1 (Supplementary Fig. 1c). Energy minimizations indicated that a subset of these compounds preferred the backwards bent conformation, even in the unbound state, and preferred hydrophobic substituents at the 2′,4′; 2′,5′; 3′,5′; 2′,4′,5′; and 2′,4′,6′ positions on the phenyl ring. These allow for favorable proximity to the hydrophobic residues of Site 2 (Supplementary Fig. 5b). Hsp90α, Hsp90β and Trap-1 were unable to accommodate these derivatives in this pose owing to their inability to expose Site 2 (Supplementary Fig. 5c).
In the X3-dependent subtype, the presence of a methyl group at the C1′ position of the N9 alkyl chain yielded compounds with greater than tenfold selectivity for Grp94 over Hsp90α, Hsp90β and Trap-1 (Supplementary Fig. 1c). Modeling indicated that the C1′ methyl group favored the placement of the 8-aryl ring into the backwards bent conformation, resulting in binding into Site 2 of Grp94 (Supplementary Fig. 5b). In contrast to the Ar-X2–dependent subtype described above, the affinity of these compounds for Grp94 was modest (60–90 μM), reflecting the less hydrophobic character of the X2-substituents (that is, three methoxy groups). Hsp90α, Hsp90β and Trap-1 could not accommodate these inhibitors (Supplementary Fig. 5c).
Ligand characteristics that confer Hsp90 selectivity
Selectivity toward Hsp90α and Hsp90β in the Type 1 chemical space was a consequence of distinct X3 substituents attached to the N9 of the purine scaffold (Supplementary Figs. 2a and 5d). We observed two X3-dependent subtypes, one characterized by a rigidifying, electron-rich component in the X3 chain and another by the presence of an oxygen atom two carbons away from N9 that was either followed by or part of a larger substituent (Supplementary Fig. 5d). These conferred approximately 1-log selectivity for Hsp90α and Hsp90β over Grp94 and Trap-1 (Supplementary Fig. 2c).
The X3 chain sits near a conserved exit channel that interacts distinctly with the ligands. Several pocket residues in Grp94 and Trap-1 imposed constraints on the N9 chain (Supplementary Fig. 5e). The two chains disfavored by Grp94 and Trap-1, in contrast, established favorable interactions with Hsp90α. Introducing unsaturation after two carbons increased Hsp90α binding by enhancing hydrophobic interactions with Leu107, and introducing a heteroatom (O) into the N9 chain enhanced water-mediated hydrogen bonding (Supplementary Fig. 5f).
Testing of selective paralog inhibition in cells
The selectivity of the Type 1 and Type 2 compounds measured in vitro with recombinant protein may differ from their effects within cells. To confirm that these compounds act in vivo through paralog-specific inhibition, we tested the selective target modulation of these compounds in cells (Supplementary Fig. 6). Type 2 Grp94-selective compounds dose-dependently inhibited IGF-II secretion (Supplementary Fig. 6a–c) and Toll-like receptor 9 trafficking (Supplementary Fig. 6d–g). Both of these are Grp94-mediated cellular events22,23. At concentrations of Type 2 compounds that inhibited Grp94 activity, we observed no Hsp70 induction or AKT degradation, both of which are hallmarks of cytosolic Hsp90 inhibition1,5 (Supplementary Fig. 6b,f,g). Conversely, treatment with Type 1 compounds led to a dose-dependent increase in Hsp70 and the degradation of AKT (Supplementary Fig. 6b,f,g) but minimally affected the Grp94 hallmarks (Supplementary Fig. 6a,e,f). Notably, at the concentrations of these compounds that elicited target modulation, neither PU-WS13 (2; Grp94-selective), PU-29F (3; Hsp90-selective) or PU-H71 (4; pan-Hsp90 inhibitor and tumor Hsp90-selective)24 was toxic to the two nonmalignant cell lines in which these analyses were performed (Supplementary Fig. 6c,f).
Hsp90 paralogs regulate HER2 in a tumor-specific manner
We next investigated the roles of Hsp90 paralogs on a classical Hsp90 client protein, HER2. HER2 is a receptor tyrosine kinase, which, when activated, leads to the stimulation of numerous cancer-driving signaling pathways25. The expression of HER2 is elevated in many tumors25. The regulation of HER2 by Hsp90 has been investigated using pan-Hsp90 inhibitors and engineered proteins and cells26–29. These studies suggested that association of Grp94 with newly synthesized HER2 regulates the trafficking of HER2 to the plasma membrane29, whereas Hsp90 maintains the stability of the plasma membrane HER2, where most HER2 is located at steady state27. This view was later revised to suggest that the stability and the intracellular trafficking of newly synthesized HER2 were also regulated by Hsp90 and not by Grp94 (ref. 28). Our paralog-specific chemical tools now allow us to reexamine the dependence of HER2 on Hsp90 in endogenous cellular systems.
Using a tool set that combined the paralog-selective inhibitors with siRNAs, we probed two breast cancer cell lines, SKBr3 (high HER2 expression) and MCF7 (low HER2 expression), for the roles of Hsp90 paralogs in HER2 regulation. Although Grp94 was not thought to have a role in mature HER2 expression or stability, we found that steady-state amounts of HER2 were sensitive to selective inhibition of Grp94. To our surprise, however, this was seen only in SKBr3 cells and not in MCF7 cells (Fig. 3a and Supplementary Fig. 7a). Knockdown of Grp94 by siRNAs mimicked the effect of the Grp94 inhibitors. In both cases, a similar reduction in the steady-state amounts of HER2 in SKBr3 cells, but not in MCF7 cells, was observed (Fig. 3a and Supplementary Fig. 7b).
Figure 3. HER2 is sensitive to Hsp90 paralog inhibition in a tumor-specific manner.

(a) SKBr3 (left graph, high HER2) and MCF7 (right graph, low HER2) cells were treated for 24 h with vehicle (DMSO), the Grp94-selective inhibitors PU-WS13 and PU-H39, the Hsp90α- and Hsp90β-selective inhibitors PU-29F and PU-20F, the pan-Hsp90 inhibitor PU-H71 or for 72 h with siRNAs specific for Grp94 or Hsp90α and Hsp90β. Amounts of HER2 are plotted for each experimental condition. Associated representative western blots (WBs) are shown in Supplementary Figure 7a,b. Control siRNA, scramble construct. Data are presented as mean ± s.d. (n = 3). (b) Representative western blot of HER2 complexes in MCF7 extracts isolated by precipitation with an antibody to HER2 (anti-HER2) or a nonspecific IgG. IP, immunoprecipitation; rIgG, rabbit IgG, an isotype control. (c) MCF7 cells treated for the indicated times with the Hsp90α- and Hsp90β-selective PU-11 (40 μM) or the Grp94-selective PU-WS13 (15 μM). Protein amounts in membrane and cytosolic fractions were plotted against the time of treatment. Data are presented as mean ± s.e.m. (n = 3). Associated representative western blots are shown in Supplementary Figure 7f. Full gels of key experiments are shown in Supplementary Note 1.
In contrast, steady-state amounts of HER2 in both cell types were sensitive to inhibition or knockdown of Hsp90α and Hsp90β. In SKBr3 cells with high HER2 expression, the amount of HER2 decreased only at high inhibitor concentrations that were indicative of simultaneous Hsp90α and Hsp90β inhibition, as seen for another Hsp90α and Hsp90β client protein, Raf-1 (ref. 1) (Supplementary Fig. 7c). This was confirmed by siRNA knockdowns, where only dual, but not individual, knockdown of Hsp90α and Hsp90β mimicked the effect of Hsp90α and Hsp90β inhibitors in this cell line (Fig. 3a and Supplementary Fig. 7b).
In MCF7 cells with low HER2 expression, however, total HER2 decreased at inhibitor concentrations that were characteristic of selective binding to Hsp90α but not Hsp90β (Supplementary Fig. 7a,c). We also found a significant correlation in MCF7 cells between HER2 degradation and the affinity of inhibitors for Hsp90α but not for Hsp90β, Grp94 or Trap-1 (Supplementary Fig. 7d; r2 = 0.83, 0.137, 0.217 and 0.005, respectively). HER2 also purified together specifically with Hsp90α in these cells (Fig. 3b). Selective reduction of Hsp90α by means of siRNA, however, failed to decrease the amount of total HER2 in MCF7 cells (Fig. 3a), possibly owing to feedback induction of Hsp90β when Hsp90α was suppressed (Supplementary Fig. 7e).
Because HER2 is located in a membrane compartment associated with either the endoplasmic reticulum and Golgi networks, the plasma membrane, or is trafficked through the cytosol, we investigated the effect of paralog-selective inhibitors on HER2 in these locations. In MCF7 cells, the amounts of cytosolic HER2 protein were rapidly reduced by the Hsp90α and Hsp90β inhibitor but not by the Grp94 inhibitor. Neither inhibitor modified membrane HER2. We observed a similar profile for other Hsp90-validated kinases (Fig. 3c). Together, these results point to a tumor-specific involvement of the Hsp90 paralogs in the chaperoning of HER2.
Grp94 regulates plasma membrane HER2 in SKbr3 cells
We detected a fraction of the total cellular Grp94, but not Hsp90, at the plasma membrane of SKBr3 cells (Fig. 4a,b and Supplementary Fig. 8a,b), which express a high density of HER2 at this location29. We found that plasma membrane–associated Grp94 localized and precipitated together with HER2. Specific complex formation was confirmed both by chemical and reciprocal immunopurification of Grp94–HER2 complexes and by affinity purification performed with the Grp94-specific chemical tool in cell lysates in which Grp94 amounts were reduced by immunopurification with Grp94-specific antibodies (Fig. 4b and Supplementary Fig. 8b,c).
Figure 4. Grp94 and Hsp90α and Hsp90β regulate distinct HER2 functions in HER2-overexpressing cancer cells.
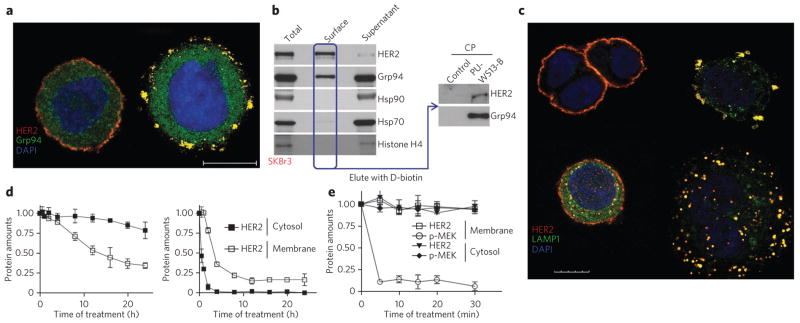
(a) Fluorescence microscopy image of SKBr3 cells treated for 4 h with DMSO (left) or PU-WS13 (right, 15 μM) and then stained with the indicated markers upon fixation and permeabilization. Scale bar, 10 μm. (b) Representative blot of surface-exposed proteins chemically labeled with biotin and purified using streptavidin columns. Histone H4 was blotted to control for membrane impermeability. Proteins eluted from the streptavidin column were affinity purified and analyzed by western blotting as indicated. Total, total cell extracts; supernatant; nonsurface proteins; PU-WS13B, PW-WS13-biotin immobilized on streptavidin beads; CP, chemical precipitation. (c) Same as in a for cells treated with DMSO (top left), PU-WS13 (top right, 15 μM) PU-29F (bottom left, 20 μM) or PU-H71 (bottom right, 1 μM). Scale bar, 10 μm. Additional stains are shown in Supplementary Figure 8d,e. (d,e) SKBr3 cells were treated for the indicated times with 20 μM of the Grp94-selective compound PU-WS13 (d, left, and e) or the Hsp90α- and Hsp90β-selective PU-29F (d, right). Proteins in membrane and cytosolic fractions were plotted against the time of treatment. Data are presented as mean ± s.e.m. (n = 3). Associated representative western blots are shown in Supplementary Figure 8g,h. Full gels of key experiments are shown in Supplementary Note 1.
We next investigated the biological significance of the association of HER2 with Grp94 at the plasma membrane. Because the Grp94 inhibitors described here target the chaperone activity of Grp94, we hypothesized that Grp94 acts on HER2 at the plasma membrane to stabilize the protein. In support of this, brief treatment of SKBr3 cells with PU-WS13 disrupted the circular architecture of HER2 at the plasma membrane, resulting in a ‘shredded’ HER2 pattern (Fig. 4a,c and Supplementary Fig. 8d,e). No such effect was observed upon direct HER2 inhibition with lapatinib, a small molecule that binds the ATP-regulatory pocket of HER2 (ref. 30) (Supplementary Fig. 8e). The effect of PU-WS13 on the HER2 surface architecture is thus mediated through Grp94.
Upon Grp94 inhibition, HER2 molecules translocated to early endosomes and plasma membrane–adjacent lysosomes (Fig. 4c and Supplementary Fig. 8d). Grp94-inhibited HER2 did not localize together with endoplasmic reticulum and Golgi structures (Supplementary Fig. 8d). Membrane but not cytosolic HER2 molecules were substantially reduced in a time-dependent manner upon Grp94 inhibition in SKBr3 cells (Fig. 4d), further demonstrating that Grp94 regulates HER2 specifically at the plasma membrane in SKBr3 cells.
In SKBr3 cells and other HER2-overexpressing breast cancer cells, the high-density HER2 formations at the cell membrane result in increased signaling and activation of several survival and proliferation-inducing signaling pathways, including Raf-MAPK, AKT and STAT3 (ref. 25). For the Raf-MAPK pathway, HER2 promotes retention of Raf-1 in the plasma membrane, resulting in prolonged activation of the MAP kinase cascade31. In further accord with a role for Grp94 in regulating HER2 function at the plasma membrane, we found that pharmacologic inactivation of Grp94 in SKBr3 cells resulted in a rapid inhibition of Raf-1–MAPK signaling at the membrane but not in the cytosol (Fig. 4e). Collectively, these findings indicate that in SKBr3 cells, Grp94 chaperoning is needed to maintain high-density HER2 architecture and signaling at the plasma membrane but not in the cytosol (Supplementary Fig. 8f).
Hsp90α and Hsp90β regulate cytosolic HER2
We next examined the role of Hsp90 in regulating cytosolic HER2. In SKBr3 cells, Hsp90α and Hsp90β inhibitors primarily modified the cytosolic HER2 species and failed to disturb the membrane HER2 architecture. As such, upon Hsp90α and Hsp90β inhibition, we observed a marked HER2 redistribution toward lysosomal and early endosomal structures that were distributed throughout the cytosol (Fig. 4c and Supplementary Fig. 8d). In addition, by 30 min after Hsp90α and Hsp90β inhibition, steady-state amounts of cytosolic but not membrane-associated HER2 greatly decreased (Fig. 4d), similar to what we have seen in MCF7 cells (Fig. 3c). Following cytosolic HER2 depletion, we noted a decrease in plasma membrane–associated HER2 (Fig. 4d), confirming the previously proposed role of Hsp90 in the trafficking and regulation of the cytosolic HER2 species26,27,32.
These data point to distinct Hsp90 paralog requirements for HER2 regulation that are dictated by proteome alterations in the cell (Fig. 5). In HER2-overexpressing cells, where maintenance of a high-density and high-signaling HER2 species is a mechanism for its oncogenic properties, the cell seems to use Hsp90α, Hsp90β and Grp94. Cytosolic HER2 chaperoning requires both Hsp90α and Hsp90β. The aberrantly high amounts of plasma membrane HER2 require Grp94. In cells with low HER2 expression, by contrast, the activity of Hsp90α alone seems sufficient to sustain HER2 function, although Hsp90β may compensate in cases of Hsp90α depletion.
Figure 5. Schematic representation summarizing the tumor-specific regulation of HER2 by the Hsp90 paralogs.

All epithelial cells contain two copies of the HER2-encoding gene and express small amounts of the HER2 receptor on the cell surface. During oncogenic transformation, the number of gene copies per cell may increase, as in the SKBr3 cell line, leading to an increase in mRNA transcription and a 100- to 1,000-fold increase in the number of HER2 receptors on the cell surface. Our data suggest a tumor-specific involvement of the Hsp90 paralogs in the chaperoning of HER2. The Hsp90α paralog is sufficient for HER2 trafficking and function in cells with low HER2 expression such as MCF7. In contrast, in cells with excessive amounts of HER2, such as in SKBr3, all three paralogs have an important role, with Hsp90α and Hsp90β regulating cytosolic HER2 and Grp94 regulating plasma membrane HER2. Because HER2 is the major oncogene in these cells, its dependence on Grp94 renders cells addicted to Grp94, proposing Grp94 as a new target in such cancers.
Viability of HER2-overexpressing cells is Grp94 dependent
We have identified an important role for Grp94 in plasma membrane HER2 stability and function in HER2-overexpressing SKBr3 cells. We thus asked whether inactivating Grp94 reduced SKBr3 cancer cell viability. Indeed, both Grp94 inhibition (Fig. 6a) and Grp94 knockdown (Fig. 6b) impaired SKBr3 viability. This effect was not limited to SKBr3 cells as we observed that all of the other tested HER2-overexpressing breast cancer cells were sensitive to Grp94 inhibition (Fig. 6a). Upon treatment of these cells with PU-WS13, a rapid accumulation of cells in the sub-G1 phase, PARP cleavage and a substantial increase in markers of early- and late-stage apoptosis were noted (Supplementary Fig. 9a–c).
Figure 6. Grp94 inhibition alone is sufficient to induce apoptosis in and reduce the viability of HER2 overexpressing breast cancer cells.

(a,b) Viability of breast cancer cells in which Grp94 was inhibited with PU-WS13 (a) or knocked down by means of siRNA (b). Cell viability was assessed using an assay that quantifies ATP. A representative western blot indicating protein changes in SKBr3 cells upon Grp94 knockdown is also shown in b. (c) Representative western blots of cancer cells treated for 24 h with PU-WS13 (0.5 μM, 2.5 μM and 12.5 μM) or vehicle. Full gels of key experiments are shown in Supplementary Note 1.
Notably, unlike the pan-Hsp90 and the cytosolic Hsp90 inhibitors, PU-WS13 failed to activate a feedback heat-shock response, as evidenced by little to no Hsp70 induction (Supplementary Figs. 7a and 9b). Hsp90α and Hsp90β inhibition alone, despite substantially depleting HER2, was less effective at killing these cells and instead elicited mostly cytostatic effects (Supplementary Fig. 9d,e). Neither inhibitor led to a substantial increase in Grp78, the endoplasmic reticulum Hsp70 paralog (Supplementary Fig. 9b). Downregulation of Grp94 also failed to induce Grp78 in SKBr3 cells (Supplementary Fig. 7b).
Discussion
In this report, we have identified therapeutically relevant, paralog-specific, purine-scaffold Hsp90 inhibitors and provided a rational basis for their selectivity. These results demonstrate that paralogs of Hsp90, although similar, interact with structurally related inhibitors in a very different manner. This apparent incongruity is explained by the fact that differences in protein conformation and pocket flexibility determine the ligand pose and, additionally, that preferred ligand poses influence the pocket conformation. This ligand-protein crosstalk, which was not anticipated by previous reports, seems to be the structural basis for selective inhibition.
The ligands we have identified show greater than 100-fold binding selectivity for Grp94 over the other three Hsp90 paralogs, a selectivity that is recapitulated in vivo. These chemical tools are sufficiently potent and selective to allow for the elucidation, in a time- and concentration-specific manner, of tumor cell–specific functions for the Hsp90 paralogs.
These tools have begun to offer new insights into the tumor- specific effects of the Grp94 paralog. Clinically, expression of Grp94 correlates with advanced stage and poor survival in a variety of cancers and is closely linked to cancer growth and metastasis7,33–35. The majority of the previous cancer-related studies on Grp94, however, have focused narrowly on the immunogenic activity of Grp94–peptide complexes33 and the involvement of this chaperone in the regulation of EGFR and HER2 in the endoplasmic reticulum, the secretion of IGF-I and IGF-II and the regulation of TLRs and integrins7,33,34. Thus, Grp94, despite being one of the most abundant chaperones, is still one of the least studied and least understood in the cancer cell context.
This state of affairs is largely due to two factors. First, there is no Grp94 homolog in a genetically tractable organism such as yeast. Second, and equally important, no small-molecule inhibitor with demonstrated selectivity for Grp94 and potential for use in cellular and animal systems has, until now, been developed, although initial efforts in this direction have been the subject of recent studies36. Approaches to the investigation of Grp94 that rely on mutant cell lines, cell assays and gene-deficient mouse studies have limitations: they address phenotypes in the complete absence of a gene and make use of an engineered cellular environment. Alternative strategies that address the role and biology of Grp94 in an endogenous cellular environment, where Grp94 is limiting but not absent, are therefore needed.
The power of the selective inhibitor–based approach to this problem is illustrated here. Grp94-specific inhibitors provide evidence for an unanticipated role for Grp94 in maintaining the architecture of high-density HER2 formations at the plasma membrane, particularly in cancer cells where HER2 is required to channel the amplified signaling through the receptor. Inhibition of Grp94 in these cells is sufficient to destabilize membrane HER2 and inhibit its signaling properties. These findings thus implicate Grp94 in regulating oncogenic signal transduction at the plasma membrane.
In contrast, no major requirement for Grp94 is found in cells with low HER2 plasma membrane expression. In this case, it is possible that other mechanisms maintain a functional HER2. But under conditions in which stress is imposed on the cell by proteome alterations (that is, HER2 overexpression), we show that the chaperoning function of Grp94 is vital for proper HER2 functioning.
As was previously reported for calreticulin, another endoplasmic reticulum chaperone, although Grp94 resides primarily in the lumen of the endoplasmic reticulum, it is observed at the cell surface under conditions of cell stress37,38. Previously, cell-surface Grp94 in cancer cells has been thought of as a peptide presentation tool for T-cell responses, an activity that is independent of its ATPase-regulated functions39. Our findings now implicate plasma membrane–translocated Grp94, in a phenotype-specific manner, in the chaperoning of a cancer protein.
Our data revise the previous notion of the role of Hsp90 paralogs in HER2 chaperoning1,26,27,29,32 and argue instead for their closer integration under conditions of cellular stress. Thus, in cells with low HER2 expression, Hsp90α alone is sufficient to maintain functional amounts of this tyrosine kinase. In contrast, HER2-overexpressing breast tumors recruit Hsp90β to buffer the increased HER2 molecules trafficked through the cytosol and Grp94 to stabilize the high-density HER2 formations at the plasma membrane. These results, revealed by the paralog-specific tool set we have developed, demonstrate the integration of the Hsp90 chaperone system, where distinct members are brought in to regulate a protein as dictated by the cellular need.
The HER2–Grp94 dependence we describe does not occur in all HER2-expressing cells but rather is a characteristic only of cells such as SKBr3 where high-density HER2 expression at the plasma membrane is needed to maintain the malignant phenotype. Our findings are thus in agreement with earlier studies showing that in other cells Hsp90 is sufficient to regulate cytosolic HER2 function26–28.
HER2 chaperoning by Grp94 in HER2-positive breast cancer has important clinical implications. Treatment with pan-Hsp90 inhibitors in HER2-positive breast cancer patients resulted in a 59% overall clinical benefit rate in trastuzumab-refractory disease40. Given our current results, these effects are most likely mediated in large part by Grp94 inhibition. Grp94 inhibition thus presents an exciting therapeutic opportunity because the advantage of the pan-Hsp90 inhibitors may be obtained without feedback upregulation of anti-apoptotic and resistance-mediating heat shock proteins, such as Hsp70 (refs. 40,41).
In conclusion, our work provides a new tool set of Grp94- and Hsp90-selective inhibitors that we show are specific for their targets both in biochemical and in cellular systems. These represent unique chemical tools to investigate Hsp90 paralog mechanisms where Hsp90s are limiting but not absent. We expect that they will allow for the identification of new cancer mechanisms, as exemplified here not only by the identification of a new role for Grp94 in regulating HER2 but also by the discovery of a cell-specific and proteome alteration–driven regulation of HER2 by Hsp90 and Grp94. The mechanisms we detail here set the ground for new treatments of HER2-positive breast cancers with a Grp94 inhibitor. Finally, our studies are also first to uncover a new, therapeutically viable binding site in Grp94 that could lead to the design of even more potent and selective inhibitors.
Online Methods
Sequence alignment
Sequences were aligned and shown as Percentage Identity view using the program of T-Coffee Multiple Sequence Alignment in Jalview 2.7 (http://www.tcoffee.org/Projects/tcoffee/).
Molecular modeling
All computations were carried out on a HP workstation xw8200 with the Ubuntu 8.10 operating system.
Homology model for Trap-1
The protein structures of the Hsp90α NTD (PDB code 2FWZ), Grp94 NTD (PDB code 3O2F) and the amino acid sequence of Trap-1 protein (Uniprot accession number Q12931) were used for model building. To create the model, the protein sequence of Trap-1 protein (accession number: Q12931) was entered as an input sequence in Prime’s Structure Preparation wizard (Schrödinger LLC., New York). The homologous protein Hsp90α (PDB code 2FWZ), with 31% identities, 47% positives and 20% gaps, and Grp94 (PDB code 3O2F), with 28% identities, 45% positives and 28% gaps, were imported. The NTD Trap-1 sequence and the templates were aligned and then edited using parameters as implemented in Prime. In the ‘Build structure’ option of Prime, amino acids 179–196 (Grp94) were selected from the structure deposited under PDB code 3O2F, whereas the remaining amino acids were from Hsp90α (PDB code 2FWZ). The structure was then built using selected sequence alignment of the template (or templates), taking solvent, ligand, force field and other contributions into account via a series of algorithms implemented in Prime. Structural discontinuities were optimized by inserting template gaps for more than 20 residues. All of the loops were refined with the default parameter settings of Prime. The obtained homology model of Trap-1 was further refined using the protein preparation wizard available in Maestro (version 8.5). Partial atomic charges were assigned according to the OLPS-AA force field. To obtain a more reliable three-dimensional structure of Trap-1, the homology model was further subjected to a series of energy minimization steps that consisted of 5,000 iterations of steepest descent (SD) and conjugate gradient (CG), until the root mean-square deviation (r.m.s. deviation) was lower than 0.001 kcal mol−1 Å−1.
Ligand preparation
All of the compounds were constructed using the fragment dictionary of Maestro (version 8.5). The geometry of compounds was optimized using the Macromodel program (version 9.6) and the OLPS-AA force field42. Resulting ligands were further prepared using the Ligprep (version 2.2) utility provided by Schrödinger LLC., New York.
Docking
The X-ray crystal structure of Hsp90α NTD in complex with PU-H71 (PDB code 2FWZ), Hsp90β NTD in complex with EC144 (PDB code 3NMQ), Grp94 NTD in complex with PU-H54 (PDB code 3O2F), ADP (PDB code 1TC6) and unliganded (PDB code 1YT0) and Trap-1 homology model were first aligned using the protein structure alignment tool and then were optimized for subsequent grid generation and docking using the default parameters in Protein Preparation Wizard provided by Schrödinger LLC. Grids were then prepared using the Receptor Grid Generation tool in Glide (version 5.0)43–45. Next, the extra precision (XP) Glide docking method was used to dock compounds flexibly into the ATP binding site of the Hsp90 paralogs. Upon completion of each docking calculation, at most 100 poses per docking were run and at most 10 poses per ligand were allowed to be generated. Top-scored docking poses (orientation plus conformation) based on the Glide scoring (GScore) function were analyzed. To validate docking parameters and experimental set-up, endogenous ligands (PU-H71, PDB code 2FWZ; EC144, PDB code 3NMQ; PU-H54, PDB code 3O2F; ADP, PDB code 1TC6) were removed from the binding site and redocked. Very good agreement was found between inhibitor pose as obtained from docking analyses and as captured in the crystal structure (r.m.s. deviation of 0.7 Å; PDB code 2FWZ, 0.9 Å; PDB code 3NMQ, 0.04 Å; PDB code 3O2F and 1.2 Å; PDB code 1TC6) between the predicted conformation and the observed X-ray crystallographic conformation, validating the docking strategy.
Binding site analysis
SiteMap46 analysis was carried out on the X-ray crystal structures of Hsp90α (PDB code 2FWZ), Hsp90β (PDB code 3NMQ) and Grp94 (PDB code 3O2F) and the refined homology model of Trap-1 using ‘Evaluate a single binding site region’ using default parameters implemented in SiteMap (version 2.2). Next, to investigate the ATP-binding site, hydrophobic and hydrophilic contour maps were constructed using default parameters as implemented in the ‘Manage surfaces’ function.
Reagents
Recombinant Hsp90α (ADI-SPP-776), Hsp90β (ADI-SPP-777) and Trap-1 (ADI-SPP-848) were purchased from Enzo Life Sciences. Grp94 was generated as previously reported12,14. The synthesis and characterization of PU-11, PU-20F, PU-29F, PU-H39, PU-H71, PU-H71 beads, cy3B-GM and PU-FITC3 were reported elsewhere10,24,47–51. The compounds were fully characterized, and structures were confirmed by direct comparison to previous reports and determined to have a purity of >98%. Geldanamycin (≥98%) was purchased from Sigma-Aldrich, and lapatinib (≥98%) was purchased from Selleck Chemicals. The synthesis and characterization of PU-H54, PU-WS13 and PU-WS13-biotin are described in Supplementary Note 2.
Cell lines
The HER2-overexpressing breast cancer cells SKBr3, BT474, MDA-MB-361, MDA-MB-453 and AU565 as well as the low HER2 breast cancer cells MCF7, BT20 and MDA-MB-231 were obtained from the American Type Culture Collection (ATCC). Cells were cultured routinely in McCoy’s 5A (10% FBS, SKBr3), DME/F12 (10% FBS, BT474 and MDA-MB-231), RPMI (10% FBS, AU565), MEM (10% FBS, MCF7 and BT20) and L-15 (20% FBS, MDA-MB-361 and MDA-MB-453) supplemented with 1% Glutamax and 1% penicillin and streptomycin (Pen/Strep). C2C12 and HEK293 cells were purchased from ATCC and cultured in DMEM in the presence of 10% FBS and 1% penicillin/streptomycin. When cultured, cells in L-15 medium were kept in a humidified atmosphere without CO2 at 37 °C, and all of the other cell lines were incubated in the humidified cell incubators with CO2 at 37 °C.
Statistical analysis
The results were analyzed by unpaired two-tailed Student’s t-tests in Prism5 (GraphPad). Data are presented as the mean ± s.d. or s.e.m. of duplicates or triplicates. Error bars represent the mean s.d. or s.e.m. When a single panel is presented, it is representative of two or three individual experiments.
Compound screening and fluorescence polarization measurements
The Hsp90 FP competition assays were performed on an Analyst GT instrument (Molecular Devices, Sunnyvale, CA) and carried out in black 96-well micro-plates (Corning, no. 3650) in a total volume of 100 μL in each well. A stock of 10 μM cy3B-GM and PU-FITC3 was prepared in DMSO and diluted with Felts buffer (20 mM Hepes (K), pH 7.3, 50 mM KCl, 2 mM DTT, 5 mM MgCl2, 20 mM Na2MoO4 and 0.01% NP40 with 0.1 mg/mL BGG). To each well was added the fluorescent dye–labeled Hsp90 ligand (6 nM cy3B-GM for Hsp90α, Hsp90β and Grp94 and 3 nM PU-FITC3 for Trap-1), protein (10 nM Hsp90α, 10 nM Hsp90β, 10 nM Grp94, 30 nM Trap-1) and tested inhibitor (initial stock in DMSO) in a final volume of 100 μL Felts buffer. Compounds were added in duplicate or triplicate wells. For each assay, background wells (buffer only), tracer controls (free, fluorescent dye–labeled Hsp90 ligand only) and bound controls (fluorescent dye–labeled Hsp90 ligand in the presence of protein) were included on each assay plate. The assay plate was incubated on a shaker at 4 °C for 24 h, and the FP values (in mP) were measured. The fraction of fluorescent dye–labeled Hsp90 ligand bound to Hsp90 was correlated to the mP value and plotted against values of competitor concentrations. The inhibitor concentration at which 50% of bound fluorescent dye–labeled Hsp90 ligand was displaced was obtained by fitting the data. For cy3B-GM, an excitation filter at 530 nm and an emission filter at 580 nm were used with a dichroic mirror of 561 nm. For PU-FITC3, an excitation filter at 485 nm and an emission filter at 530 nm were used with a dichroic mirror of 505 nm. All of the experimental data were analyzed using SOFTmax Pro 4.3.1 and plotted using Prism 5.0 (GraphPad Software Inc., San Diego, CA), and binding affinity values are given as relative binding affinity values (EC50, concentration at which 50% of fluorescent ligand was competed off by compound).
Cell fractionation and immunoblotting
Cells were either treated with DMSO (vehicle) or indicated compounds for 24 h and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate and 0.5% NP40) supplemented with cocktail protease inhibitors (Roche) to produce whole-cell lysates. Lysates for cytosol and membrane fractions were harvested using ProteoExtract Subcellular Proteome Extraction Kit (Calbiochem) following the manufacturer’s instructions. Protein concentrations were determined using the BCA kit (Pierce) according to the manufacturer’s instructions. The protein lysates (5–50 μg) were electrophoretically resolved by SDS-PAGE, transferred onto nitrocellulose membranes and probed with primary antibodies against HER2 (Zymed; 28004; 1:500), Hsp70 (Stressgen; SPA-810; 1:2,500), Grp94 (Stressgen; SPA-850; 1:2,500), Hsp90α (Abcam; Ab2928; 1:2,000), Hsp90β (StressMarq; SMC-107B; 1:2,500), Grp78 (Cell Signaling; 3183; 1:1,000), Raf-1 (Santa Cruz; sc-133; 1:500), phospho–Raf-1 (Cell Signaling; 9421; 1:500), MEK1/2 (Cell Signaling; 8727; 1:1,000), phospho–MEK1/2 (Cell Signaling; 9154; 1:500), ERK1/2 (Cell Signaling; 4695; 1:2,000), phospho–ERK1/2 (Cell Signaling; 4370; 1:2,000), AKT (Cell Signaling; 9272; 1:1,000), GM130 (Cell Signaling; 2296; 1:1,000), Flotillin 2 (Cell Signaling; 3436; 1:2,000), Histone H4 (Cell Signaling; 2592; 1:1,000), Histone H1 (Santa Cruz; sc-8030; 1:250), Caspase 3 (Cell Signaling; 9665; 1:500), cleaved PARP (Promega; G7341; 1:2,500), α-tubulin (Sigma; T5168; 1:2,000) and β-actin (Sigma; A1978; 1:3,000). After washing off the excess antibodies, the membranes were incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibody. Blots were visualized by autoradiography using the Enhanced Chemiluminescence Detection System (GE Healthcare) according to manufacturer’s instructions. For all of the gels, β-actin was used as a protein loading control.
Densitometry analysis
Films were scanned in Adobe Photoshop CS5 and quantitative densitometric analysis was performed using ImageJ (US National Institutes of Health).
Protein quantification
In all of the instances when protein quantification was performed, protein amounts were first normalized to β-actin then to the amounts of the vehicle-only experimental conditions. All of the quantified protein amounts are reported as a fraction of control (that is, the value obtained in the experimental condition was divided by the value obtained in the vehicle-treated cells).
Chemical precipitation
Agarose beads conjugated with Hsp90 inhibitors were washed three times with and finally suspended in Felts buffer (20 mM HEPES, 50 mM KCl, 5 mM MgCl2, 0.01% NP40, 20 mM Na2MoO4, pH ~7.2–7.3). The bead conjugates (50 μL) were then incubated for 4 h at 4 °C with cell lysates, and the volume was adjusted to 500 μl with Felts buffer. The complexes were then washed three times with Felts buffer, and proteins in the pulldown were analyzed by immunoblotting. For PU-WS13-biotin pulldown assays, the cell lysate was first incubated overnight at 4 °C with biotinylated PU-WS13 and then for 2 h with 50 μL High Capacity Streptavidin Beads (Thermosci). The beads were washed three times with Felts buffer, and the proteins in the pulldowns were identified by immunoblotting. Control beads containing 2-methoxyethylamine, an Hsp90-inert molecule, or D-biotin were used to control for nonspecific binding.
Immunoprecipitation
The HER2 antibody (Cell Signaling; 2165; 1:100), the Grp94 antibody (Abcam; Ab13509; 1:200) or a normal rabbit IgG (Santa Cruz Biotechnology; 1:100) was incubated with the cell lysate and with 40 μL protein A agarose beads (Roche). The mixture was incubated overnight on a rotator at 4 °C. The beads were washed three times with RIPA buffer and separated by SDS-PAGE, followed by a standard immunoblotting procedure.
Grp94 depletion assay
The Grp94 antibodies (Grp94-1; Abcam; Ab13509, 1:200; Grp94-2, Bioss; bs-0194R; 1:50) or a normal rabbit IgG (Santa Cruz Biotechnology; 1:100) was incubated with the cell lysate and with 40 μL protein A agarose beads (Roche). The mixture was incubated for 4 h on a rotator at 4 °C. The supernatants were collected after centrifugation and then incubated with the Grp94 antibody or a normal rabbit IgG and then with 40 μL protein A agarose beads to further deplete Grp94 in the cell lysate. After three rounds of antibody depletions, the supernatants were collected and incubated overnight with the PU-WS13-biotin beads at 4 °C. The beads were washed three times with Felts buffer and separated by SDS-PAGE, followed by a standard immunoblotting procedure.
siRNA knock-down of Hsp90α, Hsp90β and Grp94
Transient transfections were carried out using Lipofectamine RNAiMax reagent (Invitrogen, for SKBr3 cells) or electroporation with Neon transfection system (Life Technologies, for MCF7 cells.) according to the manufacturer’s instructions. For each target, different siRNAs were designed against the open reading frame of Hsp90α (Gene HSP90AA1), Hsp90β (Gene HSP90AB1) or Grp94 (Gene HSP90B1) and purchased from Qiagen. Control cells were transfected with scramble siRNA. Cells were transfected with 20 nM siRNA, and knockdown efficiency was evaluated at 72 h post transfection by immunoblotting. Electroporation in MCF7 was optimized, and the experiments were performed using two 1,230-V 20-ms pulses on Neon transfection system (Life Technologies). SKBr3 cells were transfected with 20 nM siRNAs for 72 h and then retransfected with 20 nM siRNAs for another 48 h before western blotting analysis.
Cell viability assessment
Cells were treated for 72 h with inhibitors or transfected with Grp94 siRNA or control siRNA, and their viability was assessed using CellTiter-Glo luminescent Cell Viability Assay (Promega), as previously described52,53. The method determines the number of viable cells in culture based on quantification of the ATP present, which signals the presence of metabolically active cells.
Immunofluorescence
Cells were seeded and grown onto culture slides (BD Falcon) for 24 h. After washing with cold PBS, cells were fixed by treating at 4 °C for 20 min with 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS containing 10% FBS for 10 min and blocked with 2% BSA for 1 h. After washing four times with PBS, primary antibodies were added onto the chambers, and then cells were incubated overnight at 4 °C and washed again with PBS, followed by incubation with the secondary antibody for 1 h at room temperature. Cell were washed and then mounted and observed under microscope (Leica Upright Confocal SP5). The primary antibodies used in the assay are against HER2 (Zymed; 28004; 1:50), Grp94 (Stressgen; SPA-850; 1:100), Hsp70 (Stressgen; SPA-810; 1:200), LAMP1-FITC (Abcam; ab25406; 1:100), EEA1 (Abcam; ab70521; 1:100), 58K Golgi-FITC (Abcam; ab27043; 1:50) and Calnexin (BD; 610523; 1:50).
Flow cytometry
Flow cytometry analysis was performed using either MACSQuant analyzer (Miltenyi Biotec) or BD LSRII flow cytometer. For surface Grp94 detection, cells were collected by centrifugation at 500g for 5 min. Excess medium was removed, and the cell pellet was resuspended in cold medium containing human AB serum for blocking. Then the primary antibody Grp94-PE (Enzo; SPA-850PE; 1:200) or an isotype control (BD Pharmingen; 557229; 1:25) was added to each tube and incubated with cells for 1 h on ice. Cells were stained on ice with 7-AAD (BD Pharmingen) for 15 min, resuspended in 1% paraformaldehyde and subjected to the analysis on the flow cytometer. Data were further analyzed by FlowJo (Ashland). Dead cells with positive 7-AAD staining were excluded from the analysis. To assess inhibition of protein trafficking by Brefeldin A, cells were treated with GolgiPlug (BD Biosciences, 555029) for 4 h according to the manufacturer’s instructions, then either processed for live-cell staining or first permeabilized with 0.1% Triton X-100 before flow analysis. For cell cycle analysis, cells were washed and fixed in cold 70% ethanol overnight at 4 °C. Fixed cells were collected and stained with PBS containing PI and DNase-free RNase A (Sigma-Aldrich) for 1 h at room temperature in the dark. DNA content was measured, and the data were further analyzed using FloJo. The chicken erythrocyte nuclei singlets (CEN, Biosure) were used as the reference. For apoptosis assessment, cells were finally stained with Annexin V-FITC and 7-AAD for 15 min at room temperature in the dark. Signals from FL1 and FL3 channels were collected and analyzed. Early apoptosis was defined as Annexin V (+)/7-AAD (−), and late apoptosis was observed as Annexin V (+)/7-AAD (+).
Assessment of cell-surface proteins
A cell surface protein isolation kit (Pierce) was used to biotinylate proteins on the cell surface according to the manufacturer’s instructions. Briefly, four 75-cm2 flasks of cells were incubated with Sulfo-NHS-SS-biotin for 30 min at 4 °C, and then the reaction was quenched, and cells were lysed. The biotinylated proteins were isolated using NeutrAvidin Agarose beads, and then eluted with Laemmli buffer and subjected to SDS-PAGE analysis and immunoblotting. Alternatively, after biotin labeling of the cell-surface proteins, the biotinylated proteins were purified by using monomeric Avidin beads, followed by elution of proteins from the beads by incubation with 2 mM biotin for 6 h at 4 °C.
C2C12 differentiation and IGF-II secretion assay
C2C12 cells were maintained and cultured in DMEM in the presence of 10% FBS and 1% penicillin/streptomycin (culture medium). C2C12 is an immortal line of mouse skeletal myoblasts originally derived from satellite cells from the thigh muscle of a 2-month-old female mouse donor. These cells differentiate well into myocytes under appropriate culture conditions22,54. Here, cells were induced to differentiate by replacing the culture medium with DMEM supplemented with 2% horse serum and 1% penicillin/streptomycin (differentiation medium) for 36–48 h. Secreted IGF-II was quantified by using IGF-II mouse ELISA kit (Abcam, AB100696) according to the manufacturer’s instructions. Briefly, after shifting the culture medium to differentiation medium, C2C12 cells were treated for 24 h with the indicated compounds. Medium from each experimental condition was then transferred into ELISA plates coated with anti–IGF-II and incubated overnight at 4 °C. The bound IGF-II was detected with a biotinylated anti–IGF-II antibody provided in the kit. After the sequential incubation with HRP-conjugated streptavidin, TMB One-step substrate reagent and the Stop solution, the absorbance was measured at 450 nm. The secreted IGF-II was quantified against a standard curve generated with recombinant IGF-II provided by the kit.
TLR9-trafficking assay
HEK 293T cells were transfected with pUNO-hTLR9-HA (Invivogen) using X-tremgene HP (Roche) according to the manufacturer’s instructions. At 24 h after transfection, cells were split onto cell culture chamber slides (Lab-Tek). Cells were then treated for 24 h with compounds at varying concentrations. After treatment, cells were fixed for 20 min in 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked with 3% BSA in PBS for 30 min, followed by staining for 1 h with anti-HA (Abcam; ab9110; 1:250) or a normal rabbit IgG (Abcam; ab37415; 1:250). Cells were washed with PBS, stained with an anti– rabbit-Cy3 antibody (Invitrogen; A10520; 1:400) and finally mounted in the dark at 4 °C with with one drop of Prolong Gold Antifade reagent (with DAPI, Life Technologies, P36935). Cells were visualized under a confocal microscope (Leica Upright Confocal SP5). Fluorescence intensity was quantified using MetaMorph Microscopy Automation and Image Analysis Software (Molecular Devices Inc.) and normalized to the cell number.
Preparation of crude plasma membranes
All of the steps were performed at 4 °C, and all of the buffers were chilled on ice before use. The cells were gently scraped in PBS, pelleted by centrifugation at 600g for 5 min and resuspended in 1 mL 1× Hypotonic Extraction Buffer (Sigma, H8412, 10 mM HEPES, pH 7.8, 1 mM EGTA, 25 mM KCl) for 20 min to allow the cells to swell. Then, cells were collected at 1,000g for 5 min, resuspended in 0.5 mL 1× Isotonic Extraction Buffer (Sigma, I3533, 10 mM HEPES, pH 7.8, 0.25 M sucrose, 1 mM EGTA, 25 mM KCl), homogenized with 20 strokes of the Dounce homogenizer and then centrifuged for 10 min at 1,000g. The supernatant with the floating lipid layer was carefully collected and layered on top of 12 mL 30% Percoll (Sigma, P4937) in Isotonic Extraction Buffer, followed by ultracentrifugation at 28,184 r.p.m. in a TH641 rotor (Thermo Scientific) for 45 min55. The crude plasma membrane fraction was visible as a ring at 5.4 cm from the bottom of the tube.
Crystallization of Grp94– and hHsp90–PU-H54 complexes
Recombinant canine Grp94NΔ41 (69–337 Δ278–327) and human Hsp90αN (1-236) were expressed as GST- and His-tagged fusions, respectively, and purified as described previously11,12. Prior to crystallization, protein-inhibitor complexes were formed by the addition of a twofold molar excess of PU-H54 to Grp94NΔ41 at 30 mg/mL or a threefold molar excess of PU-H54 to human Hsp90αN at 20 mg/ml in 10 mM Tris, pH 7.6, 100 mM NaCl and 1 mM DTT. Grp94NΔ41 crystals were grown by hanging-drop vapor diffusion at 18 °C by mixing a 1:1 ratio of protein to reservoir solution containing 14–17% isopropanol, 300–375 mM MgCl2, 0.1–1.0% glycerol and 100 mM Hepes, pH 7.4. Grp94NΔ41 complex crystals were cryoprotected by rapid passage through a solution containing 30% glycerol, 5% isopropanol and 100 mM Hepes, pH 7.4, before flash freezing in liquid nitrogen. Hsp90αN crystals were grown by hanging-drop vapor diffusion at 4 °C by mixing a 1:1 ratio of protein to reservoir solution containing (11–15% PEG 2K MME, 200 mM MgCl2 and 100 mM sodium cacodylate, pH 6.5). Hsp90αN crystals were cryoprotected by sequentially passing through reservoir solution rapidly followed by a cryoprotectant solution containing 35% PEG 2K MME, 200 mM MgCl2 and 100 mM sodium cacodylate, pH 6.5, before flash freezing in liquid nitrogen.
Data collection, structure determination and refinement
X-ray diffraction data for the Grp94NΔ41 plus PU-H54 (ps23b3a_1) and human Hsp90N plus PU-H54 (ps3c6a) co-crystals were collected on a MAR-325 CCD detector at SSRL beamline 11-1 using an X-ray wavelength of 0.979 Å. Data were reduced and scaled using HKL2000. Initial phases for the co-crystals were obtained by molecular replacement using Phaser software in the CCP4 software suite56. The search model for Grp94NΔ41 was the core region (residues 100–166 and 200–337) of Grp94NΔ41 plus ATP (PDB code 1TC0), and the search model for hHsp90 was hHsp90 plus PU-H71 (PDB code 2FWZ). Initial molecular replacement models were manually rebuilt in Coot57 and refined using Refmac 5.5 in CCP4 (ref. 58). Ligand topology files for PU-H54 were generated using the Dundee PRODRG server. For the Grp94NΔ41 plus PU-H54 structure, density modification was carried out using DM software in CCP4, and TLS parameters generated using TLSMD were applied in the final stage of refinement59,60. Final models contained 0% Ramachandran outliers with 95.1% and 97.6% of the residues appearing in Ramachandran-favored regions for the GRP94NΔ41 plus PU-H54 and Hsp90N plus PU-H54 structures, respectively.
Supplementary Material
Acknowledgments
G.C. is funded by the Breast Cancer Research Fund, R01 CA172546-01A1, U01 AG032969-01A1, R21 AI090501, R21 CA158609-01 and R01 CA155226-01. P.Y. is supported by the Translational and Integrative Medicine Research Fund of Memorial Sloan-Kettering Cancer Center. P.D.P. and R.A.S. are supported by funds and resources from St. John’s University. D.T.G. is funded by R01 CA095130. We thank Y. Argon (Children’s Hospital of Philadelphia) and C. Leifer (Cornell University College of Veterinary Medicine) for reagents.
Footnotes
Accession codes. Protein Data Bank (PDB): Structure factors and coordinates were deposited under accession codes 3O2F for Grp94N-PU-H54 and 3O0I for Hsp90N-PU-H54.
Author contributions
P.D.P., P.Y., H.J.P. and C.Y. performed the chemistry and chemical biology experiments, and T.T. and W.S. provided reagents. P.M.S., N.S.Q. and D.T.G. carried out the crystal structure determination and analysis. All of the authors participated in the design and analysis of various experiments, and G.C., D.T.G. and P.D.P. wrote the paper.
Competing financial interests
The authors declare competing financial interests: details accompany the online version of the paper.
Additional information
Supplementary information, chemical compound information and chemical probe information is available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone Hsp90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann NY Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 2.Sreedhar AS, Kalmar E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004;562:11–15. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 3.Johnson JL. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim Biophys Acta. 2012;1823:607–613. doi: 10.1016/j.bbamcr.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 4.Chène P. ATPases as drug targets: learning from their structure. Nat Rev Drug Discov. 2002;1:665–673. doi: 10.1038/nrd894. [DOI] [PubMed] [Google Scholar]
- 5.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410:439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 6.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marzec M, Eletto D, Argon Y. GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta. 2012;1823:774–787. doi: 10.1016/j.bbamcr.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen B, Piel WH, Gui L, Bruford E, Monteiro A. The HSP90 family of genes in the human genome: insights into their divergence and evolution. Genomics. 2005;86:627–637. doi: 10.1016/j.ygeno.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Schulte TW, et al. Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol Endocrinol. 1999;13:1435–1448. doi: 10.1210/mend.13.9.0339. [DOI] [PubMed] [Google Scholar]
- 10.Taldone T, et al. Experimental and structural testing module to analyze paralog-specificity and affinity in the Hsp90 inhibitors series. J Med Chem. 2013 doi: 10.1021/jm400619b. http://dx.doi.org/10.1021/jm400619b. [DOI] [PMC free article] [PubMed]
- 11.Immormino RM, Kang Y, Chiosis G, Gewirth DT. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J Med Chem. 2006;49:4953–4960. doi: 10.1021/jm060297x. [DOI] [PubMed] [Google Scholar]
- 12.Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94: basis for ligand specificity and regulation. J Biol Chem. 2003;278:48330–48338. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- 13.Immormino RM, et al. Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. J Mol Biol. 2009;388:1033–1042. doi: 10.1016/j.jmb.2009.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dollins DE, Immormino RM, Gewirth DT. Structure of unliganded GRP94, the endoplasmic reticulum Hsp90. Basis for nucleotide-induced conformational change. J Biol Chem. 2005;280:30438–30447. doi: 10.1074/jbc.M503761200. [DOI] [PubMed] [Google Scholar]
- 15.Richter K, Reinstein J, Buchner J. A Grp on the Hsp90 mechanism. Mol Cell. 2007;28:177–179. doi: 10.1016/j.molcel.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Ali MM, et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leskovar A, Wegele H, Werbeck ND, Buchner J, Reinstein J. The ATPase cycle of the mitochondrial Hsp90 analog Trap1. J Biol Chem. 2008;283:11677–11688. doi: 10.1074/jbc.M709516200. [DOI] [PubMed] [Google Scholar]
- 18.Chiosis G, Kang Y, Sun W. Discovery and development of purine-scaffold Hsp90 inhibitors. Expert Opin Drug Discov. 2008;3:99–114. doi: 10.1517/17460441.3.1.99. [DOI] [PubMed] [Google Scholar]
- 19.Chiosis G, et al. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol. 2001;8:289–299. doi: 10.1016/s1074-5521(01)00015-1. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, et al. Development of a fluorescence polarization assay for the molecular chaperone Hsp90. J Biomol Screen. 2004;9:375–381. doi: 10.1177/1087057104265995. [DOI] [PubMed] [Google Scholar]
- 21.Wright L, et al. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem Biol. 2004;11:775–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 22.Ostrovsky O, Ahmed NT, Argon Y. The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol Biol Cell. 2009;20:1855–1864. doi: 10.1091/mbc.E08-04-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–226. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moulick K, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 26.Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. 1996;271:22796–22801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- 27.Xu W, et al. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001;276:3702–3708. doi: 10.1074/jbc.M006864200. [DOI] [PubMed] [Google Scholar]
- 28.Xu W, Mimnaugh EG, Kim JS, Trepel JB, Neckers LM. Hsp90, not Grp94, regulates the intracellular trafficking and stability of nascent ErbB2. Cell Stress Chaperones. 2002;7:91–96. doi: 10.1379/1466-1268(2002)007<0091:hngrti>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chavany C, et al. p185erbB2 binds to GRP94 in vivo Dissociation of the p185erbB2/GRP94 heterocomplex by benzoquinone ansamycins precedes depletion of p185erbB2. J Biol Chem. 1996;271:4974–4977. doi: 10.1074/jbc.271.9.4974. [DOI] [PubMed] [Google Scholar]
- 30.Kim TE, Murren JR. Lapatinib ditosylate GlaxoSmithKline. IDrugs. 2003;6:886–893. [PubMed] [Google Scholar]
- 31.Zhang L, Bewick M, Lafrenie RM. EGFR and ErbB2 differentially regulate Raf-1 translocation and activation. Lab Invest. 2002;82:71–78. doi: 10.1038/labinvest.3780396. [DOI] [PubMed] [Google Scholar]
- 32.Xu W, et al. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci USA. 2002;99:12847–12852. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng H, Dai J, Stoilova D, Li Z. Cell surface targeting of heat shock protein gp96 induces dendritic cell maturation and antitumor immunity. J Immunol. 2001;167:6731–6735. doi: 10.4049/jimmunol.167.12.6731. [DOI] [PubMed] [Google Scholar]
- 34.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eletto D, Dersh D, Argon Y. GRP94 in ER quality control and stress responses. Semin Cell Dev Biol. 2010;21:479–485. doi: 10.1016/j.semcdb.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duerfeldt AS, et al. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134:9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cabanes D, et al. Gp96 is a receptor for a novel Listeria monocytogenes virulence factor, Vip, a surface protein. EMBO J. 2005;24:2827–2838. doi: 10.1038/sj.emboj.7600750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Na X, Kim H, Moyer MP, Pothoulakis C, LaMont JT. gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect Immun. 2008;76:2862–2871. doi: 10.1128/IAI.00326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ostrovsky O, Makarewich CA, Snapp EL, Argon Y. An essential role for ATP binding and hydrolysis in the chaperone activity of GRP94 in cells. Proc Natl Acad Sci USA. 2009;106:11600–11605. doi: 10.1073/pnas.0902626106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modi S, et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17:5132–5139. doi: 10.1158/1078-0432.CCR-11-0072. [DOI] [PubMed] [Google Scholar]
- 41.Whitesell L, Bagatell R, Falsey R. The stress response: implications for the clinical development of hsp90 inhibitors. Curr Cancer Drug Targets. 2003;3:349–358. doi: 10.2174/1568009033481787. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 43.Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 1 Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 44.Halgren TA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2 Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 45.Friesner RA, et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 46.Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 47.Chiosis G, Lucas B, Shtil A, Huezo H, Rosen N. Development of a purine-scaffold novel class of Hsp90 binders that inhibit the proliferation of cancer cells and induce the degradation of Her2 tyrosine kinase. Bioorg Med Chem. 2002;10:3555–3564. doi: 10.1016/s0968-0896(02)00253-5. [DOI] [PubMed] [Google Scholar]
- 48.Taldone T, et al. Design, synthesis, and evaluation of small molecule Hsp90 probes. Bioorg Med Chem. 2011;19:2603–2614. doi: 10.1016/j.bmc.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Llauger L, et al. Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90. J Med Chem. 2005;48:2892–2905. doi: 10.1021/jm049012b. [DOI] [PubMed] [Google Scholar]
- 50.He H, et al. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J Med Chem. 2006;49:381–390. doi: 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- 51.Moulick K, et al. Synthesis of a red-shifted fluorescence polarization probe for Hsp90. Bioorg Med Chem Lett. 2006;16:4515–4518. doi: 10.1016/j.bmcl.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 52.Rodina A, et al. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat Chem Biol. 2007;3:498–507. doi: 10.1038/nchembio.2007.10. [DOI] [PubMed] [Google Scholar]
- 53.Caldas-Lopes E, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci USA. 2009;106:8368–8373. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wanderling S, et al. GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol Biol Cell. 2007;18:3764–3775. doi: 10.1091/mbc.E07-03-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sokolowska I, et al. Proteomic analysis of plasma membranes isolated from undifferentiated and differentiated HepaRG cells. Proteome Sci. 2012;10:47. doi: 10.1186/1477-5956-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 58.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 59.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 60.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Crystallogr. 2006;39:109–111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.