

Abstract
We introduce the field of Hamiltonian medicine, which centres on the roles of genetic relatedness in human health and disease. Hamiltonian medicine represents the application of basic social-evolution theory, for interactions involving kinship, to core issues in medicine such as pathogens, cancer, optimal growth and mental illness. It encompasses three domains, which involve conflict and cooperation between: (i) microbes or cancer cells, within humans, (ii) genes expressed in humans, (iii) human individuals. A set of six core principles, based on these domains and their interfaces, serves to conceptually organize the field, and contextualize illustrative examples. The primary usefulness of Hamiltonian medicine is that, like Darwinian medicine more generally, it provides novel insights into what data will be productive to collect, to address important clinical and public health problems. Our synthesis of this nascent field is intended predominantly for evolutionary and behavioural biologists who aspire to address questions directly relevant to human health and disease.
Keywords: inclusive fitness, health, disease, kinship
1. Introduction
The term Darwinian medicine refers to the application of evolutionary concepts and tools to understand health and the causes and treatments of disease [1]. This field has been predicated on the roles of natural selection, and other evolutionary processes, in mediating disease risks and symptoms. In this context, Darwinian medicine focuses mainly on the interfaces of fitness maximization, by humans and their parasites, with deviations from health. Health is considered here, following the World Health Organization definition, as ‘a state of complete physical, mental and social well-being and not merely the absence of disease or infirmity’ [2, p. 1]. As described below, the correspondence of this state with evolutionary concepts of fitness remains a matter for inquiry.
Humans are, of course, expected to maximize not simply fitness, but Hamilton's inclusive fitness, which integrates individual reproduction with effects on reproduction of other individuals that may carry one's genes identical by descent [3,4]. A phenotype is thus favoured if benefits (b) multiplied by genetic relatedness (r) to another individual, minus costs (c) to self, are greater than zero (rb − c > 0), which represents Hamilton's famous rule. Inclusive fitness has been recognized by theorists and practitioners of Darwinian medicine as a fundamental metric of natural selection [4]. However, its precepts have yet to be systematically integrated into understanding the evolutionary bases of human health and disease.
Foster [5] introduced the term Hamiltonian medicine originally to describe the impacts on human infectious disease of evolution driven by social interactions among microbes. In this review, we extend and generalize Foster's concept of Hamiltonian medicine to encompass the roles of genetic relatedness in all aspects of human health and disease, including both infectious and non-infectious causes. In doing so, we describe a set of central principles for Hamiltonian medicine, which are intended to guide and structure its further development. We emphasize that this perspective on health and disease centres on empirically well-founded theoretical and analytic evolutionary tools for application in conventional, mainstream medicine, rather than representing any sort of alternative or complementary approach.
This review is aimed mainly at researchers in evolutionary and behavioural biology, who may be interested in ways to extend their interests and studies into the spheres of medicine and health. The definition and expansion of Hamiltonian medicine as a field should also, however, provide new insights into fundamental processes of evolution, given the importance of humans and microbes as well-understood systems for basic research.
2. Domains of Hamiltonian medicine
Hamilton's conception of inclusive fitness was of a quantity that individuals behave as though they are maximizing, owing to natural selection operating on differences among individuals in reproductive success summed across descendant kin and effects on collateral kin, weighted by relatedness [6]. Hamiltonian medicine, like inclusive fitness theory, commonly involves a gene-centric view of phenotypic effects and evolution, in its emphasis on genetic relatedness. Alleles thus exert phenotypic effects that modify their rates of transmission, relative to alternative alleles at the same locus, through effects on copies of themselves in descendant or collateral kin.
Our first domain of Hamiltonian medicine corresponds to interactions between cells. In this context, microbe-to-microbe social interactions modulate microbial phenotypes that affect the health of their human hosts. This domain corresponds primarily to infectious diseases, as microbes can be transmitted horizontally between human hosts. This domain also applies, however, to social interactions between cancer cells during the somatic evolution of carcinogenesis.
Analyses at the cellular level consider why and how cells cooperate and compete as functions of genetic relatedness, costs and benefits. Cooperation and competition directly affect microbial transmission, virulence and resistance to host defences. Microbe transmission, virulence and disease-related adaptations additionally depend on the population structure of their human hosts, because individual responses to infection influence risks of disease in relatives [1]. Among cancer cells, kinship-based cooperation and competition also modulate cellular behaviour and resistance to chemotherapy.
The second domain corresponds to interactions among genes. In this context, genes in the same individual can compete as well as cooperate with one another, as functions of their genetic relatednesses to social partners. Differences between genes in relatedness to interactants can be caused by variation in gene transmission (in particular, a gene transmitted to a specific sex or expressed when inherited from a specific sex) or gene matching (i.e. green beards [4]). As described in more detail below, intragenomic conflict potentiates the risks of disease and can magnify health impacts of physiological dysregulation.
Our third domain corresponds to interactions among humans. In this context, human-to-human social interactions modulate human health and disease. Analyses at this level consider why and how individuals compete and cooperate as functions of genetic relatedness to their social interactants. Such intergenomic conflict is expected whenever relatedness between an actor and recipient is less than one, which is almost always the case. As a result of such relatedness differences, the optimal phenotypes for a pair of interacting individuals diverge, which may result in an increased risk of disease for one of them or both.
These three domains delineate the overall scope of Hamiltonian medicine. We next describe a suite of core principles for this field, with reference to relevant concepts and theory, illustrative examples and consideration of how the field can be advanced further.
3. Six principles of Hamiltonian medicine
(a). Relatedness between microbes mediates their virulence, transmission dynamics and other effects on hosts
Pathogenic microbes exhibit a broad swath of cooperative social interactions that influence their means and abilities to exploit human hosts [7–9] (table 1). Such cooperation normally involves non-zero genetic relatedness, whereby altruistically or mutualistically interacting cells tend to share an allele that is associated with some behaviour, usually as a result of clonal descent. For example, there are many examples in microbes of individual cells in a clone sacrificing themselves to enhance the survival and replication of clonemates, which presumably increases the relative frequency of the altruistic allele, and the clone, in future generations [7,8].
Table 1.
Microbial social behaviour and its relevance to human health and infectious disease.
| microbial social behaviour | relevance to health | relevance to treatment | reference |
|---|---|---|---|
| altruistic or spiteful secretion of bacteriocins to kill competing strains | secretions directly mediate microbial population dynamics | develop bacteriocins as narrow or broad spectrum antibiotics | [10,11] |
| altruistic dormancy, which spares resources for relatives | dormancy may complicate clearance of infections by antibiotics | manipulate dormancy systems | [8,12] |
| kin discrimination | discrimination allows conditional adjustment of behaviour with regard to relatedness | manipulate discrimination systems to reduce virulence | [13] |
| more efficient, faster growth with higher relatedness | virulence covaries positively with microbial relatedness in an infection | increase genotypic diversity in an infection; manipulate kinship cues | [8] |
| prudent resource use under rate–efficiency trade-off and high relatedness | prudence may increase or decrease virulence or chronic persistence | manipulate mechanisms of trade-offs | [8] |
| biofilm production | biofilms protect microbes from harsh environments, enhance growth and persistence | develop means of disrupting biofilm production, maintenance | [14,15] |
| coerced horizontal transfer of virulence factor genes | transfers increase virulence | manipulate mechanisms of transfer | [16–18] |
| cooperator secretion of public goods; cheater exploitation | public goods foster virulence | inoculate with cheater strains; treat with factors that favour cheaters; target public goods rather than microbes | [19–22] |
| altruistic suicide of microbes infected with phage | suicide favours survival of clonemates | manipulate cues for suicide | [23–25] |
| quorum sensing via diffusible signalling molecules | between-cell communication enhances regulation of growth and virulence | manipulate (‘quench’) quorum sensing systems | [26–29] |
As in humans, cooperation in microbes commonly involves a socially generated resource, such as shared, secreted gene products, that can be exploited by ‘cheater’ genotypes that reap the benefits of cooperation but avoid its costs. Such cheating, in turn, selects for mechanisms that limit losses to exploitation, such as repression of cheaters, pleiotropy between cooperative and functionally essential traits, and highly conditional regulation of cooperative phenotypes [9,30,31]. These microbial behaviours modulate human infectious disease, because such social and antisocial traits directly affect microbial population dynamics, virulence, transmission and the efficacy of human strategies for reducing the health impacts of infectious diseases [7,8].
Infectious disease virulence, a major determinant of health effects, may be associated with microbial intrahost genetic relatedness in at least three different ways, from current theory. First, virulence may be inversely associated with genetic relatedness, if microbial genotypes compete more strongly to exploit host resources owing to intrahost interactions with non-relatives [32–34]. Alternatively, virulence may be higher with closer relatedness, when cooperation between related microbes increases their abilities to reproduce rapidly and better-exploit human hosts [35]. Third, the relationship between virulence and relatedness may under some circumstances be U-shaped, with lower virulence under intermediate levels of relatedness [10]. The realization of these alternative expectations in natural populations should depend, in part, upon the mechanisms of cooperation and competition, or cheating, for any given microbe–host interactions, which determine how microbial behaviours affect their population dynamics [34]. For example, higher relatedness may favour virulence when microbial replication within a host depends more strongly on cooperation, but lower relatedness may favour virulence under selection for more rapid, though less efficient, host resource use. Additionally, when lower relatedness is linked with a higher frequency of cheating, virulence may be some frequency-dependent function of cooperators and cheaters [36]. As expected, given these considerations, some microbes exhibit adaptations interpretable as recognition of clonemates [9,18,37]; in other species, adaptive virulence-related behaviour may be associated with long-term population-typical levels of relatedness within hosts, without kin recognition per se.
Extending and testing inclusive fitness theory for the evolution of virulence, and elucidating the different behavioural, ecological and evolutionary conditions that yield different relationships between virulence and genetic relatedness, remain major challenges for students of microbial sociality [35,38,39]. As for macroscopic social creatures, control of social phenotypes, and specification of relatedness salient to applying Hamilton's rule, assume central importance. For example, microbial cells commonly engage in horizontal gene transfer of plasmids or other elements that encode virulence factors [18]; such transfers may be under the control of genes in the transmitted DNA, and may coerce recipient cells into social cooperation [16,17]. Such phenomena also represent social transmission of relatedness itself, as microbes become genetically related via horizontal transfer, for the locus under consideration [17,40]. Hamilton's rule may also be more complicated in microbes owing to a high incidence of strong, non-additive selection effects, which necessitates adjustments to rb − c > 0 for prediction of behavioural phenotypes [41]. Finally, microbial social behaviour and virulence depend crucially on the spatial scales of competitive and cooperative interactions among relatives, given that microbes live in such dense, ecologically interactive groups [42]; competition among relatives may thus suppress the evolution of cooperation in some conditions or select for microbial traits, such as quiescent ‘resister’ cells or programmed cell death, that reduce its effects [8,9].
Thus far, Hamiltonian-medical studies of microbes have centred on pathogenic bacteria, and on considerations from evolutionary theory more than ecology. However, pathogenic microbes are usually close phylogenetic relatives of human commensal or mutualistic microbes [43], pathogenic and mutualistic microbes frequently exist along continua even within nominal microbial ‘species’ [43,44], and mutualistic components of the human microbiome appear to exert health impacts at least as powerful as those from pathogens [45]. Can considerations from Hamiltonian medicine help to explain cooperation and competition in mutualistic human microbes, and transitions between mutualism and pathogenicity in different ecological and evolutionary contexts [44]? How do ecological–evolutionary feedbacks, which should be especially strong among bacteria, mediate social bacterial phenotypes [46,47]? To what extent, and in what ways, do competition and cooperation among viruses influence their virulence and transmission [48,49]? Table 1 summarizes microbial social behaviours, their relevance to health, and their potential usefulness for the prevention and control of infectious disease, to motivate further progress in this key area of Hamiltonian medicine.
(b). Human kinship structure mediates symptoms, virulence and transmission of pathogens and defence mechanisms
Hamilton's key insight for understanding social evolution was that genetic relatedness mediates fitness-related behaviour in mathematically predictable ways. For analysing health from a Hamiltonian perspective, it is essential to recognize that ‘behaviour’ includes all disease-related phenotypes with effects on kin, be they immune defences or responses, disease symptoms, social interactions that modulate transmission or differential patterns of investment and trade-offs regarding growth, maintenance and reproduction.
As microbial relatedness affects human health, so human relatedness also affects infectious disease risks, transmission and symptoms. Human relatedness influences disease because in groups structured by kinship, immune system functions, host-expressed disease phenotypes and adaptations of hosts to avoid, resist or fight disease represent social traits with strong effects expected on relatives [32–34,50]. Host adaptations to reduce deleterious effects from infectious disease include resistance (avoiding infection, or investing in immune responses to clear it from the body) and tolerance (mitigating the detrimental effects of infection, without clearance). Frank [32] used an inclusive-fitness model to show that higher relatedness selects for increased investment in resistance (avoidance of infection, in this case, through induction of immunity). Such resistance strategies may include altruistic, disease-related responses and behaviour by human hosts, which reduce pathogen transmission to kin [25,51,52]. Relatedness structure is also predicted to restrict the evolution of tolerance [51,52], in part, because tolerant individuals remain infectious. Such effects may be especially important owing to widespread trade-offs of investment in immune functions with investment in growth and reproduction, the usual two contexts for consideration of maximizing inclusive fitness.
Best et al. [53] corroborated Frank's results, and also showed, using a coevolutionary model, that more-local, among-relative interactions favour reduced parasite transmissibility and virulence. These findings are important because increased admixture, as found in present-day populations, is predicted by their model to favour a combination of high pathogen virulence and transmissibility with reduced host resistance to infection, precisely the conditions that favour human pandemics with high rates of mortality.
Transmission dynamics of disease, and the evolution of alleles that influence disease resistance, are also mediated by relatedness, because alleles that protect an individual from disease will also protect their family members who share copies identical by descent. Schliekelman [54] showed that the strength of selection for resistance depends crucially on how resistance alleles influence between-family transmission rates relative to within-family transmission. Such rates have been measured, demonstrating strong effects of kinship structure; for example, transmission of Yersinia pestis (plague) between households in England during the late seventeenth century was ‘usually achieved by visiting close relatives’ [55, p. 135].
Finally, Williams & Nesse [1] pointed out that symptoms of disease that appear to represent manipulations of hosts by pathogens, such as coughing and sneezing, may exhibit large benefits to pathogens but low costs to hosts unless such symptoms differentially impact upon the host's kin. These considerations indicate that inclusive-fitness effects may influence all human disease-related phenotypes that modify rates of transmission, because humans live in kinship-structured groups. As for conflict situations within species, the outcomes of pathogen-versus-host conflicts over disease symptoms that affect transmission depend upon the strengths of selection on both parties, and the phenotypic strategies available to them; in this context, to what extent do disease symptoms represent effects of selection involving relatives?
The study of kinship effects in pathogenic microbes offers outstanding opportunities for evolutionary biologists, ecologists and behavioural biologists who are willing to train in microbiology, molecular biology or epidemiology. Research in several groups [8,9] represents exemplars for success in such interdisciplinary studies.
(c). Inclusive-fitness effects mediate growth, and resistance to treatment, of cancer cells
Applications of social–behavioural perspectives, and evolutionary medicine, to the study of carcinogenesis have thus far focused predominantly on three areas: (i) natural selection between genetically divergent cancer cell lineages for phenotypes that increase their replication rate, (ii) cancer cell competition with normal cells and (iii) cooperation among cancer cells, which has been suggested to generate ‘tumour cell societies’ [56–60]. Such studies have highlighted the causes and effects of genetic heterogeneity among cells within a tumour, and the molecular mechanisms whereby cells interact with one another positively or negatively, but explicit considerations from inclusive fitness theory have, surprisingly, yet to be applied.
Genetic relatedness among cancer cells is essentially the same as for groups of microbes: a cell harbouring an allele that controls some phenotype is related by 1 to other, recipient cells with the same allele (identical by descent or horizontal transfer [61]), and by 0 to any given cell with an alternative allele. In mixed-genotype, local groups of cells, relatedness is between 0 and 1, because the effects of a focal cell's allele impact upon cells that do, and do not, bear it.
How do more or less related cancer cells interact, in a behavioural sense? Like microbes, they express cell-surface molecules, and secrete diffusible products (public goods), that modulate the behaviour (gene expression patterns, and interactive phenotypes) of nearby cells, as well as modify their environments [62–65]. Cancer cells also, like microbes, form heterogeneous, three-dimensional structures, comprising mixtures of cells that differ in genotype and phenotype [56]. Such cells may differ phenotypically from one another in socially relevant ways: for example, tumours remain very small until some cells undergo an ‘angiogenic switch’ to specializing in the development of tissue that serves as blood vessels, providing nutrients and disposing of waste [60]. This morphogenetic variation appears to represent a form of cellular division of labour involving trade-offs between proliferation and food-provisioning, with angiogenesis as an altruistic phenotype that should be susceptible to the secondary invasion of ‘cheater’ clones that exploit the resources provided [60]. Indeed, ‘angiogenic potential relies on cooperative production of the angiogenic signal’ [60], in the same way that shared, secreted products mediate many social interactions among pathogenic microbes.
Nagy [66,67] used mathematical models of competition between cancer cell types, in relation to degrees of vascularization and overall tumour growth, to analyse the expected dynamics of such trade-offs. His models demonstrated that competition between cancer cell lineages could lead to the development of highly proliferative ‘hypertumours’, that outcompete and destroy earlier-developing cancer tissue, and then either self-destruct owing to their lack of capacity for angiogenesis, or persist with other tumour tissue at a frequency-dependent equilibrium. The somatic evolution of hypertumours is consistent with examples of spontaneous regression, and areas of necrosis, in neuroblastoma and other cancers [66], but it remains to be studied in detail.
Hypertumours represent one possible trajectory of interactions within and between cancer cell genotypes and lineages; presumably, other, less dramatic competitive and cooperative cellular–behavioural phenotypes modulate carcinogenesis from inception to regression or malignancy. Another potential example involves benefits provided to cancer stem cells from clonally related, partially differentiated cells (cancer cell ‘helpers at the nest’, as it were) [68]; this process has been demonstrated in simulations [69], and may be of considerable significance for cancer cell behaviour and evolution.
Taken together, these considerations suggest that the evolution of cancer cells resembles the evolution of microbes in fundamental ways (table 2), despite the fact that cancer cell evolution proceeds de novo in each instance of carcinogenesis and involves somatic-evolutionary cooption of genes and pathways that evolved in the contexts of normal cellular activities. Transmissible cancers in domestic dogs and Tasmanian devils [78], indeed, represent empirical bridges between infectious disease and carcinogenesis, as does the application of experimental–evolutionary methods [79], and the development of relatively ‘evolution-proof’ therapeutic agents that delay or preclude the evolution of resistance [80]. Microbes themselves may even be subjected to cancer-like overgrowth by ‘rogue lineages’ [81], whose control and dynamics should provide insights into repression of over-proliferating cells much more generally [81,82].
Table 2.
Parallels between cancer cells and microbial cells, with regard to kinship-related social behaviours that impact upon human health.
| cancer-cell social behaviour | microbial parallel | reference, for cancer cells |
|---|---|---|
| cellular secretion of chemicals that modify the survival, growth and behaviour of other cells (clonemates, other cancer cells and normal cells) | bateriocins, public goods, chemical signalling | [59,64,65,70] |
| quorum sensing modulation of patterns of cell growth, quiescence, dispersal and stem cell immortalization | quorum sensing that modulates growth, behaviour and virulence | [63,71] |
| differentiation of some cells into less-reproductive ‘helper’ cells (e.g. the angiogenic switch) | differentiated, less-reproductive microbial cells | [60] |
| mutualistic and altruistic cell–cell cooperation, usually within clones | diverse forms | [57,72] |
| mutualistic and altruistic interactions in groups of cells that express group-level phenotypes; groups may comprise clones or clonal mixtures | multicellular phenotypes in many bacteria | [56,60,63,73,74] |
| cooperative stress responses among interacting cells, mediating the evolution of drug resistance | social evolution of antibiotic resistance | [75] |
| horizontal gene transfer mediation of cell survival and growth | horizontal transfer of plasmids and other sources of DNA | [61,76,77] |
| altruistic dispersal, whereby dispersal of some cells frees up resources for clonemates (?) | may operate in context of biofilms, other groupings | n.a. |
| altruistic suicide, whereby programmed cell death frees up resources for clonemates in stressful conditions (?) | reported in some bacteria | n.a. |
Conceptualization and modelling of cancer cell population dynamics in terms of inclusive fitness theory, and relatedness-based cooperative and competitive behaviour, should lead to novel insights with direct implications for therapy. Organism-level researchers seeking to contribute to this research area will need to invest heavily in learning cancer biology, and research with the strongest impacts will probably involve empirical work by integrative teams.
(d). Intragenomic conflicts mediate disease risks and phenotypes
Intragenomic conflict follows directly from a Hamiltonian gene's eye view of evolution [83]. By this process, divergence of genetic interests is caused by differences between sets of genes in either transmission pattern or gene matching (via green-beard genes), resulting in differential relatedness of focal genes to social interactants of their carriers [4]. For example, female full sibs in eutherian mammals are related by 3/4 for X-linked genes, but by 1/2 for autosomal genes; such genomic factions [84] may thus be in conflict over some phenotype if rb − c > 0 for one party but not for the other. More generally, genes coding for a particular phenotype are in conflict when the phenotypic value that maximizes the replication of one gene is different from the phenotype that maximizes the replication of the other (differently inherited or related) gene. By this process, alleles that advance their own representation in future generations can, in some circumstances, increase in prevalence even if they increase risks of disease.
Differences in relatedness may lead individual genomes to show signs of conflict between autosomal, sex-chromosomal and mitochondrial genes (figure 1). For example, consider altruism towards female siblings. Mitochondrial genes favour the greatest level of altruism, whereas Y-chromosomal genes favour the lowest. Conflict may also exist between maternally inherited genes, paternally inherited genes, and genes that do not carry information on their parental origin. Consider, for example, altruism towards matrilineal sibs. Maternally inherited genes favour the greatest level of altruism while paternally inherited genes favour the lowest. Finally, genomes can show signs of conflict between a specific allele that recognizes itself in a social partner (a ‘green-beard gene’) and an allele that does not [4,85,86].
Figure 1.

Conflict between genes underpinning helping behaviour. The x-axis represents the benefit experienced by the recipient of help (B), while the y-axis represents the inclusive fitness of the actor (IF). The evolutionarily stable strategy corresponds to the case IF = rB–C = 0, where C is the cost experienced by the actor, and r is the coefficient of relatedness. This graphically corresponds to the point where the IF-line intersects the 0-line. When IF < 0 a gene is selected for less help, when IF > 0 a gene is selected for more help. (a) This figure corresponds to two generic genes, a and b, coding for help. Genes are under different selective pressure when their relatedness to recipients differs, i.e. ra ≠ rb. When relatedness of gene a is greater than that of gene b, i.e. ra > rb, the optimal benefit for gene a is smaller than that for gene b, i.e.  and gene a is selected for more help than gene b is. The region where genes a and b are selected for different help is a region of conflict (in grey in the figures). (b) This figure corresponds to genes coding for help directed to female siblings. Mitochondrial genes (m) are selected for more help than X-linked genes (x). X-linked genes are selected for more help than autosomal genes (a). Autosomal genes are selected for more help than Y-linked genes (y). As a result, depending on the current level of help, there can be conflict between any of these pairs of genes, namely: m versus x, x versus a, a versus y, m versus a, m versus y, x versus y. (c) This figure corresponds to genes coding for help directed to maternal siblings. Genes of maternal origin (mo) are selected for more help than those of unknown origin (uo). Genes of unknown origin are selected for more help than those of paternal origin (po). As a result, depending on the current level of help, there can be conflict between any of these pairs of genes, namely: mo versus uo, uo versus po, mo versus po.
and gene a is selected for more help than gene b is. The region where genes a and b are selected for different help is a region of conflict (in grey in the figures). (b) This figure corresponds to genes coding for help directed to female siblings. Mitochondrial genes (m) are selected for more help than X-linked genes (x). X-linked genes are selected for more help than autosomal genes (a). Autosomal genes are selected for more help than Y-linked genes (y). As a result, depending on the current level of help, there can be conflict between any of these pairs of genes, namely: m versus x, x versus a, a versus y, m versus a, m versus y, x versus y. (c) This figure corresponds to genes coding for help directed to maternal siblings. Genes of maternal origin (mo) are selected for more help than those of unknown origin (uo). Genes of unknown origin are selected for more help than those of paternal origin (po). As a result, depending on the current level of help, there can be conflict between any of these pairs of genes, namely: mo versus uo, uo versus po, mo versus po.
In the context of intragenomic conflicts, the genome can be considered as a social system of cooperating and competing, yet mutually dependent, genes and pathways comprising different factions according to their autosomal, sex-chromosomal or cytoplasmic locations, and their paternal or maternal expression [84,87]. Health and disease impacts follow directly from effects of the resulting genic behaviour on relative fitness.
The outcome or resolution of intragenomic conflict should depend on which set of genes controls the resulting phenotype. For example, control of phenotypic values should be influenced by the number of genes included in each set [4]. For this reason, many instances of conflict between autosomal and sex-chromosomal or mitochondrial genes should, in theory, be resolved to the advantage of autosomal genes [87,88]. By contrast, the number of maternally and paternally inherited genes will be almost evenly split (evenly in females, but unevenly in males who have a slightly larger number of maternally inherited genes) and thus resolution of such conflicts will depend on the specific phenotype coded for and the details of molecular interactions.
Intragenomic conflict can increase the risks of disease in several ways. First, such conflicts are expected to result in the evolution of novel, conflict-related genetic and epigenetic regulatory mechanisms. The increased complexity of these regulatory mechanisms may enhance the probability of dysregulation, compared with non-conflictual situations [89–91].
Second, intragenomic conflict may cause decanalization of developmental trajectories and functions [92]. By this mechanism, any degree of conflict results in escalated expression of antagonistic genes, or selection for other means of ‘winning’. The extent to which this escalation continues is determined not by the extent of the conflict, but by the costs of enhanced expression of each antagonistic gene. As a result, a perturbation to the dynamic equilibria established by genes in intragenomic conflict tends to result in pathologies more often, and more extreme, than perturbations to genes not in conflict [88,93]. Silver–Russell undergrowth syndrome and Beckwith–Wiedemann overgrowth syndrome represent paradigmatic examples of pathologies caused by the dysregulation of imprinted-gene conflict systems, here mediated predominantly by alterations at the imprinted domains IGF2/H19 and CDKN1C/KCNQ1OT1 [94,95].
Third, intragenomic conflicts should involve antagonistic selection for effects as early in development as possible, and selection for control over the dynamics of stem cells. Such effects are expected because during development, power over phenotypes is commonly (i) some function of time, because events earlier in ontogeny have larger, more-cascading effects, and (ii) some function of control over rates and patterns of cell and tissue proliferation, because this is how organisms grow and differentiate. This prediction is well supported by recent studies showing that in a wide range of human tissues, imprinted genes orchestrate stem cell replication versus inhibition [96–98], and by the observation that imprinted genes exert especially pervasive effects on placental development, at the start of fetal growth and development [99,100]. Imprinted-gene conflicts will therefore be especially important in mediating causes of variation in physical health, because so many human health outcomes are highly dependent on early-life events and phenotypes such as placental function and birth weight [101], and on the roles of stem cells in tissue renewal and repair.
Consider, as an example, intragenomic conflict between maternally and paternally expressed imprinted genes, in a fetus, over taking resources from the mother. Maternally inherited genes in the fetus are selected to extract less resources from the mother than are paternally inherited genes, owing to their evolutionary history of higher relatedness to matrilineal kin under some degree of multiple paternity. This conflict has two effects. First, within a locus it drives the silencing of the gene coding for a lower level of expression (i.e. the maternally inherited copy if the gene codes for extraction of maternal resources, and the paternally inherited copy if the gene codes for preserving maternal resources) [102]. Second, between loci, the conflict drives an escalation in the expression of loci coding for opposing phenotypes.
For the specific phenotype ‘maternal blood pressure’ during mid to late pregnancy, a higher value translates into a greater amount of nutrients reaching the fetus. A locus coding for higher blood pressure is thus expected to be paternally expressed only, as a result of intralocus conflict. A locus coding for the opposite phenotype, lower blood pressure, is expected to be maternally expressed. Both loci will be selected to escalate their levels of expression as a result of interlocus conflict [103]. A loss of function mutation in an allele at a non-conflict locus should have small phenotypic effects, as the other allele would be expressed. However, at a conflict locus, there would be twofold effects: (i) because the locus is functionally haploid, the gene product would be missing when the mutation is maternally inherited, and (ii) because there is a dynamic, tug-of-war equilibrium driven by tension between high and low blood pressure loci, a reduction in the production of gene products favouring low blood pressure should result in large-scale pathology, here leading potentially to pre-eclampsia. A set of pre-eclampsia-related genes are indeed imprinted [104], and moderately but not overly high maternal blood pressure results in higher than normal birth weight [105].
In contrast to situations involving the mother and fetus, mathematical modelling suggests that maternally inherited genes expressed at adult stages may in some circumstances be less related to infants and juveniles in their social group than are paternally inherited genes [106,107]. Paternally inherited genes in adults may thus be selected to promote more altruism (e.g. communal parental care) towards infants and juveniles in their social group than are maternally inherited genes. For the phenotype ‘female fertility’, this situation translates into higher production of one's own offspring and thus less provision of communal care versus production of fewer offspring and thus greater provision of communal care.
A locus coding for higher fertility may be predicted to be paternally silenced, as a result of intragenomic conflict. However, a locus coding for the opposite phenotype, lower fertility, is expected to be maternally silenced [108]. Both loci will be selected to escalate their levels of expression as a result of interlocus conflict. A loss of function mutation at a locus coding for fertility is expected to have the twofold effect usual in conflictual loci: loss of that particular gene product when maternally inherited owing to functionally haploidy, and a cascade of pathologies related to low fertility (including premature ovarian failure) owing to disequilibria between genes coding for higher and lower fertility [108]. These examples also illustrate how for imprinted genes, theory can predict the forms of pathological phenotypes expected under dysregulation, which provides for tight links between theory and clinical medicine.
Imprinted genes are strikingly understudied from evolutionary perspectives, despite the support for the kinship theory of imprinting as regards growth, and the considerable importance of such genes for growth, cancer and metabolic disease [89,109]. The perspective that genes themselves exhibit conflictual and cooperative behaviour [110] should motivate behavioural and evolutionary biologists to analyse the molecular mechanisms by which they socially interact, and thereby mediate health and disease.
(e). Resource-related conflicts between relatives, especially mother–offspring conflict, potentiate and modulate disease risks
Cooperation between genetically related individuals represents the original context of Hamilton's inclusive fitness theory, which was developed to help understand the otherwise-paradoxical evolution of altruism. Trivers [111] was the first to emphasize that close relatives are also commonly close competitors, and that many parent–offspring and sib–sib interactions can be interpreted in the context of more or less restrained conflicts. Considering a particular phenotype resulting from the interaction of two individuals, the phenotypic value that maximizes the inclusive fitness of one individual might be different from the value that maximizes the inclusive fitness of the other individual. There is thus a region of the phenotypic space where changes in value increase the inclusive fitness of one individual at the expense of the other. Which phenotypic value will be ultimately expressed is conditional on the strengths of selection on the two parties, and which individual has greater control over the phenotype. Thus, one individual may end up gaining control of phenotypic expression, owing to physical and physiological asymmetries. Such conflicts are based on identical relatedness to social interactants across non-imprinted autosomal genes, the numerically predominant agents in genomes, which are expected to agree on tactics of social behaviour towards kin.
Conflicts between kin are relevant from a medical perspective because they may increase the risks of developing diseases that are associated with changes to inclusive fitness. Such changes commonly involve control over resources vital to growth and development. For example, Haig [112] demonstrated that many of the major disorders of human pregnancy, including gestational diabetes, pre-eclampsia and intrauterine growth restriction, could be understood to result, in part, from mother–offspring conflicts over maternal resources during pregnancy. Considering, for example, the phenotype ‘glucose levels in maternal blood’, the fetus is selected to maintain higher levels of glucose (by placental release of chemicals into maternal blood that increase her resistance to insulin), than the mother is selected to maintain. The mother therefore produces more insulin. Health sequelae result if this evolved tug-of-war system becomes dysregulated, or if the fetus ‘wins’ such that the mother develops gestational diabetes. Comparable conflicts between relatives are expected whenever they compete for fitness-related resources, with the outcomes of conflict mediating vulnerability and phenotypes of disease.
(f). Social interactions with kin early in life generate and exacerbate risks of mental disorders
Mother–offspring conflicts may be psychological as well as physiological. Parturition represents a transition point between conflicts over physical resource investment, with an offspring advantage in some regards, and conflicts over behavioural and psychological interactions, with predominant maternal control over most activities. As the placenta and breast provide energetic resources to the developing offspring, psychological interactions, especially with the mother, provide cognitive and emotional resources that support early childhood psychological development, with lifelong impacts on psychological health and well-being.
John Bowlby's concept of attachment—child psychological bonding that promotes optimal development of social cognition and emotion—serves as a primary construct and metric for understanding mother–offspring interactions and their consequences [113,114]. Child attachment may be (i) secure, such that the child develops an ongoing expectation of investment from the mother, or insecure, either (ii) anxious–insecure, whereby children suffering unmet solicitation of needs express increased distress and contact-seeking, combined with anger and ambivalence or (iii) avoidant–insecure, whereby children with unsatisfied expectations come to avoid and reject carers [115]. The former category can be interpreted as active attempts to improve a suboptimal situation for the child, whereas the latter may represent behaviour designed to avoid making a bad situation worse [114,116]. Despite the fact that such insecure attachment is strongly associated with a broad set of psychological and psychiatric disorders [117,118], its incidence is remarkably high, on the order of one-third of children [119].
How do these considerations involve inclusive fitness theory, and impact upon health? Attachment theory, although explicitly based on the integration of evolutionary and ethological principles, has yet to incorporate conflictual inclusive-fitness effects into its core precepts. Attachment centrally involves psychological solicitation by the child, and response (or not) by the carer, usually the mother. As described above, offspring autosomal genes have been subjected to selection for soliciting more physical investment from mothers than they have been selected to provide, and paternally expressed imprinted genes favour phenotypes that likewise increase solicitation whereas maternally expressed imprinted genes favour the opposite. These considerations also apply to behavioural and psychological investment, especially in humans for whom early social-brain development assumes paramount importance for mental health. For any given mother–offspring dyad, child psychological health in the context of attachment should thus be some function of the matching between child solicitation and maternal supply, and the resulting levels of maternal investment combined with its effectiveness [114].
This system of demand and supply exhibits two key features, from an inclusive-fitness standpoint. First, presuming predominant maternal control over attachment-related phenotypes, and trade-offs affecting maternal allocation of resources in this context, insecure attachment is not unexpected for a substantial proportion of cases, as documented above. This asymmetry is a simple consequence of maternal control, which allows mothers to achieve their optimum as regards attachment-related levels of investment. Second, developing attachment interactions may be perturbed, by genetic, epigenetic and environmental factors mediated by evolved conflict systems, in either of two opposite directions: (i) towards insecure attachment owing to under-solicitation, under-provision, or both, or (ii) towards ‘overly secure’ attachment owing to some combination of over-solicitation and over-provision. Moreover, the proximate mechanisms should involve conflictual inclusive-fitness effects, and the direction and magnitude of such perturbations should be directly and causally connected with specific psychological and psychiatric health outcomes. For a child, paternal biases in brain imprinted-gene expression are thus predicted to be associated with over-solicitation; by contrast, maternal biases should be associated with reduced solicitation [114]. How well are these expectations met?
Attachment systems originally evolved in stem placental mammals, with the advent of viviparity, extensive maternal care, lactation, relatively large brains and genomic imprinting [83]. The neuropeptide oxytocin, which also originated around this time, serves as a primary molecular mechanism for mother–offspring bonding, in both directions, in addition to its roles in labour-induction, lactation and infant sucking [120]. Evidence from mice and humans indicates that knockouts of any of a suite of paternally expressed imprinted genes lead to substantial reductions in numbers of oxytocin-producing neurons in the hypothalamus, and impairments of sucking by neonates [120–124]. Moreover, two of the genes involved, PEG3 and NDN, represent central hubs of an imprinted-gene coexpression network described by Varrault et al. [125]. These findings indicate that imprinted-gene expression exerts strong effects on oxytocinergic system development in the brain, which impact upon costs imposed by offspring on mothers in the direction predicted by the kinship theory of imprinting. As such, intragenomic conflicts are also implicated in the psychological phenotypes mediated by oxytocin in humans, which include prosociality, empathy, trust, ingroup–outgroup conceptualizations, perception of kinship, and altruism [126–131].
These considerations suggest that oxytocin may serve as a key hormonal mediator of inclusive-fitness maximizing, such that closer degrees of kinship, or perceived inclusive-fitness salience and benefit, elicit stronger oxytocin release and response. As such, alterations to the oxytocinergic system (and interacting dopaminergic, serotoninergic and other endocrine systems) should have disproportionately large effects on psychological health, construed in the context of ability to unconsciously maximize one's inclusive fitness. Direct evidence for oxytocin effects, and imprinting effects, on attachment and its associated psychiatric and psychological phenotypes in humans comes from studies of three neurogenetic disorders involving imprinted-gene dysregulation.
First, Prader–Willi syndrome is cause by the loss of paternally biased imprinted-gene expression for a set of genes on chromosome 15 (causing a maternal bias to development); it is characterized in early childhood by low levels of solicitation expressed as reduced and weak crying, extended sleep, reduced sucking and weak attachment to the mother [93,132]. Numbers of oxytocin neurons in the hypothalamus, and serum oxytocin, are substantially reduced in this condition [133]. In adulthood, a high proportion of individuals with Prader–Willi syndrome also develop forms of schizophrenia or affective psychosis (psychosis that includes depression), making it among the most penetrant causes of psychotic-affective conditions [134]; moreover, insecure childhood attachment is strongly associated with other psychotic-affective conditions, including schizophrenia [117]. The causal links between insecure attachment and psychotic-affective conditions require further study, but they may include under-development of the hypothalamic-centred, limbic ‘paternal brain’ relative to the neocortical, mentalistic ‘maternal brain’ [135], as well as childhood neglect, maltreatment and abuse that dysregulate cognitive–affective development [114,117,118].
Second, Angelman syndrome is caused by loss of function for the maternally expressed imprinted gene UBE3A, which thus produces a bias towards paternal imprinted gene interests in development [135]. During early childhood, individuals with Angelman syndrome show excessive degrees of solicitation to parents, as implicated by extreme levels of crying, hyperactivity and sleeplessness; they also, however, show high levels of positive mood, notably in social interactions [136,137]. Taken together, these traits have been interpreted as essentially the psychological opposite of Prader–Willi syndrome, whereby increased paternal imprinted-gene influences favour increased solicitation via activity and positive mood. Levels of serotonin and dopamine are elevated in several brain regions of UBE3A-knockout mice [138], but oxytocin has not been studied; similarly, attachment to the mother has yet to be investigated.
Most children with Angelman syndrome, and with the strongly overlapping disorders Rett syndrome and Pitt–Hopkins syndrome, exhibit autism spectrum disorders [139], with reduced development of mentalistic, neocortical, social–cognitive skills. Surprisingly, among individuals with relatively high-functioning autism, the frequency of insecure attachment is not increased over normative levels [140]. Children with autism certainly tend to impose higher time and energy demands on parents, especially the mother, but the degree to which less extreme and non-pathological autism spectrum phenotypes involve ‘overly secure’ attachment, which retains elements of early childhood, psychologically simple dependency on the mother [141], remains to be evaluated.
Third, Williams syndrome is caused by a small chromosomal deletion that includes the transcription factor GTF2I, a gene that shows a strong parent-specific expression bias indicative of genomic imprinting [142]. This syndrome involves hypersocial behaviour, whereby affected children exhibit high levels of approach, social engagement and eye contact [143,144]. GTF2I has been implicated in the social–behavioural alterations in Williams syndrome, possibly through association with the approximately threefold higher levels of oxytocin found in this condition [145]. Given that GTF2I shows a maternal-gene expression bias [146], reduced dosage of its gene product (as in Williams syndrome) generates a paternal expression bias. Williams syndrome is thus similar to Angelman syndrome in that its major behavioural phenotypes can be interpreted in the context of exaggerated attachment-related behaviours, in conjunction with paternal biases to imprinted gene expression. Despite the hypersocial solicitation found in Williams syndrome, this syndrome has also, like Angelman syndrome, been associated with reduced social skills and autism spectrum phenotypes [146]. By contrast, duplications of the Williams syndrome genomic region, which can be predicted to involve reduced levels of oxytocin and a maternal-gene bias, have recently been associated with increased risk of schizophrenia [147], in apparent parallel with Prader–Willi syndrome as described above.
Neurogenetic disorders such as Prader–Willi syndrome, Angelman syndrome and Williams syndrome involve major alterations to evolved systems of cognition and affect (mood), but their phenotypes and genetic causes represent extreme ends of continua that are expected to grade into normality. As such, variation in imprinted-gene expression and effects, components of the oxytocinergic system, and other causes of diversity in patterns of childhood attachment and early social development are expected to exert strong effects on the development of cooperation, altruism, empathy and liability to alterations of sociality and behaviour expressed in personality disorders and psychiatric conditions. For example
(1) Dependent personality disorder involves extreme levels of dependence-related and attachment-related behaviour in conjunction with maladaptively high expression of the five-factor model personality dimension of agreeableness [148]. This condition has been interpreted as involving pathologically elevated levels of altruism and self-sacrifice [148,149], and may be mediated by ‘overly secure’ attachment. By contrast, narcissistic and antisocial personality disorders can be interpreted in the context of pathologically high levels of selfishness (and low agreeableness) [150], at an opposite extreme. These considerations suggest the possibility of psychological ‘disorders of inclusive fitness maximizing’, involving either hyperaltruism (with behaviour expressed even if rb −c < 0) or hypoaltruism (with
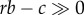 required for behavioural expression);
required for behavioural expression);(2) Borderline personality disorder, which is strongly mediated by insecure attachment and childhood maltreatment, involves dysregulation of the oxytocinergic system [151] but enhancement of empathy-related, ‘mind-reading’ skills, compared with controls [152];
(3) Neuroticism, anxiety and depression in adolescent and young adult females may commonly result from high levels of altruism-related empathy and social sensitivity, coupled with stressful or abusive family environments [153,154].
Such examples are important given the continuity between normal personality variation, personality disorders and psychiatric disorders [155], which indicates that considerations from inclusive fitness theory—a key to understanding altruism and cooperation—can provide strong links between normal and dysfunctional behaviour.
A final consideration from inclusive fitness theory for human psychological health is intragenomic conflicts expressed in the brain that affect cognition, affect and behaviour in contexts other than mother–child interactions [156,157]. This topic, mentioned above in the contexts of Prader–Willi, Angelman and Williams syndromes, has been discussed extensively [157], based on the expectation that some psychiatric disorders, especially autism and psychotic-affective disorders, including schizophrenia, represent, in part, extreme and maladaptive effects of biases towards either paternal imprinted-gene expression (towards a more self-oriented, less social ‘paternal brain’), or maternal (towards an other-oriented, social ‘maternal brain’). Empirically, this question can be addressed by studying the effects of genetic and epigenetic variation in imprinted genes (and their expression levels) on psychological traits related to sociality and modular brain architecture and functions, especially in non-clinical populations where pathology does not confound the results. For example, the imprinted schizophrenia-risk gene GABRB2 harbours a SNP allele that is associated with both more severe psychosis in schizophrenia, and with higher degrees of altruism in healthy individuals [158]; such associations of imprinting with altruism were predicted from models developed by Úbeda & Gardner [106,107,159]. Similarly, SNPs of the imprinted gene LRRTM1 influence both schizophrenia risk and handedness [160], which suggests that the largest scale of modularity in the human brain, left–right hemispheric differentiation for language and other social traits, is modulated by effects of imprinting and intragenomic conflict.
Studying mental illness from inclusive-fitness perspectives requires careful integration of psychology, neuroscience and psychiatry with evolutionary biology. For students of social behaviour and evolution, it is important to realize that psychiatric conditions can be conceptualized and studied as disorders of usually adaptive social behaviour, such that they can be analysed using theory and methods for understanding typical human and animal sociality.
4. Discussion
Hamiltonian medicine represents a central field within evolutionary medicine because the primary agents involved in human health—microbes, human cells, genes and human individuals—are all social, and are all governed by the same general rules and mechanisms for maximizing inclusive fitness. The key to developing and applying Hamiltonian medicine thus becomes the forging of connections of health with the causes and consequences of conflictual and cooperative interactions among kin, including altruism, mutualism, parasitism and spite. In this context, intragenomic and intergenomic conflicts mediate health through the evolution of conflict-related systems that become targets for dysregulation, through deviations from human phenotypic optima that follow from perpetuation or resolution of conflicts, and through conflicts between alleles or genotypes of disease microbes, other parasites or cancer cells. By contrast, cooperation can mediate health when it occurs between genetically related agents of disease, or among other related agents whose interactions influence disease risks and impacts.
Across all of the diverse principles described here, genetic relatedness represents the same variable: the probability that an entity generating a social action is genetically identical by descent or horizontal transfer, at the locus controlling the action, with the recipient, in the context of a given population. For autosomal genes in humans and other metazoans, levels of relatedness are straightforward products of meiotic links. However, for non-autosomal genes in metazoans, and for microbes or cancer cells, relatedness and inclusive-fitness effects must be conceptualized more carefully, in terms of an allele's effects on replication of copies of itself (strictly, relative to alternative alleles at the locus) in other entities, be they groups of pure or mixed-clone bacterial cells [9], more or less genetically divergent human cells within an individual [79], or interacting humans themselves. Across all of these contexts, Hamilton's rule can also be expanded to include forms of social interaction based on trait-sharing (‘kind’ selection) and feedback of social trait effects on the actor's fitness (‘kith’ selection) [161]; this formalization allows the application of Hamilton's rule across an even wider array of situations affecting fitness and health, from genes to groups.
A second key commonality that emerges from the principles of Hamiltonian medicine is the importance of control over kin-selected phenotypes that influence health, because patterns and degrees of control mediate resolution or continuance that may impact upon the health of all parties concerned. Thus, one party may ‘win’ to the health detriment of the other [112], a dynamic ‘tug-of-war’ may ensue with actual or potential health costs to both parties [88], conflict may be repressed by some agent that is more powerful than the competing parties (with possible costs to suppression) [162] or conflicts may escalate, with increasing costs and potential for dysregulatory effects [91,92,112]. Outcomes of such conflicts are likely to be predictable only on a case-by-case basis, because they depend on the proximate mechanisms of interaction. Such considerations should compel close collaboration between medical and evolutionary scientists, and cross-disciplinary training.
Genes may be more or less powerful in competitive or cooperative interactions through such effects as earlier timing of expression during development, regulation of stem cells, a higher degree of pleiotropy, or mutualistic and synergistic interactions with genes that share their inclusive-fitness interests. Imprinted genes appear to represent instances of just such effects, as described above, and X chromosomal genes may exhibit similar patterns in expression of genomic influence [84,88]. Despite such conflicts, the overwhelming autosomal majority of genes in the human genome suggests that autosomes, and genomes, should evolve towards repression of health-related intragenomic conflicts, through such effects as canalization of development or stabilization of conflictual interactions between gene products.
Cells may also vary in power, as in microbes or tumours, through signals to other cells that modulate their gene expression, survival, replication and differentiation patterns, or through secretion of products that directly modify cellular behaviour. Here, examples include the diverse forms of microbial social interaction described above, and cancer cells that secrete growth factors and other compounds to stimulate cell division, differentiation or death [60]. Ability to efficiently accrue resources by such means, and exhibit relatively rapid clonal expansion, should represent the most common targets of selection for microbes and cancer cells, although other evolutionary–ecological situations appear to favour cells that can persist through unfavourable or harsh conditions, such as treatment with therapeutic agents.
Human individuals and groups obviously differ in power, be it physiological (as between mother and fetus), physical (as for parties that differ in strength or mobility), social (through coalitions, information or leverage), institutional (collectively established) or military (conflictual) [163]. In humans, social–behavioural effects on health thus depend not just on divergences between related individuals or groups in optimal inclusive-fitness maximizing phenotypes, but also on asymmetries in ability to reach one's optima.
A third noteworthy aspect of Hamiltonian medicine is its emphasis on relatedness to non-descendent as well as descendent kin, which adds a new dimension to health because it expands the scope for altruistic behaviour that, by definition, imposes phenotypic, potentially health-related, costs on actors. This consideration further highlights the overlap and divergences between optimal health (whereby a phenotypic state is maximized) and maximization of inclusive fitness (whereby alleles are selected for by causing phenotypic states that increase their success in replication). Good health is thus expected to lead to maximal inclusive fitness to the extent that it allows pursuit of the most effective means to do so. However, given the natural selection of senescence, deviations from an optimal health state are increasingly expected as residual reproductive value declines following young adulthood. Reduced health may, of course, commonly also be a direct result of maximizing inclusive fitness through investments in offspring and other kin. Inclusive fitness and health may thus sometimes trade-off, but individuals are expected to strive for maximal inclusive fitness nonetheless.
What is optimal mental health, from such an inclusive-fitness perspective? Natural selection is predicted to maximize not happiness or psychological well-being [164], but continual efforts to increase inclusive fitness via aspects of cognition, affect and behaviour, subject to each individual's capabilities and environment. From this viewpoint, deviations from happiness are not unexpected and should take particular, predictable forms. For example, mild depression can be interpreted as a psychological response to perceived or actual impediments that hinder success in striving [165–167], whereas mania in bipolar disorder can be construed as risky, runaway striving [168]. In both cases, the psychiatric disorders represent extreme, maladaptive expressions of inclusive-fitness maximizing mechanisms that are beneficial in relatively mild forms, although antithetical at least to short-term mental well-being.
The primary implication of this perspective is that mental health treatments that smooth an individual's optimal path to maximizing inclusive fitness should be more effective than treatments that attempt to increase happiness or well-being by any other means. Most generally, psychological health may be regarded in terms of ongoing success in inclusive-fitness striving, which should engender deviations from happiness only to the extent that motivation trades off with contentment. The fundamental distinction between the neurological systems for ‘wanting’ compared with ‘liking’ [169], and their associations with integrated operation of the oxytocin, dopamine and serotonin systems, dovetail directly with this inclusive-fitness perspective, and provide clear avenues for synthesizing proximate with ultimate approaches.
What insights have the principles of Hamiltonian medicine provided in this article, and what medical and evolutionary studies should it strongly motivate? Hamiltonian medicine has been most thoroughly developed thus far in the context of social interactions among infectious human disease agents, especially bacteria. However, most major human pathogens remain virtually unstudied from inclusive-fitness perspectives, with regard to (i) genetic relatedness among interacting microbial cells, (ii) the diversity of cooperative, competitive and socially exploitative microbial behaviours displayed across microbial taxa (and their effects on virulence) and (iii) how symptoms and transmission mechanisms of infectious disease are modulated by human interactions within and between families and larger social groups. In addition, despite the emerging importance of mutualistic human microbial symbionts to the maintenance of human health, none appear to have been studied, as yet, from inclusive-fitness perspectives.
Evolutionary and ecological approaches to understanding and fighting cancer have burgeoned over the last ten years. However, despite empirically based studies by Gloria Heppner in the 1980s and 1990s that described ‘tumour cell societies’ as key to cancer progression [56], none of this work has proceeded from basic, established inclusive-fitness principles for understanding and analysing interactions between more or less related entities. We have sketched out a framework for such studies, drawing on the extensive social–behavioural parallels, but also differences, between microbial cells and cancer cells.
Human social interactions, especially among kin in early life, appear to represent among the most potent and pervasive determinants of mental health throughout the lifespan, both directly and through gene-by-environment interactions. We have sought to integrate inclusive-fitness considerations into attachment theory, the primary theoretical framework for understanding early social–behavioural interactions that shape brain development and lifetime risks for personality and psychiatric disorders. In doing so, a potential role for the neuropeptide oxytocin has emerged as a metric or indicator for socially cooperative and altruistic or mutualistic inclusive-fitness salience. Does oxytocin generate the pleasures of cooperation and social solidarity, especially with kin, while other hormones such as vasopressin and testosterone serve more as mechanisms to modulate cognitive and emotional conceptions of behavioural costs and benefits? How do genomic conflicts involving the oxytocinergic system modulate human psychological health?
W. D. Hamilton took a sceptical view of modern medicine that he believed could, in the long run, lead to excessive dependence on technology [170]. We suggest that Hamiltonian medicine itself, which centres on evolved mechanisms for maximizing human health through its focus on the kinship-related social behaviours of cells, genes and individuals, represents a useful way forward—one that we think Hamilton would have appreciated. This approach will be demanding because it requires evolutionary-, ecological- and behavioural-minded biologists to master challenging subfields of medicine. Such studies, however, will have the added benefit of accelerating insights into the major unresolved questions in evolutionary biology, ecology and behaviour, given that microbes and humans also represent our best-understood research systems from proximate, mechanistic perspectives.
Acknowledgements
We are grateful to Sam Brown, Andy Gardner, Randy Nesse, Kyle Summers and Jon Wilkins for helpful comments, to Ashley Griffith, Andy Gardner and Stuart West for inviting us to contribute this article, and to NSERC for support.
References
- 1.Williams GW, Nesse RM. 1991. The dawn of Darwinian medicine. Q. Rev. Biol. 66, 1–22. ( 10.1086/417048) [DOI] [PubMed] [Google Scholar]
- 2.World Health Organisation. 2006. Basic documents, 45th edn Geneva, Switzerland: WHO. [Google Scholar]
- 3.Hamilton WD. 1964. The genetical evolution of social behaviour, I and II. J. Theor. Biol. 7, 1–52. ( 10.1016/0022-5193(64)90038-4) [DOI] [PubMed] [Google Scholar]
- 4.Grafen A. 2006. Optimization of inclusive fitness. J. Theor. Biol. 238, 541–563. ( 10.1016/j.jtbi.2005.06.009) [DOI] [PubMed] [Google Scholar]
- 5.Foster KR. 2005. Hamiltonian medicine: why the social lives of pathogens matter. Science 308, 1269–1270. ( 10.1126/science.1108158) [DOI] [PubMed] [Google Scholar]
- 6.West SA, Gardner A. 2013. Adaptation and inclusive fitness. Curr. Biol. 23, R577–R584. ( 10.1016/j.cub.2013.05.031) [DOI] [PubMed] [Google Scholar]
- 7.Crespi BJ. 2001. The evolution of social behavior in microorganisms. Trends Ecol. Evol. 16, 178–183. ( 10.1016/S0169-5347(01)02115-2) [DOI] [PubMed] [Google Scholar]
- 8.West SA, Diggle SP, Buckling A, Gardner A, Griffin AS. 2007. The social lives of microbes. Annu. Rev. Ecol. Evol. Syst. 38, 53–77. ( 10.1146/annurev.ecolsys.38.091206.095740) [DOI] [Google Scholar]
- 9.Strassmann JE, Gilbert OM, Queller DC. 2011. Kin discrimination and cooperation in microbes. Annu. Rev. Microbiol. 65, 349–367. ( 10.1146/annurev.micro.112408.134109) [DOI] [PubMed] [Google Scholar]
- 10.Gardner A, West SA, Buckling A. 2004. Bacteriocins, spite and virulence. Proc. R. Soc. Lond. B 271, 1529–1535. ( 10.1098/rspb.2004.2756) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cotter PD, Ross RP, Hill C. 2013. Bacteriocins: a viable alternative to antibiotics? Nat. Rev. Microbiol. 11, 95–105. ( 10.1038/nrmicro2937) [DOI] [PubMed] [Google Scholar]
- 12.Ratcliff WC, Hoverman M, Travisano M, Denison RF. 2013. Cryptic kin selection in the evolution of microbial dormancy. Am. Nat. 182, 147–156. ( 10.1086/670943) [DOI] [PubMed] [Google Scholar]
- 13.Reece SE, Drew DR, Gardner A. 2008. Sex ratio adjustment and kin discrimination in malaria parasites. Nature 453, 609–614. ( 10.1038/nature06954) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadell CD, Xavier JB, Foster KR. 2009. The sociobiology of biofilms. FEMS Microbiol. Rev. 33, 206–224. ( 10.1111/j.1574-6976.2008.00150.x) [DOI] [PubMed] [Google Scholar]
- 15.Boyle KE, Heilmann S, van Ditmarsch D, Xavier JB. 2013. Exploiting social evolution in biofilms. Curr. Opin. Microbiol. 16, 207–212. ( 10.1016/j.mib.2013.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith J. 2001. The social evolution of bacterial pathogenesis. Proc. R. Soc. Lond. B 268, 61–69. ( 10.1098/rspb.2000.1330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nogueira T, Rankin DJ, Touchon M, Taddei F, Brown SP, Rocha EP. 2009. Horizontal gene transfer of the secretome drives the evolution of bacterial cooperation and virulence. Curr. Biol. 19, 1683–1691. ( 10.1016/j.cub.2009.08.056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGinty SÉ, Lehmann L, Brown SP, Rankin DJ. 2013. The interplay between relatedness and horizontal gene transfer drives the evolution of plasmid-carried public goods. Proc. R. Soc. B 280, 20130400 ( 10.1098/rspb.2013.0400) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiricny N, Diggle SP, West SA, Evans BA, Ballantyne G, Ross-Gillespie A, Griffin AS. 2010. Fitness correlates with the extent of cheating in a bacterium. J. Evol. Biol. 23, 738–747. ( 10.1111/j.1420-9101.2010.01939.x) [DOI] [PubMed] [Google Scholar]
- 20.Mellbye B, Schuster M. 2011. The sociomicrobiology of antivirulence drug resistance: a proof of concept. mBio 2, e00131 ( 10.1128/mBio.00131-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pepper JW. 2012. Drugs that target pathogen public goods are robust against evolved drug resistance. Evol. Appl. 5, 757–761. ( 10.1111/j.1752-4571.2012.00254.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diard M, Garcia V, Maier L, Remus-Emsermann MN, Regoes RR, Ackermann M, Hardt WD. 2013. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature 494, 353–356. ( 10.1038/nature11913) [DOI] [PubMed] [Google Scholar]
- 23.Tanouchi Y, Pai A, Buchler NE, You L. 2012. Programming stress-induced altruistic death in engineered bacteria. Mol. Syst. Biol. 8, 626 ( 10.1038/msb.2012.57) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berngruber TW, Lion S, Gandon S. 2013. Evolution of suicide as a defence strategy against pathogens in a spatially structured environment. Ecol. Lett. 16, 446–453. ( 10.1111/ele.12064) [DOI] [PubMed] [Google Scholar]
- 25.Refardt D, Bergmiller T, Kümmerli R. 2013. Altruism can evolve when relatedness is low: evidence from bacteria committing suicide upon phage infection. Proc. R. Soc. B 280, 20123035 ( 10.1098/rspb.2012.3035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hentzer M, Eberl L, Nielsen J, Givskov M. 2003. Quorum sensing: a novel target for the treatment of biofilm infections. Biodrugs 17, 241–250. ( 10.2165/00063030-200317040-00003) [DOI] [PubMed] [Google Scholar]
- 27.André J-B, Bernard G. 2005. Multicellular organization in bacteria as a target for drug therapy. Ecol. Lett. 8, 800–810. ( 10.1111/j.1461-0248.2005.00783.x) [DOI] [Google Scholar]
- 28.Dong Y-H, Wang L-Y, Zhang L-H. 2007. Quorum-quenching microbial infections: mechanisms and implications. Phil. Trans. R. Soc. B 362, 1201–1211. ( 10.1098/rstb.2007.2045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swem LR, Swem DL, O'Loughlin CT, Gatmaitan R, Zhao B, Ulrich SM, Bassler BL. 2009. A quorum-sensing antagonist targets both membrane-bound and cytoplasmic receptors and controls bacterial pathogenicity. Mol. Cell. 35, 143–153. ( 10.1016/j.molcel.2009.05.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunny GM, Brickman TJ, Dworkin M. 2008. Multicellular behavior in bacteria: communication, cooperation, competition and cheating. Bioessays 30, 296–298. ( 10.1002/bies.20740) [DOI] [PubMed] [Google Scholar]
- 31.Travisano M, Velicer GJ. 2004. Strategies of microbial cheater control. Trends Microbiol. 12, 72–78. ( 10.1016/j.tim.2003.12.009) [DOI] [PubMed] [Google Scholar]
- 32.Frank SA. 1992. A kin selection model for the evolution of virulence. Proc. R. Soc. Lond. B 250, 195–197. ( 10.1098/rspb.1992.0149) [DOI] [PubMed] [Google Scholar]
- 33.Frank SA. 1996. Models of parasite virulence. Q. Rev. Biol. 71, 37–78. ( 10.1086/419267) [DOI] [PubMed] [Google Scholar]
- 34.Lively CM. 2009. Local host competition in the evolution of virulence. J. Evol. Biol. 22, 1268–1274. ( 10.1111/j.1420-9101.2009.01743.x) [DOI] [PubMed] [Google Scholar]
- 35.Buckling A, Brockhurst MA. 2008. Kin selection and the evolution of virulence. Heredity (Edinb.) 100, 484–488. ( 10.1038/sj.hdy.6801093) [DOI] [PubMed] [Google Scholar]
- 36.Ross-Gillespie A, Gardner A, West SA, Griffin AS. 2007. Frequency dependence and cooperation: theory and a test with bacteria. Am. Nat. 170, 331–342. ( 10.1086/519860) [DOI] [PubMed] [Google Scholar]
- 37.Eldar A. 2011. Social conflict drives the evolutionary divergence of quorum sensing. Proc. Natl Acad. Sci. USA 108, 13 635–13 640. ( 10.1073/pnas.1102923108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rumbaugh KP, Trivedi U, Watters C, Burton-Chellew MN, Diggle SP, West SA. 2012. Kin selection, quorum sensing and virulence in pathogenic bacteria. Proc. R. Soc. B 279, 3584–3588. ( 10.1098/rspb.2012.0843) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lion S. 2013. Multiple infections, kin selection and the evolutionary epidemiology of parasite traits. J. Evol. Biol. 26, 2107–2122. ( 10.1111/jeb.12207) [DOI] [PubMed] [Google Scholar]
- 40.Mitri S, Foster KR. 2013. The genotypic view of social interactions in microbial communities. Annu. Rev. Genet. 47, 247–273. ( 10.1146/annurev-genet-111212-133307) [DOI] [PubMed] [Google Scholar]
- 41.Smith J, Van Dyken JD, Zee PC. 2010. A generalization of Hamilton's rule for the evolution of microbial cooperation. Science 328, 1700–1703. ( 10.1126/science.1189675) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Griffin AS, West SA, Buckling A. 2004. Cooperation and competition in pathogenic bacteria. Nature 430, 1024–1027. ( 10.1038/nature02744) [DOI] [PubMed] [Google Scholar]
- 43.Dethlefsen L, McFall-Ngai M, Relman DA. 2007. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449, 811–818. ( 10.1038/nature06245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown SP, Cornforth DM, Mideo N. 2012. Evolution of virulence in opportunistic pathogens: generalism, plasticity, and control. Trends Microbiol. 20, 336–342. ( 10.1016/j.tim.2012.04.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madupu R, Szpakowski S, Nelson KE. 2013. Microbiome in human health and disease. Sci. Prog. 96, 153–170. ( 10.3184/003685013X13683759820813) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alizon S, de Roode JC, Michalakis Y. 2013. Multiple infections and the evolution of virulence. Ecol. Lett. 16, 556–567. ( 10.1111/ele.12076) [DOI] [PubMed] [Google Scholar]
- 47.Allen B, Nowak MA. 2013. Cooperation and the fate of microbial societies. PLoS Biol. 11, e1001549 ( 10.1371/journal.pbio.1001549) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blower TR, Evans TJ, Przybilski R, Fineran PC, Salmond GP. 2012. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet. 8, e1003023 ( 10.1371/journal.pgen.1003023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nonacs P, Kapheim KM. 2012. Modeling disease evolution with multilevel selection: HIV as a quasispecies social genome. J. Evol. Med. 1, 235553 ( 10.4303/jem/235553) [DOI] [Google Scholar]
- 50.Cotter SC, Kilner RM. 2010. Personal immunity versus social immunity. Behav. Ecol. 21, 663–668. ( 10.1093/beheco/arq070) [DOI] [Google Scholar]
- 51.Débarre F, Lion S, van Baalen M, Gandon S. 2012. Evolution of host life-history traits in a spatially structured host-parasite system. Am. Nat. 179, 52–63. ( 10.1086/663199) [DOI] [PubMed] [Google Scholar]
- 52.Horns F, Hood ME. 2012. The evolution of disease resistance and tolerance in spatially structured populations. Ecol. Evol. 2, 1705–1711. ( 10.1002/ece3.290) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Best A, Webb S, White A, Boots M. 2011. Host resistance and coevolution in spatially structured populations. Proc. R. Soc. B 278, 2216–2222. ( 10.1098/rspb.2010.1978) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schliekelman P. 2007. Kin selection and evolution of infectious disease resistance. Evolution 61, 1277–1288. ( 10.1111/j.1558-5646.2007.00122.x) [DOI] [PubMed] [Google Scholar]
- 55.Scott S, Duncan C. 2004. Return of the black death: the world’s greatest serial killer. West Sussex, UK: John Wiley and Sons. [Google Scholar]
- 56.Heppner GH. 1993. Cancer cell societies and tumor progression. Stem Cells 11, 199–203. ( 10.1002/stem.5530110306) [DOI] [PubMed] [Google Scholar]
- 57.Axelrod R, Axelrod DE, Pienta KJ. 2006. Evolution of cooperation among tumor cells. Proc. Natl Acad. Sci. USA 103, 13 474–13 479. ( 10.1073/pnas.0606053103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michor F, Polyak K. 2010. The origins and implications of intratumor heterogeneity. Cancer Prev. Res. (Phila.) 3, 1361–1364. ( 10.1158/1940-6207.CAPR-10-0234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wagstaff L, Kolahgar G, Piddini E. 2013. Competitive cell interactions in cancer: a cellular tug of war. Trends Cell. Biol. 23, 160–167. ( 10.1016/j.tcb.2012.11.002) [DOI] [PubMed] [Google Scholar]
- 60.Nagy JD, Armbruster D. 2012. Evolution of uncontrolled proliferation and the angiogenic switch in cancer. Math. Biosci. Eng. 9, 843–876. ( 10.3934/mbe.2012.9.843) [DOI] [PubMed] [Google Scholar]
- 61.Trejo-Becerril C, et al. 2012. Cancer progression mediated by horizontal gene transfer in an in vivo model. PLoS ONE 7, e52754 ( 10.1371/journal.pone.0052754) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Archetti M. 2013. Evolutionary dynamics of the Warburg effect: glycolysis as a collective action problem among cancer cells. J. Theor. Biol. 341C, 1–8. [DOI] [PubMed] [Google Scholar]
- 63.Hickson J, Diane Yamada S, Berger J, Alverdy J, O'Keefe J, Bassler B, Rinker-Schaeffer C. 2009. Societal interactions in ovarian cancer metastasis: a quorum-sensing hypothesis. Clin. Exp. Metastasis 26, 67–76. ( 10.1007/s10585-008-9177-z) [DOI] [PubMed] [Google Scholar]
- 64.Ghosh S, Elankumaran S, Puri IK. 2011. Mathematical model of the role of intercellular signalling in intercellular cooperation during tumorigenesis. Cell Prolif. 44, 192–203. ( 10.1111/j.1365-2184.2011.00739.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee TH, D'Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. 2011. Microvesicles as mediators of intercellular communication in cancer: the emerging science of cellular ‘debris’. Semin. Immunopathol. 33, 455–467. ( 10.1007/s00281-011-0250-3) [DOI] [PubMed] [Google Scholar]
- 66.Nagy JD. 2004. Competition and natural selection in a mathematical model of cancer. Bull. Math. Biol. 66, 663–687. ( 10.1016/j.bulm.2003.10.001) [DOI] [PubMed] [Google Scholar]
- 67.Nagy JD. 2008. Hypertumors in cancer can be caused by tumor phosphorus demand. Proc. Appl. Math. Mech. 7, 1121703 ( 10.1002/pamm.200700761) [DOI] [Google Scholar]
- 68.Aktipis CA, Nesse RM. 2013. Evolutionary foundations for cancer biology. Evol. Appl. 6, 144–159. ( 10.1111/eva.12034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sprouffske K, Aktipis CA, Radich JP, Carroll M, Nedelcu AM, Maley CC. 2013. An evolutionary explanation for the presence of cancer non-stem cells in neoplasms. Evol. Appl. 6, 92–101. ( 10.1111/eva.12030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kosaka N, Iguchi H, Yoshioka Y, Hagiwara K, Takeshita F, Ochiya T. 2012. Competitive interactions of cancer cells and normal cells via secretory microRNAs. J. Biol. Chem. 287, 1397–1405. ( 10.1074/jbc.M111.288662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Agur Z, Kogan Y, Levi L, Harrison H, Lamb R, Kirnasovsky OU, Clarke RB. 2010. Disruption of a quorum sensing mechanism triggers tumorigenesis: a simple discrete model corroborated by experiments in mammary cancer stem cells. Biol. Direct. 5, 20 ( 10.1186/1745-6150-5-20) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krepkin K, Costa J. 2011. Defining the role of cooperation in early tumor progression. J. Theor. Biol. 285, 36–45. ( 10.1016/j.jtbi.2011.06.035) [DOI] [PubMed] [Google Scholar]
- 73.Heppner GH. 1989. Tumor cell societies. J. Natl. Cancer Inst. 81, 648–649. ( 10.1093/jnci/81.9.648) [DOI] [PubMed] [Google Scholar]
- 74.Ben-Jacob E, Coffey DS, Levine H. 2012. Bacterial survival strategies suggest rethinking cancer cooperativity. Trends Microbiol. 20, 403–410. ( 10.1016/j.tim.2012.06.001) [DOI] [PubMed] [Google Scholar]
- 75.Lambert G, Estévez-Salmeron L, Oh S, Liao D, Emerson BM, Tlsty TD, Austin RH. 2011. An analogy between the evolution of drug resistance in bacterial communities and malignant tissues. Nat. Rev. Cancer 11, 375–382. ( 10.1038/nrc3039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bergsmedh A, Szeles A, Henriksson M, Bratt A, Folkman MJ, Spetz AL, Holmgren L. 2001. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl Acad. Sci. USA 98, 6407–6411. ( 10.1073/pnas.101129998) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. 2011. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2, 180 ( 10.1038/ncomms1180) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murchison EP. 2008. Clonally transmissible cancers in dogs and Tasmanian devils. Oncogene 27, S19–S30. ( 10.1038/onc.2009.350) [DOI] [PubMed] [Google Scholar]
- 79.Sprouffske K, Merlo LMF, Gerrish PJ, Maley CC, Sniegowski PD. 2012. Cancer in light of experimental evolution. Curr. Biol. 22, R762–R771. ( 10.1016/j.cub.2012.06.065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Avner BS, Fialho AM, Chakrabarty AM. 2012. Overcoming drug resistance in multi-drug resistant cancers and microorganisms: a conceptual framework. Bioengineered 3, 262–270. ( 10.4161/bioe.21130). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frank SA. 2013. Microbial evolution: regulatory design prevents cancer-like overgrowths. Curr. Biol. 23, R343–R346. ( 10.1016/j.cub.2013.03.046) [DOI] [PubMed] [Google Scholar]
- 82.Dejosez M, Ura H, Brandt VL, Zwaka TP. 2013. Safeguards for cell cooperation in mouse embryogenesis shown by genome-wide cheater screen. Science 341, 1511–1514. ( 10.1126/science.1241628) [DOI] [PubMed] [Google Scholar]
- 83.Hamilton WD. 1967. Extraordinary sex ratios. A sex-ratio theory for sex linkage and inbreeding has new implications in cytogenetics and entomology. Science 156, 477–488. ( 10.1126/science.156.3774.477) [DOI] [PubMed] [Google Scholar]
- 84.Haig D. 2006. Intragenomic politics. Cytogenet. Genome Res. 113, 68–74. ( 10.1159/000090816) [DOI] [PubMed] [Google Scholar]
- 85.Jansen VAA, van Baalen M. 2006. Altruism through beard chromodynamics. Nature 440, 663–666. ( 10.1038/nature04387) [DOI] [PubMed] [Google Scholar]
- 86.Biernaskie JM, West SA, Gardner A. 2011. Are greenbeards intragenomic outlaws? Evolution 65, 2729–2742. ( 10.1111/j.1558-5646.2011.01355.x) [DOI] [PubMed] [Google Scholar]
- 87.Stencel A, Crespi B. 2013. What is a genome? Mol. Ecol. 22, 3437–3443. ( 10.1111/mec.12355) [DOI] [PubMed] [Google Scholar]
- 88.Frank SA, Crespi BJ. 2011. Pathology from evolutionary conflict, with a theory of X chromosome versus autosome conflict over sexually antagonistic traits. Proc. Natl Acad. Sci. USA 108, 10 886–10 893. ( 10.1073/pnas.1100921108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crespi BJ. 2010. The origins and evolution of genetic disease risk in modern humans. Ann. N.Y. Acad. Sci. 1206, 80–109. ( 10.1111/j.1749-6632.2010.05707.x) [DOI] [PubMed] [Google Scholar]
- 90.Úbeda F, Wilkins JF. 2008. Imprinted genes and human disease: an evolutionary perspective. In Genomic imprinting (ed. Wilkins JF.), pp. 101–113. New York, NY: Springer. [PubMed] [Google Scholar]
- 91.Wilkins JF, Úbeda F. 2011. Diseases associated with genomic imprinting. Prog. Mol. Biol. Transl. Sci. 101, 401–445. ( 10.1016/B978-0-12-387685-0.00013-5) [DOI] [PubMed] [Google Scholar]
- 92.Wilkins JF. 2011. Genomic imprinting and conflict-induced decanalization. Evolution 65, 537–553. ( 10.1111/j.1558-5646.2010.01147.x) [DOI] [PubMed] [Google Scholar]
- 93.Úbeda F. 2008. Evolution of genomic imprinting with biparental care: implications for Prader–Willi and Angelman syndromes. PLoS Biol. 6, e208 ( 10.1371/journal.pbio.0060208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eggermann T, Eggermann K, Schönherr N. 2008. Growth retardation versus overgrowth: Silver–Russell syndrome is genetically opposite to Beckwith–Wiedemann syndrome. Trends Genet. 24, 195–204. ( 10.1016/j.tig.2008.01.003) [DOI] [PubMed] [Google Scholar]
- 95.Soejima H, Higashimoto K. 2013. Epigenetic and genetic alterations of the imprinting disorder Beckwith–Wiedemann syndrome and related disorders. J. Hum. Genet. 58, 402–409. ( 10.1038/jhg.2013.51) [DOI] [PubMed] [Google Scholar]
- 96.Andersen DC, Laborda J, Baladron V, Kassem M, Sheikh SP, Jensen CH. 2013. Dual role of delta-like 1 homolog (DLK1) in skeletal muscle development and adult muscle regeneration. Development 140, 3743–3753. ( 10.1242/dev.095810) [DOI] [PubMed] [Google Scholar]
- 97.Venkatraman A, et al. 2013. Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature 500, 345–349. ( 10.1038/nature12303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schmidt-Edelkraut U, Hoffmann A, Daniel G, Spengler D. 2013. Zac1 regulates astroglial differentiation of neural stem cells through socs3. Stem Cells 31, 1621–1632. ( 10.1002/stem.1405) [DOI] [PubMed] [Google Scholar]
- 99.Renfree MB, Suzuki S, Kaneko-Ishino T. 2013. The origin and evolution of genomic imprinting and viviparity in mammals. Phil. Trans. R. Soc. B 368, 20120151 ( 10.1098/rstb.2012.0151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frost JM, Moore GE. 2010. The importance of imprinting in the human placenta. PLoS Genet. 6, e1001015 ( 10.1371/journal.pgen.1001015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barker DJ, Lampl M, Roseboom T, Winder N. 2012. Resource allocation in utero and health in later life. Placenta 33, e30–e34. ( 10.1016/j.placenta.2012.06.009) [DOI] [PubMed] [Google Scholar]
- 102.Úbeda F, Haig D. 2003. Dividing the child. Genetica 117, 103–110. ( 10.1023/A:1022320801661) [DOI] [PubMed] [Google Scholar]
- 103.Wilkins JF, Haig D. 2001. Genomic imprinting of two antagonistic loci. Proc. R. Soc. Lond. B 268, 1861–1867. ( 10.1098/rspb.2001.1651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Choudhury M, Friedman JE. 2012. Epigenetics and microRNAs in preeclampsia. Clin. Exp. Hypertens. 34, 334–341. ( 10.3109/10641963.2011.649931) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Naeye RL. 1981. Maternal blood pressure and fetal growth. Am. J. Obstet. Gynecol. 141, 780–787. [DOI] [PubMed] [Google Scholar]
- 106.Úbeda F, Gardner A. 2011. A model for genomic imprinting in the social brain: adults. Evolution 65, 462–475. ( 10.1111/j.1558-5646.2010.01115.x) [DOI] [PubMed] [Google Scholar]
- 107.Úbeda F, Gardner A. 2012. A model for genomic imprinting in the social brain: elders. Evolution 66, 1567–1581. ( 10.1111/j.1558-5646.2011.01517.x) [DOI] [PubMed] [Google Scholar]
- 108.Úbeda F, Ohtsuki H, Gardner A. 2013. Ecology drives intra-genomic conflict over menopause. Ecol. Lett. 17, 165–174. ( 10.1111/ele.12208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Crespi B. 2011. The evolutionary biology of child health. Proc. R. Soc. B 278, 1441–1449. ( 10.1098/rspb.2010.2627) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Springer SA, Crespi BJ, Swanson WJ. 2011. Beyond the phenotypic gambit: molecular behavioral ecology and the evolution of genetic architecture. Mol. Ecol. 20, 2240–2257. ( 10.1111/j.1365-294X.2011.05116.x) [DOI] [PubMed] [Google Scholar]
- 111.Trivers RL. 1974. Parent–offspring conflict. Am. Zool. 14, 249–264. ( 10.1093/icb/14.1.249) [DOI] [Google Scholar]
- 112.Haig D. 1993. Genetic conflicts in human pregnancy. Q. Rev. Biol. 68, 495–532. ( 10.1086/418300) [DOI] [PubMed] [Google Scholar]
- 113.Bowlby J. 1999. Attachment: attachment and loss, 2nd edn., vol. 1 New York, NY: Basic Books. [Google Scholar]
- 114.Crespi BJ. 2011. The strategies of the genes: genomic conflicts, attachment theory, and development of the social brain. In Brain, behavior and epigenetics (eds Petronis A, Mill J.), pp. 143–168. Heidelberg, Berlin: Springer. [Google Scholar]
- 115.Shaver PR, Mikulincer M. 2002. Attachment-related psychodynamics. Attach. Hum. Dev. 4, 133–161. ( 10.1080/14616730210154171) [DOI] [PubMed] [Google Scholar]
- 116.Chisholm JS. 1996. The evolutionary ecology of attachment organization. Hum. Nat. 7, 1–38. [DOI] [PubMed] [Google Scholar]
- 117.Benoit D. 2004. Infant–parent attachment: definition, types, antecedents, measurement and outcome. Paediatr. Child Health 9, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Berry K, Barrowclough C, Wearden A. 2007. A review of the role of adult attachment style in psychosis: unexplored issues and questions for further research. Clin. Psychol. Rev. 27, 458–475. ( 10.1016/j.cpr.2006.09.006) [DOI] [PubMed] [Google Scholar]
- 119.Prior V, Glaser D. 2006. Understanding attachment and attachment disorders: theory, evidence and practice. London, UK: Jessica Kingsley Publishers. [Google Scholar]
- 120.Schaller F, Watrin F, Sturny R, Massacrier A, Szepetowski P, Muscatelli F. 2010. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 19, 4895–4905. ( 10.1093/hmg/ddq424) [DOI] [PubMed] [Google Scholar]
- 121.Champagne FA, Curley JP, Swaney WT, Hasen NS, Keverne EB. 2009. Paternal influence on female behavior: the role of Peg3 in exploration, olfaction, and neuroendocrine regulation of maternal behavior of female mice. Behav. Neurosci. 123, 469–480. ( 10.1037/a0015060) [DOI] [PubMed] [Google Scholar]
- 122.Muscatelli F, Abrous DN, Massacrier A, Boccaccio I, Le Moal M, Cau P, Cremer H. 2000. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader–Willi syndrome. Hum. Mol. Genet. 9, 3101–3110. ( 10.1093/hmg/9.20.3101) [DOI] [PubMed] [Google Scholar]
- 123.Plagge A, Gordon E, Dean W, Boiani R, Cinti S, Peters J, Kelsey G. 2004. The imprinted signaling protein XLαs is required for postnatal adaptation to feeding. Nat. Genet. 36, 818–826. ( 10.1038/ng1397) [DOI] [PubMed] [Google Scholar]
- 124.Curley JP. 2011. Is there a genomically imprinted social brain? Bioessays 33, 662–668. ( 10.1002/bies.201100060) [DOI] [PubMed] [Google Scholar]
- 125.Varrault A, et al. 2006. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 11, 711–722. ( 10.1016/j.devcel.2006.09.003) [DOI] [PubMed] [Google Scholar]
- 126.Feldman R. 2012. Oxytocin and social affiliation in humans. Horm. Behav. 61, 380–391. ( 10.1016/j.yhbeh.2012.01.008) [DOI] [PubMed] [Google Scholar]
- 127.Veening JG, Olivier B. 2013. Intranasal administration of oxytocin: behavioral and clinical effects, a review. Neurosci. Biobehav. Rev. 37, 1445–1465. ( 10.1016/j.neubiorev.2013.04.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.De Dreu CK, Greer LL, Van Kleef GA, Shalvi S, Handgraaf MJ. 2011. Oxytocin promotes human ethnocentrism. Proc. Natl Acad. Sci. USA 108, 1262–1266. ( 10.1073/pnas.1015316108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fischer-Shofty M, Levkovitz Y, Shamay-Tsoory SG. 2013. Oxytocin facilitates accurate perception of competition in men and kinship in women. Soc. Cogn. Affect. Neurosci. 8, 313–317. ( 10.1093/scan/nsr100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fischer-Shofty M, Brüne M, Ebert A, Shefet D, Levkovitz Y, Shamay-Tsoory SG. 2013. Improving social perception in schizophrenia: the role of oxytocin. Schizophr. Res. 146, 357–362. ( 10.1016/j.schres.2013.01.006) [DOI] [PubMed] [Google Scholar]
- 131.Preston SD. 2013. The origins of altruism in offspring care. Psychol. Bull. 139, 1305–1341. ( 10.1037/a0031755) [DOI] [PubMed] [Google Scholar]
- 132.Haig D, Wharton R. 2003. Prader–Willi syndrome and the evolution of human childhood. Am. J. Hum. Biol. 15, 320–329. ( 10.1002/ajhb.10150) [DOI] [PubMed] [Google Scholar]
- 133.Swaab DF, Purba JS, Hofman MA. 1995. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader–Willi syndrome: a study of five cases. J. Clin. Endocrinol. Metab. 80, 573–579. [DOI] [PubMed] [Google Scholar]
- 134.Soni S, Whittington J, Holland AJ, Webb T, Maina EN, Boer H, Clarke D. 2008. The phenomenology and diagnosis of psychiatric illness in people with Prader–Willi syndrome. Psychol. Med. 38, 1505–1514. ( 10.1017/S0033291707002504) [DOI] [PubMed] [Google Scholar]
- 135.Mabb AM, Judson MC, Zylka MJ, Philpot BD. 2011. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303. ( 10.1016/j.tins.2011.04.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Oliver C, Horsler K, Berg K, Bellamy G, Dick K, Griffiths E. 2007. Genomic imprinting and the expression of affect in Angelman syndrome: what's in the smile? J. Child Psychol. Psychiatry 48, 571–579. ( 10.1111/j.1469-7610.2007.01736.x) [DOI] [PubMed] [Google Scholar]
- 137.Bernard D. 2008. Angelman syndrome. Hoboken, NJ: Wiley-Blackwell. [Google Scholar]
- 138.Farook MF, DeCuypere M, Hyland K, Takumi T, LeDoux MS, Reiter LT. 2012. Altered serotonin, dopamine and norepinepherine levels in 15q duplication and Angelman syndrome mouse models. PLoS ONE 7, e43030 ( 10.1371/journal.pone.0043030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jedele KB. 2007. The overlapping spectrum of Rett and Angelman syndromes: a clinical review. Semin. Pediatr. Neurol. 14, 108–117. ( 10.1016/j.spen.2007.07.002) [DOI] [PubMed] [Google Scholar]
- 140.Rutgers AH, Bakermans-Kranenburg MJ, van Ijzendoorn MH, van Berckelaer-Onnes IA. 2004. Autism and attachment: a meta-analytic review. J. Child. Psychol. Psychiatry 45, 1123–1134. ( 10.1111/j.1469-7610.2004.t01-1-00305.x) [DOI] [PubMed] [Google Scholar]
- 141.Crespi B. 2013. Developmental heterochrony and the evolution of autistic perception, cognition and behavior. BMC Med. 11, 119 ( 10.1186/1741-7015-11-119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Collette JC, Chen XN, Mills DL, Galaburda AM, Reiss AL, Bellugi U, Korenberg JR. 2009. William's syndrome: gene expression is related to parental origin and regional coordinate control. J. Hum. Genet. 54, 193–198. ( 10.1038/jhg.2009.5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dilts CV, Morris CA, Leonard CO. 1990. Hypothesis for development of a behavioral phenotype in Williams syndrome. Am. J. Med. Genet. Suppl. 6, 126–131. [DOI] [PubMed] [Google Scholar]
- 144.Järvinen A, Korenberg JR, Bellugi U. 2013. The social phenotype of Williams syndrome. Curr. Opin. Neurobiol. 23, 414–422. ( 10.1016/j.conb.2012.12.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dai L, Carter CS, Ying J, Bellugi U, Pournajafi-Nazarloo H, Korenberg JR. 2012. Oxytocin and vasopressin are dysregulated in Williams Syndrome, a genetic disorder affecting social behavior. PLoS ONE 7, e38513 ( 10.1371/journal.pone.0038513) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tordjman S, et al. 2012. Autistic disorder in patients with Williams–Beuren syndrome: a reconsideration of the Williams–Beuren syndrome phenotype. PLoS ONE 7, e30778 ( 10.1371/journal.pone.0030778) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mulle JG, et al. 2013. Reciprocal duplication of the Williams-Beuren Syndrome deletion on chromosome 7q11.23 is associated with schizophrenia. Biol. Psychiatry. 75, 371–377. ( 10.1016/j.biopsych.2013.05.040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Widiger TA, Presnall JR. 2012. Clinical application of the five-factor model. J. Pers. 81, 515–527. ( 10.1111/jopy.12004) [DOI] [PubMed] [Google Scholar]
- 149.Samuel DB, Gore WL. 2012. Maladaptive variants of conscientiousness and agreeableness. J. Pers. 80, 1669–1696. ( 10.1111/j.1467-6494.2012.00770.x) [DOI] [PubMed] [Google Scholar]
- 150.Widiger TA, Costa PT., Jr 2012. Integrating normal and abnormal personality structure: the five-factor model. J. Pers. 80, 1471–1506. ( 10.1111/j.1467-6494.2012.00776.x) [DOI] [PubMed] [Google Scholar]
- 151.Bartz J, Simeon D, Hamilton H, Kim S, Crystal S, Braun A, Vicens V, Hollander E. 2011. Oxytocin can hinder trust and cooperation in borderline personality disorder. Soc. Cogn. Affect. Neurosci. 6, 556–563. ( 10.1093/scan/nsq085) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dinsdale N, Crespi BJ. 2013. The borderline empathy paradox: evidence and conceptual models for empathic enhancements in borderline personality disorder. J. Pers. Disord. 27, 172–195. ( 10.1521/pedi.2013.27.2.172) [DOI] [PubMed] [Google Scholar]
- 153.Zahn-Waxler C, Crick N, Shirtcliff EA, Woods K. 2006. The origins and development of psychopathology in females and males. In Developmental psychopathology, vol. 1 (eds Cicchetti D, Cohen DJ.), pp. 76–138. Hoboken, NJ: Wiley. [Google Scholar]
- 154.Zahn-Waxler C, Shirtcliff EA, Marceau K. 2008. Disorders of childhood and adolescence: gender and psychopathology. Annu. Rev. Clin. Psychol. 4, 275–303. ( 10.1146/annurev.clinpsy.3.022806.091358) [DOI] [PubMed] [Google Scholar]
- 155.Samuel DB, Carroll KM, Rounsaville BJ, Ball SA. 2013. Personality disorders as maladaptive, extreme variants of normal personality: borderline personality disorder and neuroticism in a substance using sample. J. Pers. Disord. 27, 625–635. ( 10.1521/pedi.2013.27.5.625) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Haig D. 2003. On intrapersonal reciprocity. Evol. Hum. Behav. 24, 418–425. ( 10.1016/S1090-5138(03)00063-1) [DOI] [Google Scholar]
- 157.Crespi B, Badcock C. 2008. Psychosis and autism as diametrical disorders of the social brain. Behav. Brain Sci. 31, 241–261. ( 10.1017/S0140525X08004214) [DOI] [PubMed] [Google Scholar]
- 158.Tsang SY, et al. 2013. Social cognitive role of schizophrenia candidate gene GABRB2. PLoS ONE 8, e62322 ( 10.1371/journal.pone.0062322) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ubeda F, Gardner A. 2010. A model for genomic imprinting in the social brain: juveniles. Evolution 64, 2587–2600. ( 10.1111/j.1558-5646.2010.01015.x) [DOI] [PubMed] [Google Scholar]
- 160.Francks C, et al. 2007. LRRTM1 on chromosome 2p12 is a maternally suppressed gene that is associated paternally with handedness and schizophrenia. Mol. Psychiatry 12, 1129–1139. ( 10.1038/sj.mp.4002053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Queller DC. 2011. Expanded social fitness and Hamilton's rule for kin, kith, and kind. Proc. Natl Acad. Sci. USA 108, 10 792–10 799. ( 10.1073/pnas.1100298108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Frank SA. 2003. Repression of competition and the evolution of cooperation. Evolution 57, 693–705. [DOI] [PubMed] [Google Scholar]
- 163.Boehm C, Flack J. 2010. The emergence of simple and complex power structures through social niche construction. In The social psychology of power (ed. Guinote A.), pp. 46–87. London, UK: Guilford. [Google Scholar]
- 164.Nesse RM. 2004. Natural selection and the elusiveness of happiness. Phil. Trans. R. Soc. Lond. B 359, 1333–1347. ( 10.1098/rstb.2004.1511) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Keller MC, Nesse RM. 2005. Is low mood an adaptation? Evidence for subtypes with symptoms that match precipitants. J. Affect. Disord. 86, 27–35. ( 10.1016/j.jad.2004.12.005) [DOI] [PubMed] [Google Scholar]
- 166.Nettle D. 2004. Evolutionary origins of depression: a review and reformulation. J. Affect. Disord. 81, 91–102. ( 10.1016/j.jad.2003.08.009) [DOI] [PubMed] [Google Scholar]
- 167.Andrews PW, Thomson JA., Jr 2009. The bright side of being blue: depression as an adaptation for analyzing complex problems. Psychol. Rev. 116, 620–654. ( 10.1037/a0016242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Johnson SL, Fulford D, Carver CS. 2012. The double-edged sword of goal engagement: consequences of goal pursuit in bipolar disorder. Clin. Psychol. Psychother. 19, 352–362. ( 10.1002/cpp.1801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Berridge KC, Robinson TE, Aldridge JW. 2009. Dissecting components of reward: ‘liking’, ‘wanting’, and learning. Curr. Opin. Pharmacol. 9, 65–73. ( 10.1016/j.coph.2008.12.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Segerstrale U. 2013. Nature’s oracle: the life and work of W. D. Hamilton. Oxford, UK: Oxford University Press. [Google Scholar]