

Abstract
Background
Earlier work identified specific tumor-promoting abnormalities that are shared between lung cancers and adjacent normal bronchial epithelia. We sought to characterize the yet unknown global molecular and adjacent airway field cancerization (FC) in early-stage non–small cell lung cancer (NSCLC).
Methods
Whole-transcriptome expression profiling of resected early-stage (I–IIIA) NSCLC specimens (n = 20) with matched tumors, multiple cytologically controlled normal airways with varying distances from tumors, and uninvolved normal lung tissues (n = 194 samples) was performed using the Affymetrix Human Gene 1.0 ST platform. Mixed-effects models were used to identify differentially expressed genes among groups. Ordinal regression analysis was performed to characterize site-dependent airway expression profiles. All statistical tests were two-sided, except where noted.
Results
We identified differentially expressed gene features (n = 1661) between NSCLCs and airways compared with normal lung tissues, a subset of which (n = 299), after gene set enrichment analysis, statistically significantly (P < .001) distinguished large airways in lung cancer patients from airways in cancer-free smokers. In addition, we identified genes (n = 422) statistically significantly and progressively differentially expressed in airways by distance from tumors that were found to be congruently modulated between NSCLCs and normal lung tissues. Furthermore, LAPTM4B, with statistically significantly increased expression (P < .05) in airways with shorter distance from tumors, was upregulated in human immortalized cells compared with normal bronchial epithelial cells (P < .001) and promoted anchorage-dependent and -independent lung cancer cell growth.
Conclusions
The adjacent airway FC comprises both site-independent profiles as well as gradient and localized airway expression patterns. Profiling of the airway FC may provide new insights into NSCLC oncogenesis and molecular tools for detection of the disease.
Earlier work by Slaughter et al. in patients with oral premalignant and cancer lesions suggested that histologically normal-appearing tissues adjacent to lesions display tumor-associated molecular abnormalities (1). Notably, Auerbach et al. demonstrated that cigarette smoke induces widespread histological changes and premalignant lesions in the bronchial epithelia in the lungs of smokers, suggestive of a field effect (2). This phenomenon, coined field cancerization (FC), was shown to be evident in various epithelial malignancies, including gastric, esophageal, hepatic, cervical, skin, and lung cancers (3–6) and was proposed to precede and explain the development of multiple primary and locally recurrent cancer (3,7).
Previously, an analysis of histologically normal epithelium and premalignant and malignant epithelia from lung squamous cell carcinoma (SCC) patients indicated that multiple, sequentially occurring allele-specific chromosomal deletions commence early in the multistage pathogenesis of SCCs (8,9). Notably, 31% of histologically normal epithelium specimens had clones of cells with allelic loss at one or more regions examined, including loss of heterozygosity at chromosomal regions 3p and 9p (8,10). Belinsky et al. identified promoter methylation of p16, a common aberration in lung tumors (11), in at least one bronchial epithelial site from 44% of lung cancer cases examined (12). In addition, our group and others have demonstrated that mutations in the epidermal growth factor receptor (EGFR) and KRAS oncogenes were also found in histologically normal tissue adjacent to lung tumors (13,14).
Global expression profiles have been described in bronchial epithelium of smokers, a portion of which exhibited cancer diagnostic properties (15–18), as well as in the field of injury of early-stage non–small cell lung cancer (NSCLC) patients who had their tumors surgically resected before airway transcriptome analysis (19). However, the adjacent airway FC in NSCLC has not yet been characterized at a whole-transcriptome level. In this study, we performed expression profiling of matched NSCLCs, uninvolved normal lung tissue, and multiple airways with varying distances from tumors to define the transcriptomic architecture of the adjacent airway FC.
Methods
Lung Tumor Resected FC Specimens and Airway Epithelial Cell Collection
The FC specimens, comprised of lung tumors, uninvolved normal lung parenchyma, and multiple normal-appearing airways with varying distances from tumors, were obtained from early stage (I–IIIA) patients at MD Anderson Cancer Center. Tumor stage was classified as described previously (20). The study was approved by the institutional review boards, and all participants provided written informed consent. Malignant and paired normal lung tissues from each case patient were obtained snap-frozen, preserved in RNAlater or by surface brushing. For each tissue sample, the percentage of malignant tissue was calculated by histological examination (J. Fujimoto) after hematoxylin and eosin staining. All malignant samples contained more than 40% tumor cells. Twenty NSCLC FC case patients were included in the study and, along with their clinicopathological information, are summarized in Supplementary Table 1 (available online).
Airway epithelia were obtained by brushing three to five sequential bronchiolar structures with varying distances from tumors (Supplementary Figure 1, available online) using sterile Cytosoft cytology brushes (Medical Packaging Corporation, Camarillo, CA). The spatial distance between two consecutive airway brushings was similar (approximately 2cm). Airways were denoted by numbers 1 (relatively closest from tumor) to 5 (relatively farthest). The relative distance of an airway brushing (eg, airway 1) from the adjacent NSCLC tumor was similar across all case patients. Airway brushings were placed in Qiazol lysis buffer (Qiagen, Valencia, CA) in dry ice and immediately stored at −80°C. Confirmation of epithelial cell collection by pan-cytokeratin immunohistochemical analysis, as well as lack of neoplastic or preneoplastic cells (Supplementary Figure 1, available online), was performed as described in the Supplementary Methods (available online).
Microarray Data Analysis
RNA samples were processed for microarray expression profiling using the Affymetrix Human Gene 1.0 ST platform (Affymetrix, Santa Clara, CA) (Supplementary Methods, available online). Raw data were quantified using background correction, quantile normalization, and robust multichip analysis (21) probe-level models and summarization methods. Minimum Information About a Microarray Experiment (MIAME)-compliant data were submitted to the Gene Expression Omnibus under series GSE44077 (samples GSM1077844–GSM1078069). Basic quality control was assessed by graphical summaries of array intensities and Bland-Altman (M vs A) plots (Supplementary Methods, Sweave Report 1, available online). Linear mixed-effects models were used to characterize gene features concordantly differentially expressed (in the same direction) between both NSCLCs and airways compared with normal lung tissues (site-independent analysis). The different groups were set as fixed effects, whereas patients were set as random factors in the mixed-effects models. An ordinal logistic regression model (22–24) was applied to identify gene features that were statistically significantly decreased or increased in airways by distance from tumors (site-dependent analysis). The model assumes that each FC case comprises only one airway brushing at a specific spatial location (eg, airway 1) and fits expression patterns that change in a gradient fashion with different airway spatial locations (airways 1–5) classified as categorical ordered labels. One-sided t tests were used to determine whether site-dependent differential airway expression exhibits similar directional patterns between NSCLCs and matched normal lung tissues. A site-dependent FC score was generated to collectively signify and plot gene features that are differentially modulated with respect to proximity from tumors across all airways by distance from tumors (airways 1–5) and between paired NSCLCs and uninvolved normal lung tissues. The score was calculated by the sum of gene features upregulated in airways with shorter distance from tumors minus the sum of gene features downregulated in airways with shorter distance from tumors. To adjust for multiple testing in all analyses, beta-uniform mixture models were used to estimate false discovery rates as previously described (25). Pathways analysis was performed using ingenuity pathways analysis.
Statistical Analysis
Fisher exact test was used to determine statistical significance of the difference in airway type (cancer vs no cancer) between clusters after hierarchical clustering analysis. Kruskal–Wallis test was used to test for statistical significance of differences in quantitative real-time polymerase chain reaction (QRTPCR)–based expression of genes among NSCLCs, airways, and normal lung tissues. Analysis of variance was used to test for statistical significance of differences in QRTPCR-based expression of genes among airways with respect to proximity from tumors. Student t test was used to assess for statistical significance among different groups in the in vitro experiments. All statistical tests were two-sided, except where noted. Additional methods including all details and R codes of the microarray statistical analysis are included in the Supplementary Methods and in the Supplementary Sweave Reports (available online).
Results
Identification of Adjacent Airway FC Profiles in Early-Stage NSCLC
The FC in the airway adjacent to NSCLC has not been characterized at the global transcriptomic level before. We analyzed the transcriptomes of cytologically controlled NSCLC tumors, paired uninvolved normal lung tissue, and brushings of normal airway epithelia collected at sequentially varying distances from the tumors (n = 194 samples) (Supplementary Figure 1). Samples were obtained from specimens surgically resected from 20 patients (n = 9 women and 11 men; n = 5 never-smokers and 15 smokers) with stages I to III NSCLC (n = 14 adenocarcinomas, 5 SCCs, and 1 not otherwise specified NOS NSCLC) (Supplementary Table 1, available online).
A schematic of the study’s design and different analyses is represented in Figure 1. We first sought to characterize global FC profiles in the normal-appearing airway adjacent to the NSCLC. We identified 1661 gene features (n = 457 upregulated and 1204 downregulated) (Supplementary Table 2, available online) that were statistically significantly and concordantly differentially expressed between both NSCLCs and airways compared with normal lung tissue (false discovery rate–corrected P < .01; fold-change > 1.5) (Figure 2A; Supplementary Methods, Sweave Report 2, available online). Further analysis identified statistically significantly differentially expressed gene features between the airway of adenocarcinomas and SCCs (n = 415) and between the airway of never-smoker and smoker NSCLCs (n = 51) (Supplementary Methods, Sweave Report 2, available online). Pathway analysis of the 1661 gene features by ingenuity pathways analysis revealed modulation of key cancer-associated pathways and gene-interaction networks (all P < .05) (Figure 2B). The most statistically significantly (P < .001) modulated pathway and the gene network with highest number of differentially expressed and closely related (G-protein coupled receptors) interacting genes are represented in Figure 2, C and D, respectively. These findings highlight expression patterns and pathways that are typically deregulated in overt tumors but are also prevalent in histologically normal airway epithelia.
Figure 1.

Schematic of the transcriptomic analysis of the airway field cancerization (FC) in non–small cell lung cancer (NSCLC). FC samples comprised of matched lung tumors, multiple cytologically controlled normal airways with varying proximity from tumors, and uninvolved normal lung tissues were obtained from 20 resected early-stage (I–IIIA) NSCLC specimens. Gene expression profiling of all the samples (n = 194) was performed to characterize global FC profiles (differentially expressed in the same direction between both tumors and airways compared with normal lung tissues) in the normal-appearing airway adjacent to NSCLCs (site-independent analysis). Adjacent FC profiles were then analyzed by gene set enrichment analysis (GSEA) in a set of large airways of smokers (n = 129) with and without lung cancer (17) to identify FC profiles that can distinguish lung cancer among smokers. Global expression profiling was also used to delineate FC profiles that are modulated in the airway with respect to proximity from corresponding NSCLCs (site-dependent analysis). Quantitative real-time polymerase chain reaction (PCR) was used to confirm the differential expression of select airway FC markers.
Figure 2.
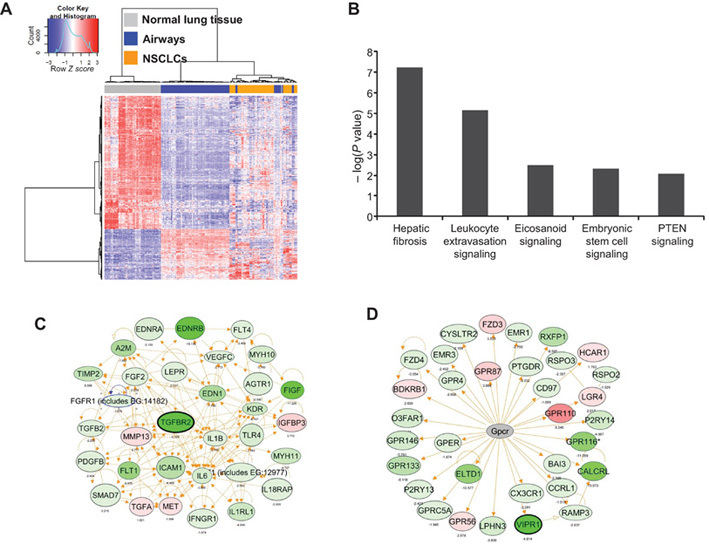
Identification of adjacent airway field cancerization (FC) profiles in non–small cell lung cancer (NSCLC). A) Hierarchical clustering of gene features statistically significantly differentially expressed between both NSCLCs and airways compared with normal lung tissues (n = 1661). Columns represent samples (n = 194 samples from 20 case patients), and rows represent gene features (red = upregulated; blue = downregulated). B) Functional pathways analysis using ingenuity pathways analysis (IPA) of the differentially expressed genes. Statistical significance of the identified overrepresented canonical pathways is indicated by the negative log of the P values. Functional pathways and interaction network analysis by IPA depicting the most statistically significantly (P < .001) modulated pathway (C) and the gene network with highest number of differentially expressed and closely related (G-protein coupled receptors) interacting genes (D); red = higher experession; green = lower expression. Genes selected for subsequent confirmation by quantitative real-time polymerase chain reaction are highlighted by black margins.
Analysis of Adjacent Airway FC Profiles in Smoker Patients With Suspected Lung Cancer
We then sought to ascertain whether the adjacent FC profiles may be indicative of lung cancer among smokers. We examined the expression of the adjacent airway FC profile in a cohort from Spira et al. (17) comprised of 129 large airway samples from smokers with and without lung cancer. After matching gene features between the two studies by common Entrez Gene identifiers, 261 upregulated and 749 downregulated adjacent airway FC gene features remained, which were used to perform gene set enrichment analysis (GSEA) with identifiers ranked according to the Student t statistic between smokers with and without lung cancer. This analysis demonstrated that the gene features that were downregulated in the adjacent airway FC were statistically significantly (P < .001) enriched within gene features downregulated in large airways of smokers with lung cancer compared with airways of cancer-free smokers, although such a statistically significant connection was not observed for the upregulated genes. Leading edge gene sets comprised of 299 genes (Supplementary Table 3, available online) that were concordantly modulated between NSCLCs and airways compared with normal lung tissue in the adjacent airway FC and between large airways of smokers with and without lung cancer (P < .001) (Figure 3) were then derived [as described previously (26)]. These data suggest that the adjacent airway FC harbors potential markers for detection of lung cancer among smokers.
Figure 3.
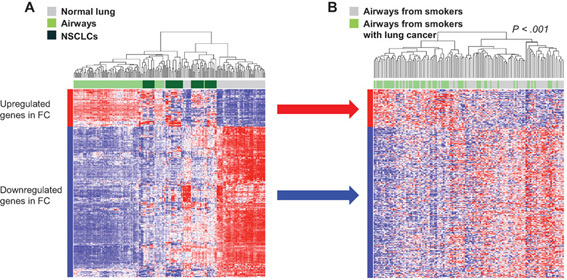
Analysis of adjacent airway field cancerization (FC) profiles in large airways of patients with suspected lung cancer. Gene set enrichment analysis (GSEA) was performed, as described in the Supplementary Methods (available online), to identify genes that were upregulated and downregulated in the adjacent airway FC that were also concordantly enriched between genes differentially expressed between histologically normal airways of smokers with and without lung cancer (17). Hierarchical cluster analysis using the 299 leading edge genes (n = 59 upregulated and 240 downregulated) was performed side by side in the adjacent airway FC (A) and in airways of smokers with and without lung cancer (B). Columns represent samples, and rows represent gene features (red = upregulated; blue = downregulated). P value showing statistical significance of separate clustering of airways of smokers with lung cancer from those of healthy smokers was obtained by the two-sided Fisher exact test.
Site-Dependent Analysis of the Adjacent Airway FC by Distance From Tumors
We then sought to determine whether the adjacent airway FC transcriptome varies with respect to tumor proximity. We performed ordinal logistic regression analysis of the transcriptomes of airway samples (n = 81) obtained at varying distances from the tumors. Using a 5% false discovery rate, we identified 422 gene features (n = 335 upregulated and 87 downregulated with shorter distance from the tumors) (Supplementary Table 4 and Supplementary Methods, Sweave Report 3, available online) and key cancer-associated signaling pathways that were differentially expressed in airways with respect to tumor proximity (Figure 4, A and B). We then derived a quantitative score (Supplementary Methods, available online) to signify the extent of the site-dependent effect (Figure 4C). It is worthwhile to mention that the site-dependent effect was more pronounced in lung SCCs than in adenocarcinomas (Supplementary Figure 2, A and B, available online).
Figure 4.
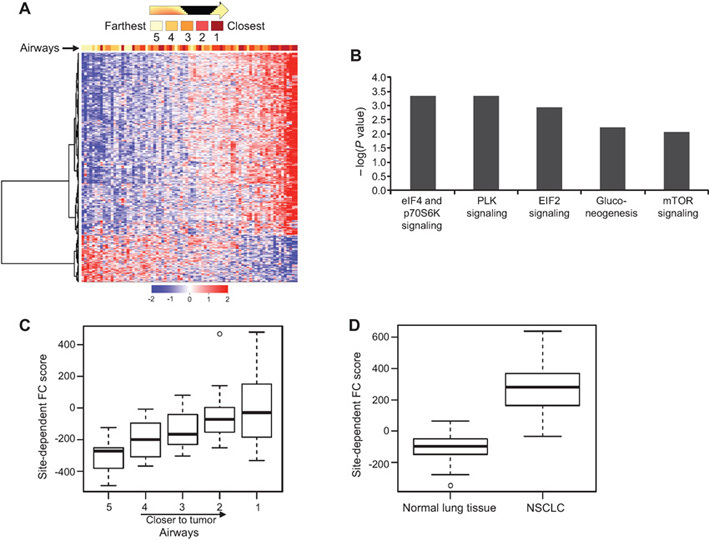
Analysis of airway expression profiles by distance from corresponding non–small cell lung cancers (NSCLCs). Ordinal logistic regression analysis of airways was performed, as described in the Supplementary Methods (available online), and identified 422 gene features with statistically differential expression in the airway with respect to tumor proximity (false discovery rate < 5%). A) Clustering analysis was performed as described in the Supplementary Sweave Reports (available online), and airway samples were arranged by difference in the expression values of the site-dependent genes between the upregulated and downregulated gene clusters. Columns represent samples, and rows represent gene features (red = upregulated; blue = downregulated). B) Functional pathways analysis by ingenuity pathways analysis of the site-dependent differentially expressed genes. Statistical significance of the identified overrepresented canonical pathways is indicated by the negative log of the P values. The site-dependent effect in the adjacent field cancerization (FC) was quantified as described in the Supplementary Methods and Sweave Reports (available online). Box plots depicting site-dependent FC score in airways (C) and between corresponding paired NSCLCs and normal lung tissue after statistical analysis by one-sided t tests (D). Heavy lines indicate medians, and whiskers indicate maximum and minimum FC scores. Airway distance from tumors is numerically indicated with a range of 1 (closest) to 5 (farthest).
We then examined whether the 422 gene features were modulated concordantly between NSCLCs and normal lung tissues. We performed one-sided t tests of the gene features between NSCLC and normal lung tissues to identify those that are modulated in the same direction between the tumors and uninvolved normal lung tissues. We found that 291 of the 335 genes that were increased and 53 of the 87 that were decreased in airways with shorter distance from tumors were also upregulated and downregulated, respectively, in NSCLCs compared with normal lung tissues (Supplementary Table 5 and Supplementary Methods, Sweave Report 4, available online). In addition, the site-dependent airway FC score was statistically significantly and concordantly modulated between NSCLCs and paired uninvolved normal lung tissues (Figure 4D). These findings suggest that the molecular airway FC in NSCLC is in part localized and modulated by distance from tumors and that this gradient site-dependent effect in the airway recapitulates NSCLC expression patterns.
QRTPCR Analysis of the Differential Expression of Adjacent Airway FC Markers
We performed QRTPCR analysis of the expression of transforming growth factor beta receptor II (TGFBR2) and vasoactive intestinal peptide receptor 1 (VIPR1), which were selected based on pathway-based analysis of the site-independent adjacent airway FC profile (Figure 2, C and D) and of neuropilin (NRP) and tolloid (TLL)-like 2 (NETO2) and lysosomal protein transmembrane 4 beta (LAPTM4B), which were among the FC markers (Supplementary Table 4, available online) with statistically significantly (P < .01) increased expression in airways with shorter distance from tumors. VIPR1 also exhibited statistically significant site-dependent expression modulation in the FC (Supplementary Table 4, available online). QRTPCR demonstrated that TGFBR2 and VIPR1 mRNA levels were statistically significantly decreased and NETO2 and LAPTM4B levels increased in NSCLCs and airways compared with normal lung tissue (P < .001) (Figure 5, A–D, top panels). QRTPCR also revealed that VIPR1 (P = .02) but not TGFBR2 exhibited statistically significantly decreased expression in airways with respect to tumor proximity (Figure 5, A and B, middle panels), whereas NETO2 and LAPTM4B levels were statistically significantly increased in the airway with shorter distance from tumors (both P < .05) (Figure 5, C and D, middle panels). Microarray and QRTPCR-based expression of the four genes were statistically correlated (P < .001) (Figure 5, A–D, lower panels). These findings highlight the confirmed differential expression of markers in the adjacent airway FC.
Figure 5.

Quantitative real-time polymerase chain reaction (QRTPCR) analysis of airway field cancerization (FC) markers. Expression of TGFBR2 (A), VIPR1 (B), NETO2 (C), and LAPTM4B (D) was analyzed by QRTPCR in 18 of 20 NSCLC FC case subjects studied with sufficient RNA from airway samples left over after expression profiling. Expression of the indicated genes is depicted by group (NSCLCs, airways, and normal lung tissues; upper panels) and across airway samples based on distance from corresponding NSCLCs (1 = airway closest to tumors; 5 = airways relatively farthest from tumors; middle panels). Relative mRNA expression was assessed by QRTPCR, normalized to that of TBP, and quantified using the 2-ΔΔCT relative quantification method as detailed in the Supplementary Methods (available online). PCR reactions for each FC sample were carried out in duplicate. Boxes indicate ± standard error of the mean; error bars indicate standard deviation. Statistical significance of differences in expression among NSCLC, airway, and normal lung groups was assessed by the Kruskal–Wallis test (upper panels), and statistical significance of differences among different airways was assessed by analysis of variance (middle lanes). Correlation between expression of the indicated genes quantified by microarray and QRTPCR analyses was statistically assessed by Spearman rank (lower panels). LAPTM4B = lysosomal protein transmembrane 4 beta; NETO2 = neuropilin (NRP) and tolloid (TLL)-like 2; TGFBR2 = transforming growth factor beta receptor II; VIPR1 = vasoactive intestinal peptide receptor 1; TBP = TATA box binding protein. *P < .05; ** P < .001; NS, not significant. All statistical tests were two-sided.
Effect of the FC Gene LAPTM4B on Lung Cancer Cell Growth
We were prompted to study the relevance of LAPTM4B, a lysosome-associated transmembrane putative oncogene (27) with no known role in lung carcinogenesis, to the lung malignant phenotype. We first found that LAPTM4B expression was statistically significantly increased in BEAS-2B immortalized lung epithelial cells compared with normal bronchial cells (P < .001) (Supplementary Figure 3, available online) and in NSCLC cell lines (data not shown) compared with bronchial epithelial cells after in silico analysis of a publicly available dataset of human NSCLC and bronchial epithelial cell lines [Gene Expression Omnibus dataset GSE4824 (28)]. Transient knockdown of LAPTM4B expression in immortalized and malignant lung epithelial cell lines effectively and statistically significantly reduced LAPTM4B expression (all P < .001) and cell growth (all P < .05) (Supplementary Figures 4 and 5, available online). Stable knockdown of LAPTM4B in Calu-6 lung cancer cells statistically significantly suppressed LAPTM4B expression (relative expression compared with empty vector, mean ± standard deviation [SD]: shLAPTM4B clone 1: 0.21±0.02; shLAPTM4B clone 2: 0.31±0.03; P < .001) (Figure 6A) concomitant with statistically significantly reduced cell growth (relative cell numbers × 104 compared to 0 hours, mean ± SD: empty vector: 5.83±0.38; scrambled shRNA: 5.69±0.75; shLAPTM4B clone 1: 3.72±0.19, P = .01; shLAPTM4B clone 2: 3.67±0.60, P = .02) (Figure 6B) and anchorage-dependent (mean colony numbers ± SD: empty vector: 31.0±9.17; scrambled shRNA: 30.3±2.89; shLAPTM4B clone 1: 10.0±1.0, P < .001; shLAPTM4B clone 2: 11.67±3.79, P = .002) (Figure 6C) and -independent colony number (mean colony numbers ± SD: empty vector: 30.3±1.53; scrambled shRNA: 29.0±2.0; shLAPTM4B clone 1: 15.3±4.04, P = .003; shLAPTM4B clone 2: 17.0±2.65, P = .006) (Figure 6D). These data demonstrate that LAPTM4B is a positive mediator of immortalized and malignant lung epithelial cell growth.
Figure 6.

Effect of RNA interference-mediated knockdown of LAPTM4B on lung cancer cell anchorage-dependent and -independent growth and colony formation. Calu-6 lung cancer cells were stably transfected with empty vectors, vectors containing scrambled short hairpin RNA (shRNA), as well as vectors with LAPTM4B-specific shRNA sequences as described in the Methods section. A) Quantitative real-time polymerase chain reaction analysis depicting statistically significantly reduced LAPTM4B relative expression in two Calu-6 sublines (shLAPTM4B-1 and shLAPTM4B-2) stably transfected with two different LAPTM4B-specific shRNA sequences compared with Calu-6 cells stably transfected with empty and scrambled shRNA-containing vectors. B) The indicated stably transfected Calu-6 cells were seeded in triplicates (5×104 cells/well) in 12-well plates for 72 hours, after which the number of cells in each well was computed by the trypan blue exclusion method. Cells were seeded in triplicates at a seeding density of 250 cells per well in six-well culture plates or 150 cells per well on soft agar for assessment of anchorage-dependent (C) and -independent (D) colony formation, respectively. Cell colonies were then quantified as described in the Methods section. All experiments were done in triplicate. Representative images of cell colonies are depicted in the lower panels and were obtained with a phase-light microscope. Error bars indicate standard deviation. *P < .05; **P < .001 by the two-sided Student t test.
Discussion
In this study we characterized the airway FC transcriptome adjacent to NSCLC. We identified gene features that were statistically significantly and concordantly modulated between both NSCLCs and airways compared with normal lung tissue, a subset of which was indicative of lung cancer among smokers. Moreover, we revealed that the adjacent airway FC exhibits gradient site-dependent expression patterns with respect to tumor proximity, which effectively predicted NSCLC profiles, pointing to their possible roles in lung cancer pathogenesis. In addition, LAPTM4B, whose expression was increased in the airway with shorter distance from the tumor, was elevated in NSCLC and in immortalized lung epithelial cells and promoted anchorage-dependent and -independent lung cancer cell growth in vitro.
Our analyses pointed to the statistically significant differential expression of FC markers—namely, TGFBR2, VIPR1, NETO2 and LAPTM4B. TGFBR2, a transmembrane receptor serine threonine kinase that mediates TGF-β signaling (29), was previously reported to be downregulated in invasive adenocarcinomas compared with bronchioalveolar carcinomas (30), and loss of the murine counterpart was shown to mediate progression and development of invasive adenocarcinomas in a mouse model of Kras-induced lung cancer (31). VIPR1 mRNA expression was shown to be highest in normal lung tissue compared with various human normal epithelial tissues examined and to peripheral blood leukocytes (32). NETO2 expression was reported to be upregulated in proliferating hemangiomas (33) and associated with invasiveness and motility of cancer cells (34). Furthermore, LAPTM4B was shown to mediate prosurvival autophagy and chemoresistance in breast tumor cells (35) and was found to be upregulated and associated with poor prognosis in ovarian and hepatocellular carcinomas (36,37). In this study we showed that LAPTM4B, whose role in lung cancer was previously unknown, was upregulated in NSCLCs and promoted anchorage-dependent and -independent lung cancer cell growth. Our findings suggest that detailed interrogation of the airway FC may be a useful approach to highlight potential uncharacterized mechanisms and molecules involved in lung cancer pathogenesis.
Our group has recently portrayed the spatial and temporal molecular field of injury in early-stage NSCLC patients by expression profiling of large airways after definitive surgery (19). It is important to mention that in our previous study, normal airway epithelia were collected by endoscopic bronchoscopy brushings within 12 months after surgical removal of the tumor and when NSCLC tumors were not present in situ at time of collection (19). In our current study, we performed expression profiling of multiple normal-appearing airways at various distances from tumors in conjunction with paired NSCLCs and normal lung tissues that were still in situ at the time of airway epithelia collection. It is important to note that although the identified adjacent airway FC profiles were, in part, enriched in large proximal airways, they were substantially dissimilar in the normal lung tissue samples (Figures 2 and 3). It is reasonable to suggest that the identified field effects are more readily discerned in samples (eg, airways) with a higher fraction of epithelial cell content and raises the notion that some of the changes in the adjacent airway FC may be related to cell type. However, this notion is also applicable to the comparison of NSCLCs with normal lung tissue, an analysis that is commonly performed. In addition and through analysis of normal airways alone, we identified gradient and localized site-dependent expression profiles within the adjacent airway FC that predicted NSCLC profiles. It is intriguing to presume that gradient site-dependent airway field effects may be associated with the development of NSCLC tumors in a particular lobe or anatomical region of the lung. However, it cannot be discerned whether these airway FC effects are a cause or consequence of NSCLC development, although it is not unlikely that they are involved in lung carcinogenesis, as embodied by the effect of the site-dependent airway FC marker, LAPTM4B, on lung cancer cell growth. This supposition can be addressed in future studies by assessment of the FC in lung cancer patients before and after surgery.
Analysis of a dataset of airways from smokers with suspected lung cancer (17) revealed that a subset of genes in the adjacent airway FC profile was able to distinguish lung cancer patients among smokers with suspicion of the disease. It is important to mention, however, that the airway FC markers we had identified to be modulated with respect to spatial proximity from tumors were not able to statistically significantly identify lung cancer among smokers. This observation may be attributable to the possible strong association of the gradient site-dependent profiles with the compartments that are adjacent or local to the tumor (6,10). It is plausible to surmise that the adjacent airway FC harbors molecular cancerization profiles that are clinically relevant to both the detection and (chemo)prevention of lung cancer and those that are biologically relevant to understanding the early pathogenesis of this malignancy.
It is worthwhile to mention that our study is not without limitations. Further analysis to fully characterize how the adjacent airway FC varies by NSCLC histology (squamous vs nonsquamous) and smoking status (never-smoker vs smoker) was hindered by the limited number of FC case patients. Moreover, although we compared and contrasted the gradient and site-dependent airway FC profile (n = 422 gene features) that we had derived from NSCLC case patients, among different subgroups (eg, by histology), identification of site-dependent profiles unique to a histological or smoking subtype of NSCLC was also impeded by the small number of FC case patients. Additionally, we could not connect, at the present time, adjacent FC profiles of nonsmoker adenocarcinoma patients to the field effect in the large airway because of the paucity of such airway samples from nonsmoker lung cancer patients. Furthermore, it is reasonable to speculate that RNA sequencing (38), compared with gene expression profiling technology that we used, would provide a more thorough characterization (eg, identification of novel tissue-specific transcripts) of the transcriptomic architecture of the airway FC. Nonetheless, our study represents the first attempt to characterize the global adjacent airway FC in NSCLC, and efforts are underway to expand this working model into different subtypes of lung cancer, including never-smoker adenocarcinomas, and to apply more advanced technologies and platforms (eg, RNA sequencing) for studying the airway FC.
In conclusion, our gene expression profiling efforts revealed that the adjacent and molecular airway FC in NSCLC is comprised of markers that can identify lung cancer among smokers as well as gradient and localized site-dependent expression patterns that recapitulate NSCLC profiles. Our findings suggest that profiling of the airway FC in conjunction with NSCLCs may provide additional insights into the biology of NSCLC and the development of molecular tools for the detection of the malignancy.
Funding
This work was funded in part by a grant from the Lung Cancer Research Foundation (to HK), Jimmy Lane Hewlett Fund for Lung Cancer Research (to IIW), NCI lung cancer SPORE P50 CA70907 (to IIW), Depart of Defense grants W81XWH-04-1-0142 (to WKH and IIW) and W81XWH-10-1-1007 (to HK, WKH, and IIW), and by the institutional Cancer Center Support grant CA16672.
References
- 1. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6(5):963–968 [DOI] [PubMed] [Google Scholar]
- 2. Auerbach O, Stout AP, Hammond EC, et al. Changes in bronchial epithelium in relation to cigarette smoking and in relation to lung cancer. N Engl J Med. 1961;265 (6):253–267 [DOI] [PubMed] [Google Scholar]
- 3. Braakhuis BJ, Tabor MP, Kummer JA, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63(8):1727–1730 [PubMed] [Google Scholar]
- 4. Gomperts BN, Spira A, Massion PP, et al. Evolving concepts in lung carcinogenesis. Semin Respir Crit Care Med. 2011;32(1):32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Graham TA, McDonald SA, Wright NA. Field cancerization in the GI tract. Future Oncol. 2011;7(8):981–993 [DOI] [PubMed] [Google Scholar]
- 6. Kadara H, Wistuba II. Field cancerization in non-small cell lung cancer: implications in disease pathogenesis. Proc Am Thorac Soc. 2012;9(2):38–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gold KA, Kim ES, Lee JJ, et al. The BATTLE to personalize lung cancer prevention through reverse migration. Cancer Prev Res (Phila). 2011;4(7):962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wistuba II, Behrens C, Milchgrub S, et al. Sequential molecular abnormalities are involved in the multistage development of squamous cell lung carcinoma. Oncogene. 1999;18(3):643–650 [DOI] [PubMed] [Google Scholar]
- 9. Wistuba II, Behrens C, Virmani AK, et al. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res. 2000;60(7):1949–1960 [PubMed] [Google Scholar]
- 10. Wistuba II, Gazdar AF. Lung cancer preneoplasia. Annu Rev Pathol. 2006;1:331–348 [DOI] [PubMed] [Google Scholar]
- 11. Belinsky SA, Nikula KJ, Palmisano WA, et al. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci U S A. 1998;95(20):11891–11896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Belinsky SA, Palmisano WA, Gilliland FD, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002;62(8):2370–2377 [PubMed] [Google Scholar]
- 13. Nelson MA, Wymer J, Clements N., Jr Detection of K-ras gene mutations in non-neoplastic lung tissue and lung cancers. Cancer Lett. 1996;103(1):115–121 [DOI] [PubMed] [Google Scholar]
- 14. Tang X, Shigematsu H, Bekele BN, et al. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res. 2005;65(17):7568–7572 [DOI] [PubMed] [Google Scholar]
- 15. Beane J, Sebastiani P, Liu G, et al. Reversible and permanent effects of tobacco smoke exposure on airway epithelial gene expression. Genome Biol. 2007;8(9):R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spira A, Beane J, Shah V, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004;101(27):10143–10148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spira A, Beane JE, Shah V, et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat Med. 2007;13(3):361–366 [DOI] [PubMed] [Google Scholar]
- 18. Gustafson AM, Soldi R, Anderlind C, et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci Transl Med. 2010;2(26):26ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kadara H, Shen L, Fujimoto J, et al. Characterizing the molecular spatial and temporal field of injury in early-stage smoker non-small cell lung cancer patients after definitive surgery by expression profiling. Cancer Prev Res (Phila). 2013;6(1):8–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Behrens C, Lin HY, Lee JJ, et al. Immunohistochemical expression of basic fibroblast growth factor and fibroblast growth factor receptors 1 and 2 in the pathogenesis of lung cancer. Clin Cancer Res. 2008;14(19):6014–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264 [DOI] [PubMed] [Google Scholar]
- 22. Bull SB. Sample size and power determination for a binary outcome and an ordinal exposure when logistic regression analysis is planned. Am J Epidemiol. 1993;137(6):676–684 [DOI] [PubMed] [Google Scholar]
- 23. Molaei M, Mansoori BK, Mashayekhi R, et al. Mucins in neoplastic spectrum of colorectal polyps: can they provide predictions? BMC Cancer. 2010;10(October 2010):537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perez-Ortiz M, Gutierrez PA, Hervas-Martinez C. Projection-based ensemble learning for ordinal regression. IEEE Trans Cybern. 2013; 10.1109/TCYB.2013.2266336 [DOI] [PubMed] [Google Scholar]
- 25. Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics. 2003;19(10):1236–1242 [DOI] [PubMed] [Google Scholar]
- 26. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li L, Wei XH, Pan YP, et al. LAPTM4B: a novel cancer-associated gene motivates multidrug resistance through efflux and activating PI3K/AKT signaling. Oncogene. 2010;29(43):5785–5795 [DOI] [PubMed] [Google Scholar]
- 28. Zhou BB, Peyton M, He B, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10(1):39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaklamani VG, Pasche B. Role of TGF-beta in cancer and the potential for therapy and prevention. Expert Rev Anticancer Ther. 2004;4(4):649–661 [DOI] [PubMed] [Google Scholar]
- 30. Borczuk AC, Kim HK, Yegen HA, et al. Lung adenocarcinoma global profiling identifies type II transforming growth factor-beta receptor as a repressor of invasiveness. Am J Respir Crit Care Med. 2005;172(6):729–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borczuk AC, Sole M, Lu P, et al. Progression of human bronchioloalveolar carcinoma to invasive adenocarcinoma is modeled in a transgenic mouse model of K-ras-induced lung cancer by loss of the TGF-beta type II receptor. Cancer Res. 2011;71(21):6665–6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sreedharan SP, Huang JX, Cheung MC, et al. Structure, expression, and chromosomal localization of the type I human vasoactive intestinal peptide receptor gene. Proc Natl Acad Sci U S A. 1995;92(7):2939–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Calicchio ML, Collins T, Kozakewich HP. Identification of signaling systems in proliferating and involuting phase infantile hemangiomas by genome-wide transcriptional profiling. Am J Pathol. 2009;174(5):1638–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Horak CE, Lee JH, Elkahloun AG, et al. Nm23-H1 suppresses tumor cell motility by down-regulating the lysophosphatidic acid receptor EDG2. Cancer Res. 2007;67(15):7238–7246 [DOI] [PubMed] [Google Scholar]
- 35. Li Y, Zhang Q, Tian R, et al. Lysosomal transmembrane protein LAPTM4B promotes autophagy and tolerance to metabolic stress in cancer cells. Cancer Res. 2011;71(24):7481–7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang H, Xiong F, Qi R, et al. LAPTM4B-35 is a novel prognostic factor of hepatocellular carcinoma. J Surg Oncol. 2010;101(5):363–369 [DOI] [PubMed] [Google Scholar]
- 37. Yin M, Li C, Li X, et al. Over-expression of LAPTM4B is associated with poor prognosis and chemotherapy resistance in stages III and IV epithelial ovarian cancer. J Surg Oncol. 2011;104(1):29–36 [DOI] [PubMed] [Google Scholar]
- 38. Mutz KO, Heilkenbrinker A, Lonne M, et al. Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol. 2013;24(1):22–30 [DOI] [PubMed] [Google Scholar]