

Abstract
As methodology development matures it can be difficult to discern the most effective ways of performing certain transformations from the rest. This review summarizes the most important contributions leading to asymmetric hydrogenations of simple unsaturated-acid and ester substrates, with the objective of highlighting at least the best types of catalysts for each. Achievements in the area are described and these reveal situations where further efforts should be worthwhile, and ones where more research is only likely to give diminishing returns. In general, our conclusions are that the most useful types of catalysts for unsaturated-acids and -esters tend to be somewhat different, simple substrates have been studied extensively, and the field is poised to address more complex reactions. These could be ones involving alternative, particularly cyclic, structures, chemoselectivity issues, and more complex substrate stereochemistries.
Keywords: stereoselective, hydrogenations, homogeneous, alkene-esters, alkene-acids
A. Introduction
Most alkene substrates for asymmetric hydrogenations are relatively hindered tri- and tetra-substituted ones. These are shielded to hydrogenation catalysts unless they bear coordinating functional groups (CFGs) for the metals typically involved, ie Rh(1+)/Rh(3+), Ir(1+)/Ir(3+). Two important effects arise from substrates having strong-CFGs in catalytic hydrogenation reactions. First, the substrate is drawn into the proximity of the metal, lowering the activation energies for hydrogenations. Second, binding of CFGs in catalytic transition states will synergize with chiral ligands tending to give relatively rigid, diastereomeric forms that lead to stereocontrol. For instance, enamides contain strong CFGs because they can give unstrained stable chelates (Figure 1). Four- membered ring chelates from α,β-unsaturated carboxylic acids are presumably less stable because of ring-strain. Esters that are α,β-unsaturated are not widely regarded as coordinating for Rh(1+)/Rh(3+), hence these are seen as simply functional groups; however, there is a “grey zone” between CFGs and FGs in asymmetric hydrogenations and esters can sometimes fall in this area.
Figure 1.
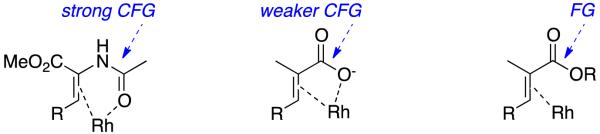
Coordinating functional groups (CFGs) and FGs in asymmetric hydrogenations.
This review compares asymmetric hydrogenations of alkenyl carboxylic -acids and -esters that do not have other CFGs. Hydrogenations of 1,1-disubstituted alkenes1,2 are excluded because these are relatively unhindered and amenable to hydrogenations by a range of catalyst systems. Thus we attempt to identify the best catalysts known for asymmetric hydrogenations of simple tri- and tetra-substituted alkenyl-carboxylic acids and -esters. Our goal is to compare catalysts that are used for each substrate type, then to look for similarities and differences for the two.
B. Carboxylic Acids
Categories
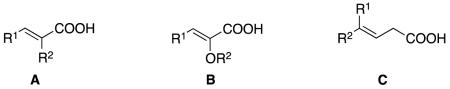
Most of the alkenyl carboxylic acids reported as substrates for asymmetric hydrogenations are represented by one of the generic structures A - C. Alkenes A where R1 and R2 are alkyl substituents (eg tiglic acid, R1 = R2 = Me)are relatively difficult to hydrogenate with high enantiocontrol, but the task is markedly easier if one or both R-groupsis aryl. Alkenes B are more electron-rich and have steric features that are highly variable according to the substituents involved. Competing double bond migration processes could complicate attempts to achieve high enantioface selectivities for the β,γ-unsaturated systems C.
Ruthenium-mediated (Noyori-type) Hydrogenations
Noyori’s first report of asymmetric hydrogenations of type A alkenes3 featured a range of different substituents and gave products with enantiomeric excesses in the 83 – 95 % range. For tiglic acid (reaction 1) it was only necessary to use hydrogen pressures of 4 atm, but substrates with slightly bulkier substituents required around 100 atm. Throughout, the catalyst loadings used were very low.
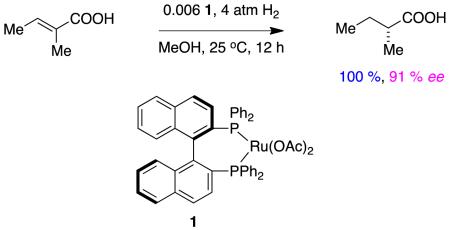 |
reaction 1 |
Halpern4 and Takaya5 considered kinetic and stereochemical aspects of illustrative reactions in this class. They concluded dihydrogen is split by oxidative addition to the metal giving a ruthenium hydride I and protons (Figure 2);6 this process tends not to be perturbed by addition of bases but it is suppressed when the medium is made acidic. It was postulated that the reaction then proceeds via insertion of the coordinated alkene into the Ru-H bond to give a metallocyclopentane II; alternative pathways involving conjugate additions of hydride to form enolates were ruled out via labeling experiments. Protonation of species II to give the penultimate intermediate in the catalytic cycle, the product-containing dicarboxylate III, precedes exchange with starting-material carboxylates to close the catalytic cycle.
Figure 2.

Halpern’s postulate for hydrogenations of unsaturated carboxylic acids with Ru(2+)bisphosphine complexes.
The studies cited above did not comment on the stereochemistry of the protonation step, except that the addition to the alkene was cis overall. We postulate that protonation of the anionic intermediates II on the metal would explain the cis-addition and how hydrogen atoms from the methanol solvent were incorporated into this position.
An example of other atropisomeric ligands applied in ways that follow Noyori’s work on unsaturated acids comes from Albert Chan’s lab.7 Somewhat exceptionally in this type of transformation, higher enantioselectivities for the tiglic acid system were obtained relative to the analogous phenyl-substituted alkenes (Figure 3).
Figure 3.

Hydrogenations of α,β-unsaturated alkenes mediated by a dipyridyl phosphine ligand.
Rhodium Catalysts
Carboxylates coordinateto cationic rhodium complexes, and alkenyl groups in these substrates can be stereoselectively hydrogenated using chiral bisphosphine Rh-complexes.8 However, there are relatively few reports wherein high enantioselectivities have been achieved using rhodium-based catalysts in these types of reactions. For instance, a maximum of 82 % ee was obtained for hydrogenation of a type A alkene (specifically, R1 = Ph, R2 = Me) using a typical Rh(1+) precursor, Rh(NBD)2BF4, and a series of ferrocenyl based bisphosphines (Figure 4).9
Figure 4.
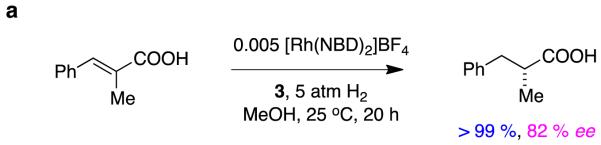

Hydrogenation of unsaturated carboxylic acids using ferrocenyl-based bisphosphines: a without-; and, b with, pendant amines.
An intriguing way to constrain intermediates in hydrogenations of unsaturated carboxylates is to ion-pair them with amines on the bisphosphine ligand.10,11 Thus when type A substrates (R = Ar) were hydrogenated with a bisferrocenyl ligand containing appropriately oriented amine groups, high enantioselectivities were obtained. Ion paired intermediates like IV were postulated to be involved in these transformations (Figure 4b).11
Reaction 2 shows another example of ion-paired catalysis in hydrogenations of α,β-unsaturated carboxylic acids where good enantioselectivities were obtained. This reaction is unusual because it is the only tetra-substituted alkene substrate that we encountered for acid substrates; it is one where hydrogenation is favored by strong factors relating to partial relief from strain in the cyclopropene system.12
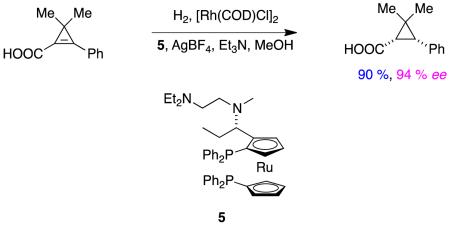 |
reaction 2 |
Rapid optimization of stereoselectivities in asymmetric hydrogenations of α,β-unsaturated carboxylic acids has been achieved by testing combinations of chiral monodentate ligands.13,14 This methodology works exceptionally well for these substrates, as shown in reaction 3.15 Comparison of this transformation with those in Figure 4 and Reaction 2suggests there is at least the possibility that carboxylic acids in these substrates ion pair with the nitrogen on the phosphoramidite ligands, but this possibility has not been discussed or explored in the literature.
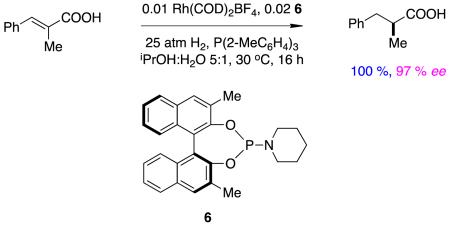 |
reaction 3 |
An example of asymmetric hydrogenation of an alkoxy-substituted (type B) substrate was demonstrated in a process synthesis of a medicinally active compound. Reaction 4 shows the system developed after screening over 250 catalyst-ligand combinations; however, even after all that effort,a maximum of only 92 % ee was obtained.16 In the context of the previous discussion, it is interesting that these somewhat disappointing results correspond to a bisphosphine that does not contain amine groups to ion pair.
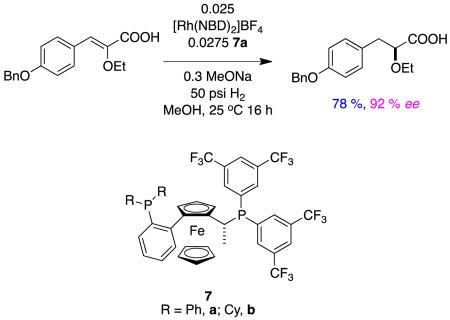 |
reaction 4 |
Chiral Analogs Of Crabtree’s Catalyst
Hydrogenations mediated by chiral analogs of Crabtree’s catalyst have at least the potential to involve oxidative additions of two hydrogen molecules giving Ir(5+) intermediates. Several groups have asserted this mechanism based on DFT calculations.17-21 However, to the best of our knowledge these calculations have never been performed for unsaturated carboxylic acid substrates, so involvement of Ir(5+) in this particular case is unresolved {calculations on unsaturated esters22 are discussed later}.
Qi-Lin Zhou’s spirocyclic ligands 823 give impressive enantioselectivities in hydrogenations of some α,β-unsaturated carboxylic acids.24 Figure 5 shows several reactions in which 8a and b (benzyl and iso-propyl oxazoline substituents, respectively) shone in these transformations. Excellent ee’s were achieved for aryl-substituted systems, tiglic acid, and substrates containing simple alkyl substituents.
Figure 5.


Spirocyclic ligands in asymmetric hydrogenations of alkyl-substituted α,β-unsaturated carboxylic acids. aα-Methylcinnamate-; b, α-alkylcinnamate-; and, c dialkyl-derivatives.
The selectivities obtained by Zhou are excellent compared to most other catalysts for this transformation. For instance, the carbene oxazoline complex 922 gave relatively modest enantiomeric excesses in hydrogenations of tiglic acid (reaction 5).
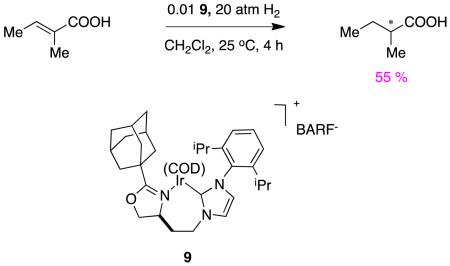 |
reaction 5 |
Recently Zhou has shown the spirocyclic catalysts 10 are also effective for hydrogenation of the same types of α,β-unsaturated carboxylic acids with very high enantioselectivities (eg Figure 6).25 These are perhaps the first examples of phosphine-amine ligands on iridium(1+) to be used in asymmetric hydrogenation reactions of any hindered alkene substrate.
Figure 6.

High enantioselectivities obtained for hydrogenations of diverse α,β-unsaturated carboxylic acids involving catalyst 10
Another groups, including Ding and Zhang, have also shown interest in hydrogenations of α,β-unsaturated carboxylic acids, and they also used a spirocyclic ligand.26 Unlike Zhou’s work however, they studied α-aryl systems, particularly ones related to an anti-diabetic drug, and their spirocyclic ligand in 11 has a less extended structure than Zhou’s. Very good enantiomeric excesses were obtained throughout, though the substrate diversity in this study was relatively narrow (Figure 7).
Figure 7.


Use of spirocyclic complex 11 in asymmetric hydrogenations of α,β-unsaturated carboxylic acids
Andersson’s group reported one example of hydrogenation of an unsaturated acid as part of a broader study on ester substrates (see below).27 The enantioselectivity and conversion (100 %) for that particular alkene was excellent using a low loading of the catalyst, reaction 6. Unlike many of the transformations discussed above, there was no need for an additive in this reaction, and dichloromethane was used as a solvent, not methanol.
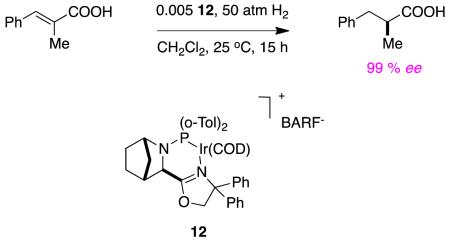 |
reaction 6 |
Zhou also applied his spirocyclic ligands to α-oxygenated-α,β-unsaturated carboxylic acids, and again the results are highly impressive.28 Several α-alkoxy-derivatives were hydrogenated with high enantiomeric excesses, as illustrated in Figure 8.
Figure 8.

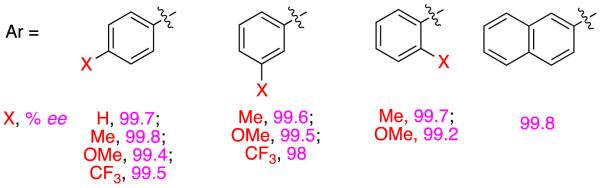
Hydrogenations of α-alkoxy- substrates.
Hydrogenations of the corresponding α-aryloxy-substrates are shown in Figure 9.29 It is impressive that such high enantiomeric excesses were obtained for the diverse set of substrates used in this study.
Figure 9.


Hydrogenations of α-aryloxy- substrates.
Zhou’s group attempted to compare their catalysts with the PHOX-system 13.30 However, only the spirocyclic ligands gave a conversion, the PHOX system failed to generate product (reaction 7).
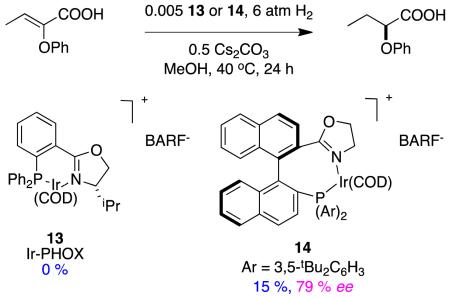 |
reaction 7 |
Zhou expanded his studies to include E-γ-aryl-γ-methyl-β,γ-unsaturated acids31 and achieved uniformly high enantioselectivities for a range of compounds with different aryl substituents, a naphthyl alkene, and a thiophenyl system. However, diminished enantioselectivities were observed when the γ-methyl substituent was substituted with a bulkier one, iPr. Isomerization of these substrates would conjugate the alkene; however, experiments with D2 addition showed double bond migration did not precede the hydrogenation step in these reactions (Figure 10).
Figure 10.

Asymmetric hydrogenations of non-conjugated alkene-carboxylic acids (type C substrates).
A direct comparison between ester and acid substrates under the conditions outlined above proved that the carboxylic acid is essential and the ester does not react. We postulate the carboxylate reactions could proceed via a chair-like intermediate V for which there is no direct parallel with esters.
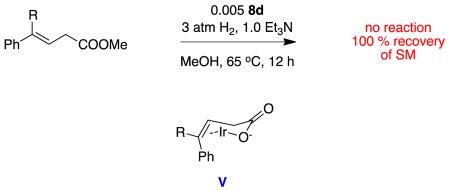 |
Summary For Hydrogenations Of Alkenyl-carboxylic Acids
Hydrogenations mediated by Ru(2+), Rh(1+)-bisphosphine, and Ir-N,P-ligands (chiral analogs of Crabtree’s catalyst) probably follow three different reaction pathways. The ruthenium reactions are suppressed by acid, the rhodium ones proceed under neutral conditions, and the iridium ones are almost always performed in the presence of added base. Somewhat longer reaction time tends to be required for the Rh systems. Catalysts based on Ir-spirocyclic-N,P-ligands give the best enantioselectivities obtained so far in the series. Exploratory work using those spirocyclic ligands must be relatively slow because the backbone chiralities have to be matched with the oxazoline, but once the ideal combination is established then this process need not to be repeated. It remains to be seen, however, how these catalysts perform on more complicated substrates; they are relatively hindered, so conversion may be a problem in these cases.
There is enough data on asymmetric hydrogenations of carboxylic acids to make a limited catalyst/substrate comparison (Table 1). Within this data set, appropriate chiral analogs of Crabtree’s catalysts give higher enantioselectivities. Less work has been reported on Ru- and Rh-systems, so this is not an even comparison, but it also could be indicative of a trend that will be reinforcedas more examples are reported. Similar conditions were used for hydrogenations mediated by all three catalysts types.
Table 1.
Enantioselectivity comparison for substrate/catalyst combinations for asymmetric hydrogenations of alkenyl-carboxylic acids.
| alkenes | Analogs of Crabtree’s Catalyst (% ee) | Ru (% ee) | Rh (% ee) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||
| 8a | 8b | 9 | 10 | 11 | 12 | 14 | 1 | 2 | 3 | 6 | |
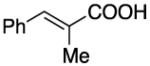
|
99 | 99 | 99 | 77 | 82 | 97 | |||||
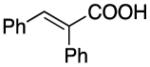
|
94 | 95 | 95 | ||||||||
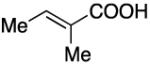
|
99 | 55 | 98 | 91 | 97 | ||||||
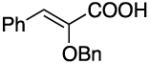
|
99 | 99 | |||||||||
|
99 | 79 | |||||||||


C. Carboxylate Esters
Categories
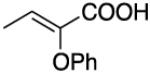
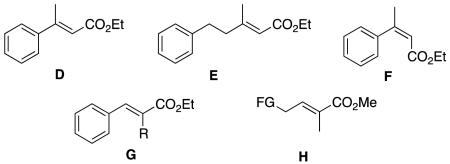
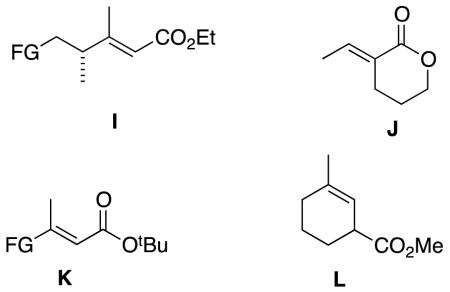
The story of asymmetric hydrogenations of unsaturated esters has an intriguing plot featuring a few main substrate-actors, and a handful of others filling peripheral roles. Generic ester D is a prima donna on which, frankly, too much attention has been lavished. Meanwhile, homologs E, geometric isomers like F, and constitutional isomers like G have received much less attention; they seem to be harder substrates to hydrogenate with high enantioselectivities. Asymmetric hydrogenations of tiglic and angelic acid esters are conspicuously under-explored, and have not been achieved with high stereoselectivities using any system. The α,ω-functionalized substrates H are also underexplored, even though these give chirons that are inherently useful because they can be homologated at either end. Hydrogenations of γ-chiral substrates I give stereochemically complex chirons that can be useful in particular cases, and these have been studied in more depth. Hydrogenations of cyclic substrates include lactones J, β,β-disubstituted K, and chiral cyclohexenyl esters L.
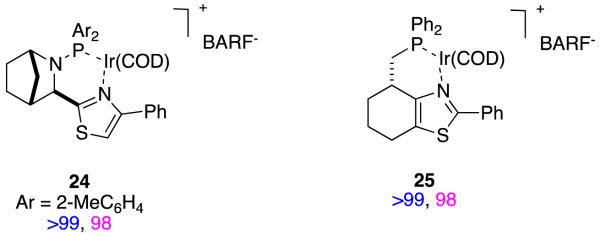
Comparisons between Rh- and Ir-catalysts in this area are informative. Overall, however, it is striking that so much time has been spent studying relatively small aspects of this area (eg D as a substrate), while some fundamental issues like isomerization preceding hydrogenations in these processes, remain unexplored.
Chiral Analogs Of Crabtree’s Catalyst
It is easy to hydrogenate substrates D with relatively high enantiomeric excesses using chiral analogs of Crabtree’s catalyst. For instance, Pfaltz’ group alone have published at least 18 research papers (excluding reviews) that feature hydrogenation of this particular substrate.30,32-48 Figure 11 shows some of the ligands that have been used to do this, the particular ester involved, and the enantiomeric excesses obtained. Figure 12 shows ligands that Andersson’s group have used in similar hydrogenations.27,49-58Stereoselectivities in these reactions are relatively tolerant of simple changes to the ester-alkyl functionality, and high conversions are the norm (Figure 13).
Figure 11.


Conversions and enantioselectivities in Pfaltz’ hydrogenations of substrates D.
Figure 12.

Conversions and enantioselectivities in Andersson’s hydrogenations of substrates D.
Figure 13.
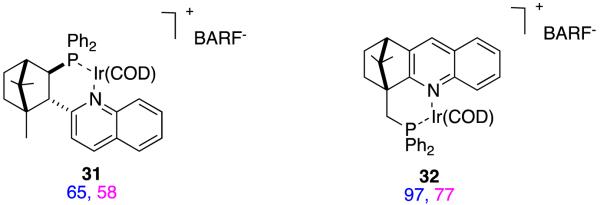

Asymmetric hydrogenation of substrate D from groups other than Pfaltz and Andersson.59-67
Substrate D has been hydrogenated by many groups, but the isomers and homologs E – G have not been studied so much. Catalysts 39,39,68 40,57 41,27 and 4237 (Figure 14) have been used for this series of substrates, and it appears that 41 and 42 are the most generally suitable. Relatively small changes to the substrate can significantly affect the conversions and enantioselectivities in these reactions. For instance, catalyst 40 is an excellent hydrogenation catalyst for substrate D, but a relatively poor one for F and G; this is also true for other catalysts that are not shown here.
Figure 14.

Asymmetric hydrogenations (conversions and enantioselectivities) of close analogs of substrate D compared.
 |
The transformations shown in Figure 15a for tiglic acid derivatives illustrates how 1-methylpropyl- chiral centers could be generated if a good catalyst had been identified, but none has emerged so far. Intriguingly, the same catalyst 9 gives opposite enantiofacial selectivities for this ester relative to similar tiglic alcohol and α-methyl stilbene derivatives. DFT calculations reveal plausible explanations for these observations.22 They indicated that one enantiomer of the product ester appears to be formed via a mechanistic pathway that parallels that for α-methyl stilbene hydrogenations by catalyst 9. However, the other enantiomer of the product apparently results via a different mechanism that involves only Ir(3+) intermediates, and coordination of the ester carbonyl to the metal (Figure 15b). This pathway is only possible if there is a carbonyl group in this position; consequently, tiglic acid, the corresponding carboxylate salt, and the Weinreb amide derivative all give this “abnormal stereoface selectivity” but tiglic alcohol and its ether derivatives do not. This is a case in which two enantiomers of a product appear to be formed via different mechanisms because the diastereomeric intermediates in the catalytic cycle favor that pathway over a competing one for only one enantiomer. That favored pathway involves an ester as a CFG.
Figure 15.

a Tiglic alcohol and acid derivatives give opposite face selectivities. b DFT calculations indicate formation of the favored enantiomer in the hydrogenations of tiglic ester derivatives occurs via coordination of the ester carbonyl.
Hydrogenations of the dienes 43 and 44 have the potential to give useful chirons with two stereocenters in one step. These reactions involve sequential asymmetric hydrogenations (at similar rates), hence high enantioselectivities are to be expected because diastereomers are the main stereochemical impurities (Horeau’s principal).69 This interesting situation should not cloud the fact that the face selectivities of the hydrogenation steps involved cannot be high. If a more selective catalyst was identified for tiglic esters, then it would be worth probing stereoselectivities in reactions 8 and 9 using that. Indeed, it would be interesting now to investigate hydrogenations of the corresponding dienyl carboxylic acids using some of Zhou’s complexes that have spirocyclic ligands since thesedo hydrogenate tiglic acid with high stereoselectivities (see above).
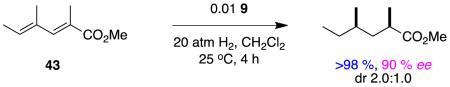 |
reaction 8 |
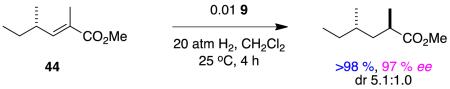 |
reaction 9 |
There are many potential applications of α,ω-functionalized substrates H (eg 45 and 46) because the termini can be elaborated via chemoselective reactions that give orthogonal stereoselectivities. It is surprising that, to the best of our knowledge, these substrates have only been investigated in reactions 10 and 11.70 The isomeric systems M have apparently not been investigated at all.
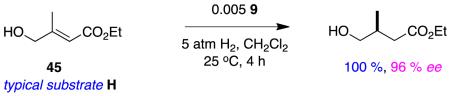 |
reaction 10 |
 |
reaction 11 |
Matching the influences of substrate and catalyst “vectors” for γ-chiral alkenes I (eg 47 - 49) can afford a range of chirons that may be used to prepare, amongst other things, polyketide-derived natural products. Figure 16 gives some examples where these reactions have been used to give 1,2- and 1,3-hydroxylmethyl, and 1,3-dimethyl motifs.22,70,71 Substrate vectors contribute to the overall stereoselectivities of these reactions; in most cases the catalyst influence is dominant (eg Figure 16a and c) but not for the γ-chiral homoallylic alcohol in Figure 16b.
Figure 16.

Hydrogenation of γ-chiral substrates to give a 1,2-hydroxylmethyl-, b 1,3-hydroxylmethyl-, and c 1,3-dimethyl-chirons.
Relatively few lactones have been reported as asymmetric hydrogenation substrates. Reactions 12 and 13 show examples featuring chiral analogs of Crabtree’s catalyst; high enantioselectivities were obtained in both cases.27
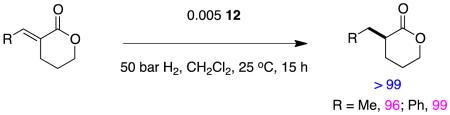 
|
reaction 12 |
 |
reaction 13 |
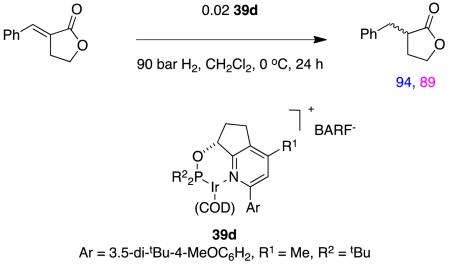
Asymmetric hydrogenations of β-alkoxy-α,β-unsaturated esters, eg 50, have been studied in one paper.72 Protons generated in hydrogenations with Crabtree’s catalyst analogs73 can lead to decomposition of enol ether substrates in competition with hydrogenation processes, but this does not happen with the carbeneoxazoline complex 9 when applied to these slightly less electron-rich alkenes.
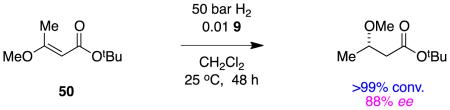 |
reaction 14 |
Hydrogenations With Other Catalysts
We cannot find reports of Noyori-style ruthenium catalysts in hydrogenations of simple (no other functionality) α,β-unsaturated carboxylate esters. A possible explanation for this is evident by reference to Halpern’s mechanism for hydrogenations of α,β-unsaturated carboxylic acids with these catalysts (Figure 2). This pathway features dissociation of saturated carboxylates and their replacement with α,β-unsaturated ones; esters simply cannot coordinate in the same way as carboxylates, and give the same types of neutral Ru(2+) complexes. However, Takaya et al have reported hydrogenation of unsaturated lactones using their ruthenium catalyst; only substrates with exocyclic alkenes gave high enantioselectivities, and the conversions were sometimes incomplete (Figure 17).74
Figure 17.

Conversions and enantioselectivities in hydrogenations of lactones using Ru-BINAP catalysts.
Some of John Brown’s early (1985) studies of alkenyl ester hydrogenations provide an excellent comparison of the relative importance of ester-coordinating effects for Crabtree’s catalyst and Rh(1+)(diphosphine) complexes.75 Specifically, his work on the achiral complexes 51 and 52 (Figure 18) show that hydrogenations with the Ir(1+) catalysts are considerably faster and more syn-selective than the Rh(1+) species. Coordination to the ester via intermediates like XI was implicated in both cases, but this effect was more significant for Crabtree’s catalyst, presumably due to transient complexation to Ir(5+) intermediates.
Figure 18.

Crabtree’s catalysts gives stronger directing effects than rhodium-diphosphines (enantioselectivities in purple).
The studies outlined in Figure 18 also featured deuteration of the same substrates. Brown et al found deuterium was incorporated onto the C1-3 positions, and only two deuterium atoms, on average, were incorporated into each molecule. This is indicative of double bond migration concurrent with the hydrogenation event.76
There are, in fact, few hydrogenations reported for α,β-unsaturated esters (no other FGs) that feature Rh(1+) catalysts. The examples that are in the literature tend to involve other functional groups that could be coordinating, eg the homoallylic alcohols in reaction 15,22 and the allylic phosphonate in reaction 16.77
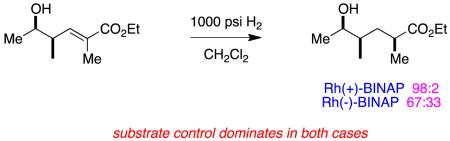 |
reaction 15 |
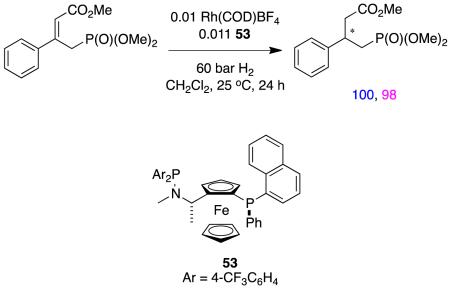 |
reaction 16 |
An exception to the observation that CFG groups tend to be required in Rh(1+) mediated catalysis of enol ether substrates K is depicted in reaction 17.78 In that transformation there are no obvious CFGs for Rh(1+) or other ways for the substrate to form out-of-sphere association with the ligand. The in situ generated catalyst involved in these reactions was probably identified from a broad screening effort.
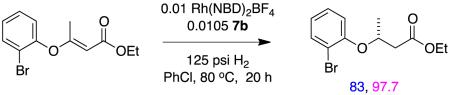 
|
reaction 17 |
Summary For Hydrogenations Of Carboxylic-esters
Brown’s early work (Figure 18), and the stereochemical reversal for tiglic esters relative to similar substrates (Figure 15), indicate that esters are coordinating functional groups in hydrogenations mediated by chiral analogs of Crabtree’s catalysts. These coordinating effects may not be as powerful as others (eg from homoallylic alcohols) but they are significant enough to promote asymmetric hydrogenations by chiral Ir-N,P catalysts making them uniquely useful for this substrate class. Ruthenium and rhodium bisphosphine complexes do not hydrogenate unsaturated esters with comparable conversions and enantioselectivities. A reason for this difference is the possible involvement of higher oxidation-state iridium species for Crabtree catalyst analogs.
Future research in this area should feature substrates that give more useful products than D does, eg the α,ω-functionalized ones H and I. Other studies should include deuteration reactions to monitor double bond migration.
D. Conclusions
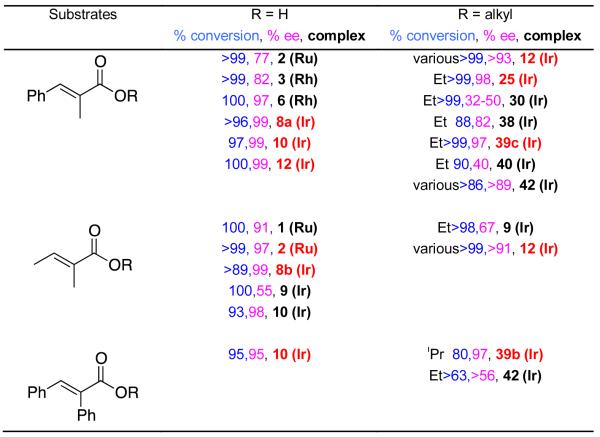
Despite all the work that has been reported on hydrogenations of unsaturated acid and ester derivatives, it is hard to make direct comparisons for the same carbon backbones. In fact, we are only able to present this for three types of substrates (Table 2). Selected iridium complexes from Zhou, featuring spirocyclic ligands, and some Noyori-based systems are preferred for the carboxylic acids. Ruthenium and rhodium based diphosphine catalysts are significantly less useful for hydrogenations of carboxylic esters. For ester substrates the preferred catalysts tend to be Ir-N,P complexes notably from Pfaltz and Andersson; Zhou’s spirocyclic systems are significantly less reactive for these substrates.
Table 2.
Hydrogenations of α-methyl cinnamate and tiglic acid derivatives compared (satisfactory system for each substrate shown in red).
|
Researchers interested in hydrogenating the simple substrates shown in Table 2 can find catalysts that have already been shown to reduce them with high stereoselectivities and conversions; there are no pressing needs to search further for other catalysts, but arguments might be made in favor of new designs that are more accessible. It might also be useful in some cases if a catalyst could be found to reduce unsaturated esters and acids with high stereoselectivities and conversions; at present, Andersson’s complex 12 comes closest to meeting this aim.
The next phase in this research is hydrogenations of more complex substrates. These include alkenes with other functionalities to gauge chemoselectivity issues and the effects of potential CFGs on stereoselectivities. Endo cyclic alkene-lactones and carbocyclic rings with peripheral acid groups also deserve more attention. More work on hydrogenation of chiral substrates to give stereochemically complex materials is warranted.
Figure 15 and the discussion around it indicate esters are CGFs in asymmetric hydrogenations mediated by Crabtree’s catalyst analogs. There has been a tendency to rationalize face-selectivities for hydrogenations by these catalysts in terms of empirical “quadrant” models. These cannot be totally reliable in situations for which a supposedly innocuous group like an ester can coordinate to the metal; consequently, models like this should be evaluated with care. It is even possible that phenyl groups and aliphatic C-H bonds have weak coordinating effects with Crabtree’s catalyst analogs that go unnoticed in most reactions, and they may perturb facial selectivities.
Overall, it is time to identify and prioritize the most important substrates to hydrogenate. Finally, Brown performed deuteration studies in his hydrogenation work over 30 years ago to test for competing double bond migration reactions. This type of experiment is rarely repeated in contemporary studies (though there are examples as outlined above), but these informative experiments could be used more in optimization reactions to distinguish alkenes that are being isomerized from those that are being hydrogenated directly.
Acknowledgements
We thank The National Institutes of Health (GM087981), and The Robert A. Welch Foundation (A-1121) for financial support.
References
- (1).Scrivanti A, Bovo S, Ciappa A, Matteoli U. Tetrahedron Lett. 2006;47:9261–9265. [Google Scholar]
- (2).Sun X, Zhou L, Wang C-J, Zhang X. Angew. Chem., Int. Ed. 2007;46:2623–2626. doi: 10.1002/anie.200604810. [DOI] [PubMed] [Google Scholar]
- (3).Ohta T, Takaya H, Kitamura M, Nagai K, Noyori R. J. Org. Chem. 1987;52:3174–3176. [Google Scholar]
- (4).Ashby MT, Halpern J. J. Am. Chem. Soc. 1991;113:589–594. [Google Scholar]
- (5).Takaya H, Ohta T, Mashima K, Noyori R. Pure & Appl. Chem. 1990;62:1135–1138. [Google Scholar]
- (6).Clapham SE, Hadzovic A, Morris RH. Coord. Chem. Rev. 2004;248:2201–2237. [Google Scholar]
- (7).Qiu L, Li Y-M, Kwong FY, Yu W-Y, Fan Q-H, Chan ASC. Adv. Syn. Catal. 2007;349:517–520. [Google Scholar]
- (8).Brown JM, Parker D. J. Org. Chem. 1982;47:2722–2730. [Google Scholar]
- (9).Sturm T, Weissensteiner W, Spindler F. Adv. Synth. Catal. 2003;345:160–164. [Google Scholar]
- (10).Yamada I, Yamaguchi M, Yamagishi T. Tetrahedron Asymmetry. 1996;7:3339–3342. [Google Scholar]
- (11).Chen W, McCormack PJ, Mohammed K, Mbafor W, Roberts SM, Whittall J. Angew. Chem. Int. Ed. 2007;46:4141–4144. doi: 10.1002/anie.200604493. [DOI] [PubMed] [Google Scholar]
- (12).Kawamura N. Jpn. Kokai Tokkyo Koho. 1996;125:57975. JP 08073400 A219960319, CAN. [Google Scholar]
- (13).Reetz M, Sell T, Meiswinkel A, Mehler G. Angew. Chem. Int. Ed. 2003;42:790–793. doi: 10.1002/anie.200390209. [DOI] [PubMed] [Google Scholar]
- (14).Reetz MT. Angew. Chem., Int. Ed. 2008;47:2556–2588. doi: 10.1002/anie.200704327. [DOI] [PubMed] [Google Scholar]
- (15).Hoen R, Boogers JAF, Bernsmann H, Minnaard AJ, Meetsma A, Tiemersma-Wegman TD, de Vries AHM, de Vries JG, Feringa BL. Angew. Chem. Int. Ed. 2005;44:4209–4212. doi: 10.1002/anie.200500784. [DOI] [PubMed] [Google Scholar]
- (16).Houpis IN, Patterson LE, Alt CA, Rizzo JR, Zhang TY, Haurez M. Org. Lett. 2005;7:1947–1950. doi: 10.1021/ol050367e. [DOI] [PubMed] [Google Scholar]
- (17).Brandt P, Hedberg C, Andersson P. Chem. Eur. J. 2003;9:339–347. doi: 10.1002/chem.200390029. [DOI] [PubMed] [Google Scholar]
- (18).Cui X, Fan Y, Hall MB, Burgess K. Chem. Eur. J. 2005;11:6859–6868. doi: 10.1002/chem.200500762. [DOI] [PubMed] [Google Scholar]
- (19).Fan Y, Cui X, Burgess K, Hall MB. J. Am. Chem. Soc. 2004;126:16688–16689. doi: 10.1021/ja044240g. [DOI] [PubMed] [Google Scholar]
- (20).Church TL, Rasmussen T, Andersson PG. Organometallics. 2010;29:6769–6781. [Google Scholar]
- (21).Hopmann KH, Bayer A. Organometallics. 2011;30:2483–2497. [Google Scholar]
- (22).Zhou J, Ogle JW, Fan Y, Banphavichit V, Zhu Y, Burgess K. Chem. Eur. J. 2007;13:7162–7170. doi: 10.1002/chem.200700390. [DOI] [PubMed] [Google Scholar]
- (23).Xie J-H, Zhou Q-L. Acc. Chem. Res. 2008;41:581–593. doi: 10.1021/ar700137z. [DOI] [PubMed] [Google Scholar]
- (24).Li S, Zhu S-F, Zhang C-M, Song S, Zhou Q-L. J. Am. Chem. Soc. 2008;130:8584–8585. doi: 10.1021/ja802399v. [DOI] [PubMed] [Google Scholar]
- (25).Zhu S.-f., Yu Y-B, Li S, Wang L-X, Zhou Q-L. Angew. Chem. Int. Ed. 2012;51 doi: 10.1002/anie.201204363. in press. [DOI] [PubMed] [Google Scholar]
- (26).Zhang Y, Han Z, Li F, Ding K, Zhang A. Chem. Commun. 2010;46:156–158. doi: 10.1039/b919902k. [DOI] [PubMed] [Google Scholar]
- (27).Li J-Q, Quan X, Andersson PG. Chem.--Eur. J. 2012;18:10609–10616. doi: 10.1002/chem.201200907. [DOI] [PubMed] [Google Scholar]
- (28).Li S, Zhu S-F, Xie J-H, Song S, Zhang C-M, Zhou Q-L. J. Am. Chem. Soc. 2010;132:1172–1179. doi: 10.1021/ja909810k. [DOI] [PubMed] [Google Scholar]
- (29).Yang S, Zhu S-F, Zhang C-M, Song S, Yu Y-B, Li S, Zhou Q-L. Tetrahedron. 2012;68:5172–5178. [Google Scholar]
- (30).Lightfoot A, Schnider P, Pfaltz A. Angew. Chem. Int. Ed. 1998;37:2897–2899. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2897::AID-ANIE2897>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- (31).Song S, Zhu S-F, Yang S, Li S, Zhou Q-L. Angew. Chem., Int. Ed. 2012;51:2708–2711. doi: 10.1002/anie.201107802. [DOI] [PubMed] [Google Scholar]
- (32).Schrems MG, Pfaltz A. Chem. Commun. 2009:6210–6212. doi: 10.1039/b912680e. [DOI] [PubMed] [Google Scholar]
- (33).Franzke A, Pfaltz A. Chem.-Eur. J. 2011;17:4131–4144. doi: 10.1002/chem.201003314. S4131/4131-S4131/4138. [DOI] [PubMed] [Google Scholar]
- (34).Franzke A, Voss F, Pfaltz A. Tetrahedron. 2011;67:4358–4363. [Google Scholar]
- (35).Woodmansee DH, Mueller M-A, Neuburger M, Pfaltz A. Chem. Sci. 2010;1:72–78. [Google Scholar]
- (36).Ganic A, Rageot D, Trondlin L, Pfaltz A. Chimia. 2012;66:187–191. doi: 10.2533/chimia.2012.187. [DOI] [PubMed] [Google Scholar]
- (37).Rageot D, Woodmansee DH, Pugin B, Pfaltz A. Angew. Chem., Int. Ed. 2011;50:9598–9601. doi: 10.1002/anie.201104105. [DOI] [PubMed] [Google Scholar]
- (38).Zalubovskis R, Hoermann E, Pfaltz A, Moberg C. ARKIVOC. 2008:58–66. [Google Scholar]
- (39).Kaiser S, Smidt SP, Pfaltz A. Angew. Chem. Int. Ed. 2006;45:5194–5197. doi: 10.1002/anie.200601529. [DOI] [PubMed] [Google Scholar]
- (40).Smidt SP, Menges F, Pfaltz A. Org. Lett. 2004;6:2023–2026. doi: 10.1021/ol049235w. [DOI] [PubMed] [Google Scholar]
- (41).Hilgraf R, Pfaltz A. Adv. Synth. Catal. 2005;347:61–77. [Google Scholar]
- (42).Bernardinelli GH, Kundig EP, Pfaltz A, Radkowski K, Zimmermann N, Neuburger-Zehnder M. Helv. Chimica Acta. 2001;84:3233–3246. [Google Scholar]
- (43).Drury WJ, III, Zimmermann N, Keenan M, Hayashi M, Kaiser S, Goddard R, Pfaltz A. Angew. Chem. Int. Ed. 2004;43:70–74. doi: 10.1002/anie.200352755. [DOI] [PubMed] [Google Scholar]
- (44).Menges F, Pfaltz A. Adv. Synth. Catal. 2002;344:40–44. [Google Scholar]
- (45).Blankenstein J, Pfaltz A. Angew. Chem. Int. Ed. 2001;40:4445–4447. doi: 10.1002/1521-3773(20011203)40:23<4445::aid-anie4445>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- (46).Cozzi PG, Zimmermann N, Hilgraf R, Schaffner S, Pfaltz A. Adv. Synth. Catal. 2001;343:450–454. [Google Scholar]
- (47).Blackmond DG, Lightfoot A, Pfaltz A, Rosner T, Schnider P, Zimmermann N. Chirality. 2000;12:442–449. doi: 10.1002/(SICI)1520-636X(2000)12:5/6<442::AID-CHIR25>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- (48).Hilgraf R, Pfaltz A. Synlett. 1999:1814–1816. [Google Scholar]
- (49).Dieguez M, Mazuela J, Pamies O, Verendel JJ, Andersson PG. Chem. Commun. 2008:3888–3890. doi: 10.1039/b806891g. [DOI] [PubMed] [Google Scholar]
- (50).Mazuela J, Norrby P-O, Andersson PG, Pamies O, Dieguez M. J. Am. Chem. Soc. 2011;133:13634–13645. doi: 10.1021/ja204948k. [DOI] [PubMed] [Google Scholar]
- (51).Mazuela J, Paptchikhine A, Pamies O, Andersson PG, Dieguez M. Chem. Eur. J. 2010;16:4567–4576. doi: 10.1002/chem.200903350. [DOI] [PubMed] [Google Scholar]
- (52).Li J-Q, Paptchikhine A, Govender T, Andersson PG. Tetrahedron Asymmetry. 2010;21:1328–1333. [Google Scholar]
- (53).Verendel JJ, Andersson PG. J. Chem. Soc., Dalton Trans. 2007:5603–5610. doi: 10.1039/b713257n. [DOI] [PubMed] [Google Scholar]
- (54).Dieguez M, Mazuela J, Pamies O, Verendel JJ, Andersson PG. J. Am. Chem. Soc. 2008;130:7208–7209. doi: 10.1021/ja801706s. [DOI] [PubMed] [Google Scholar]
- (55).Cheruku P, Paptchikhine A, Ali M, Neudoerfl J-M, Andersson PG. Org. Biomol. Chem. 2008;6:366–373. doi: 10.1039/b714744a. [DOI] [PubMed] [Google Scholar]
- (56).Kaellstroem K, Andersson PG. Tetrahedron Lett. 2006;47:7477–7480. [Google Scholar]
- (57).Hedberg C, Kaellstroem K, Brandt P, Hansen LK, Andersson PG. J. Am. Chem. Soc. 2006;128:2995–3001. doi: 10.1021/ja057178b. [DOI] [PubMed] [Google Scholar]
- (58).Kallstrom K, Hedberg C, Brandt P, Bayer A, Andersson PG. J. Am. Chem. Soc. 2004;126:14308–14309. doi: 10.1021/ja0464241. [DOI] [PubMed] [Google Scholar]
- (59).Co TT, Kim T-J. Chem. Commun. 2006:3537–3539. doi: 10.1039/b607450b. [DOI] [PubMed] [Google Scholar]
- (60).Chelucci G, Marchetti M, Malkov AV, Friscourt F, Swarbrick ME, Kocovsky P. Tetrahedron. 2011;67:5421–5431. [Google Scholar]
- (61).Metallinos C, Van Belle L. J. Organomet. Chem. 2010;696:141–149. [Google Scholar]
- (62).Lu W-J, Chen Y-W, Hou X-L. Adv. Synth. Catal. 2010;352:103–107. [Google Scholar]
- (63).Li X, Li Q, Wu X, Gao Y, Xu D, Kong L. Tetrahedron Asymmetry. 2007;18:629–634. [Google Scholar]
- (64).Liu D, Tang W, Zhang X. Org. Lett. 2004;6:513–516. doi: 10.1021/ol0362717. [DOI] [PubMed] [Google Scholar]
- (65).Bolm C, Focken T, Raabe G. Tetrahedron Asymmetry. 2003;14:1733–1746. [Google Scholar]
- (66).Bunlaksananusorn T, Polborn K, Knochel P. Angew. Chem. Int. Ed. 2003;42:3941–3943. doi: 10.1002/anie.200351936. [DOI] [PubMed] [Google Scholar]
- (67).Xu G, Gilbertson S. Tetrahedron Lett. 2003;44:953–955. [Google Scholar]
- (68).Woodmansee DH, Müller M-A, Trçndlin L, Hçrmann E, Pfaltz A. Chem. Eur. J. 2012;18:13780–13786. doi: 10.1002/chem.201202397. [DOI] [PubMed] [Google Scholar]
- (69).Vigneron J, Dhaenens M, Horeau A. Tetrahedron. 1973;29:1055–1059. [Google Scholar]
- (70).Zhao J, Burgess K. Org. Lett. 2009;11:2053–2056. doi: 10.1021/ol900308w. [DOI] [PubMed] [Google Scholar]
- (71).Zhu Y, Burgess K. J. Am. Chem. Soc. 2008;130:8894–8895. doi: 10.1021/ja802909b. [DOI] [PubMed] [Google Scholar]
- (72).Zhu Y, Burgess K. Adv. Synth. Catal. 2008;350:979–983. [Google Scholar]
- (73).Zhu Y, Fan Y, Burgess K. J. Am. Chem. Soc. 2010;132:6249–6253. doi: 10.1021/ja101233g. [DOI] [PubMed] [Google Scholar]
- (74).Ohta T, Miyake T, Seido N, Kumobayashi H, Takaya H. J. Org. Chem. 1995;60:357–363. [Google Scholar]
- (75).Brown JM, Hall SA. J. Organomet. Chem. 1985;285:333–341. [Google Scholar]
- (76).Brown JM. Angew. Chem. Int. Ed. 1987;26:190–203. [Google Scholar]
- (77).Duan Z-C, Hu X-P, Zhang C, Zheng Z. J. Org. Chem. 2010;75:8319–8321. doi: 10.1021/jo101849b. [DOI] [PubMed] [Google Scholar]
- (78).Stewart GW, Shevlin M, Yamagata ADG, Gibson AW, Keen SP, Scott JP. Org. Lett. doi: 10.1021/ol302518y. Ahead of Print. [DOI] [PubMed] [Google Scholar]