

Abstract
Treatment of cancer cell lines with high levels of nitric oxide (NO) via NO donors, such as DETANONOate, inhibits cell growth and survival pathways and sensitizes resistant tumor cells to apoptosis by chemoimmunotherapeutic drugs. In addition, we recently have reported that NO also inhibits the epithelial-to-mesenchymal transition (EMT) phenotype in metastatic cancer cell lines via dysregulation of the nuclear factor (NF)-κB/Snail/Yin Yang 1 (YY1)/Raf kinase inhibitor protein circuitry. The mechanism underlying NO-mediated dysregulation of this circuit was investigated, namely, NO-mediated inhibition of the activity of the transcription factors NF-κB, Snail, and YY1. We hypothesized that one mechanism of NO-mediated inhibition may invoke the NO-induced S-nitrosylation of these transcription factors. We demonstrate in metastatic and EMT+ human prostate carcinoma cell lines that treatment with NO results in the S-nitrosylation of NF-κB (p50), Snail, and YY1 and inhibits their activities, resulting in the reversal of the EMT phenotype into a mesenchymal-to-epithelial transition phenotype. These findings suggest that NO donors may be potential therapeutic agents in both the reversal of resistance and the inhibition of EMT and metastasis.
Keywords: nitric oxide, epithelial-to-mesenchymal transition (EMT), Yin Yang 1, Raf kinase inhibitor protein, metastasis
I. INTRODUCTION
Cancer is a disease that is characterized by uncontrolled cell growth and proliferation resulting from genetic and epigenetic alterations.1 Cancer also manifests itself to invasion and metastasis at different sites.2 Cancer consists of a large number of diseases affecting many organs, and each disease has common as well as unique biochemical, molecular, and genetic features. The tumor initially is localized and has a good prognostic outcome through various treatment modalities, including surgery, radiotherapy, chemotherapy, hormonal therapy, and immunotherapy. However, often after treatment, the tumor recurs at the primary site and metastasizes, and such metastases are consistently resistant to conventional therapies.3 Resistance is invariably the result of various strategies of tumor cell acquisition that inhibit the cytotoxic/apoptotic pathways responsive to cytotoxic therapies. Such strategies also have been implicated in the pathogenesis and progression of tumors as well as their metastatic behaviors.4–6
In metastasis, the tumor cells invade the basement membrane and the circulatory system, extravasate through the endothelial lining of the blood vessels, and form colonies at different tissue sites. The migration of the epithelial layer into the mesenchymal layer before reaching the basement membrane is known as epithelial-to-mesenchymal transition (EMT).7
EMT is induced via alterations in gene expression of the primary tumor cells and normally via extracellular growth factors. Phenotypic alterations of EMT include loss of cell adhesion, basal apical polarity, and the gain of motility and spindle cell shape.8 In addition to endowing cancer cells with the invasive phenotype, EMT also helps cancer cells to evade apoptosis and acquire resistance to both immuno- and chemotherapeutic drugs.8,9 Tumor cells that have undergone EMT exhibit several molecular alterations that can be monitored for characterization of the EMT phenotype or of its loss. Hence, during EMT, the epithelial surface molecule, E-cadherin, is lost, whereas the expression of N-cadherin is highlighted, resulting in a cadherin switch.10 For β-catenin, the intracellular localization of this protein is associated with EMT because it is localized on the cell membrane in epithelial cells. In mesenchymal cells, β-catenin is translocated into the nucleus, where it acts as a co-activator of transcription factors associated with EMT.11,12 Other markers of EMT include the extracellular proteins such as fibronectin and laminin, cytoskeletal markers such as vimentin, as well as transcription factors such as Snail and Twist.13 In EMT, the epithelial markers E-cadherin, α-catenin, and γ-catenin are repressed, whereas the mesenchymal markers vimentin, fibronectin, and N-cadherin are induced. In addition, certain microRNAs have been proposed to be key markers of EMT.14
We recently have reported the dysgregulated nuclear-factor (NF)-κB/Snail/Yin Yang (YY) 1 /Raf kinase inhibitor protein (RKIP)/phosphatase and tensin homologue (PTEN) circuitry in cancer cells, and this circuit has been shown to play a pivotal role in the EMT phenotype. 9,15–17
The role of nitric oxide (NO) in cancer has been the subject of controversy, with reports suggesting either its anti- or proneoplastic activity. This biphasic role of NO in oncology seems to be dependent on the microenvironment, the responsiveness of the tumor type, the redox state of the chemical reaction, as well as the final concentration and the duration of its intracellular exposure.18–20 This controversy might have been resolved, in large part, by the demonstration that low concentrations of NO act as an enhancer of cancer cell survival and proliferation and protect the cancer cells from apoptosis. However, high concentrations of NO exhibit, for example, direct cytotoxic effects and gene mutations, all of which contribute to tumor regression.21,22
We have reported that treatment of drug-resistant and metastatic human cancer cell lines with high levels of NO donors (e.g., DETANONOate) resulted in both the sensitization of drug-resistant tumor cells to apoptosis by chemoimmunocytotoxic drugs and the inhibition of the EMT phenotype (Fig. 1).9 On the basis of these findings, we hypothesized that NO-induced inhibition of EMT may be the result of NO-mediated inhibition of NF-κB, YY1, and Snail activities via S-nitrosylation. Here we review briefly the effect of NO on EMT via its effects on each of the factors in the circuitry (NF-κB, Snail, YY1, and RKIP) and the demonstration that each of these factors modified by NO is involved directly in the regulation of EMT.
FIGURE 1.
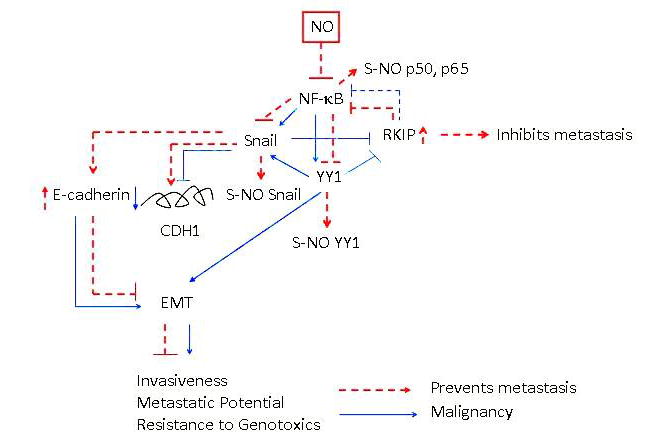
General schematic diagram demonstrating the constitutively activated nuclear factor(NF)-κB/Snail/Yin Yang (YY) 1/RKIP circuitry in tumor cells and their modification by nitric oxide (NO). In tumor cells, the NF-κB pathway is activated constitutively. It has been reported that NF-κB regulates the transcription and expression of both Snail and YY1 and that YY1 also regulates Snail transcription. Snail represses RKIP, which has been reported to inhibit NF-κB. The overexpression of NF-κB, Snail, and YY1 and the underexpression of RKIP have been shown to regulate the epithelial-to-mesenchymal transition (EMT) phenotype and metastasis. Treatment with the NO donor DETANONOate results in the inhibition of NF-κB, Snail, and YY1 and the induction of RKIP expressions. This results in the reversal of the repression of E-cadherin by Snail and upregulation of E-cadherin. In addition to NO-induced inhibition of NF-κB and its target genes, it also inhibits the DNA-binding activities of NF-κB, Snail, and YY1 via S-nitrosylation. Together, these various modifications by NO result in the inhibition of EMT and metastasis.
II. NO-MEDIATED INTERFERENCE IN THE NF-κB/SNAIL/YY1/RKIP CIRCUIT AND THE RESULTANT INHIBITION OF EMT
We have reported that treatment of EMT+ metastatic human carcinoma cell lines with DETANONOate inhibits the EMT+ phenotype through downregulation of the mesenchymal markers vimentin and fibronectin and induction of the epithelial marker E-cadherin, along with the inhibition of invasiveness. These effects were accompanied by the inhibition of NF-κB, Snail, and YY1 and the induction of RKIP.
II. A. NO-Mediated Inhibition of NF-κB Activity via S-nitrosylation of p50 and Inhibition of EMT
It has been reported that activation of NF-κB regulates positively EMT and metastasis.23–25 Treatment of EMT+ prostate carcinoma cell lines with DETANONOate inhibited NF-κB DNA-binding activity as assessed by electrophoretic mobility shift assay. Furthermore, inhibition of NF-κB by the specific NF-κB inhibitor, DHMEQ, also inhibited EMT.26 In addition, treatment with DETANONOate resulted in the S-nitrosylation of p5027 (Huerta-Yepez et al, unpublished data). The S-nitrosylation of p50 inhibited the nuclear translocation of NF-κB into the nucleus and inhibited the transcription and expression of its target genes such as Snail and YY1 (Fig. 2).
FIGURE 2.
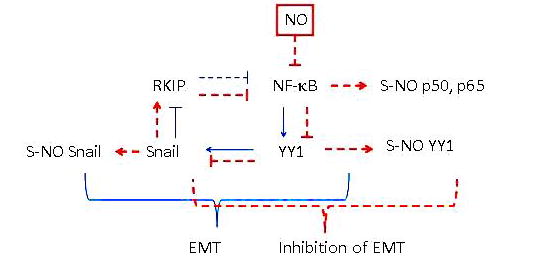
The direct role of nitric oxide (NO)–induced inhibition of nuclear factor (NF)-κB in the inhibition of the epithelial-to-mesenchymal transition (EMT). Treatment of cells with NO results in the inhibition of NF-κB expression and activity via S-nitrosylation of p50 and p65. This inhibition results in the inhibition of NF-κB target genes Yin Yang (YY) 1 and Snail expressions as well as their activities via nitrosylation by NO. The inhibition of Snail results in the de-repression of RKIP and its induction; in turn, RKIP amplifies the inhibitory activity of NF-κB. Together, NO treatment results in the inhibition of EMT.
II. B. NO-Mediated Inhibition of Snail Activity via S-nitrosylation, Leading to Inhibition of EMT
Snail is a member of the Snail superfamily of zinc finger transcription factors. It has many functions28 and plays a pivotal role in the acquisition of migratory and invasive properties and triggers EMT by repressing the expression of E-cadherin.29 The transcription of Snail is under the regulation of NF-κB and is a transcription repressor of the metastatic suppressor gene product E-cadherin and RKIP.30,31 Snail silencing reverses the EMT phenotype, whereas its overexpression induces EMT. 32,33
We also recently have reported that treatment of cancer cells with NO results in the S-nitrosylation of Snail.26 Therefore, we suggest that NO-mediated inhibition of Snail is the result of both the inhibition of its expression via the inhibition of NF-κB and directly via NO-mediated S-nitrosylation. Both the expression and the activity of Snail modified by NO are involved in the inhibition of EMT (Fig. 3).
FIGURE 3.
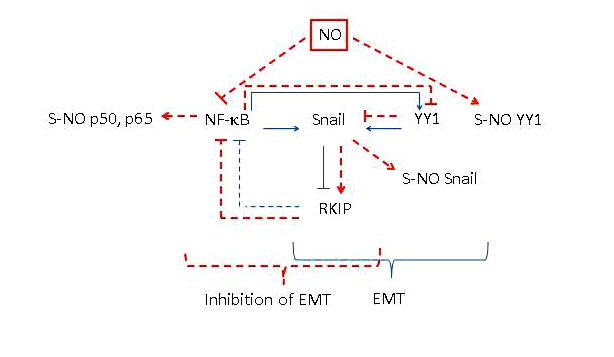
The direct role of nitric oxide (NO)–induced inhibition of Snail in the inhibition of the epithelial-to-mesenchymal transition (EMT). Treatment with NO results in the inhibition of nuclear factor (NF)-κB and its target genes Snail and Yin Yang (YY) 1. Inhibition of YY1 also inhibits Snail transcription, leading to RKIP induction. In addition, NO treatment results in the nitrosylation of NF-κB p50 and p65 and both Snail and YY1. Overexpression of Snail induces EMT and its knockdown inhibits EMT. Together, NO-induced inhibition of Snail results in the inhibition of EMT.
II. C. NO-Mediated Inhibition of YY1 via S-nitrosylation, Leading to the Inhibition of EMT
YY1 is a multifunctional, DNA-binding protein or associates with other transcription factors and functions as either an activator or a repressor.34 The transcription factor YY1 is regulated transcriptionally, in part by NF-κB and YY1; in turn, YY1 regulates the transcription of Snail.35,36 The role of YY1 in the regulation of EMT via the dysregulated NF-κB/Snail/YY1/RKIP/PTEN circuitry has been reviewed recently by our group.9 We also have reported that NO S-nitrosylates YY1 and inhibits its DNA-binding activity.27 Several lines of evidence suggest the direct role of YY1 in the regulation of EMT. Treatment of hepatic cells with YY1 siRNA reduced the invasive activity in the cells.37 Our preliminary findings show that silencing YY1 by YY1 siRNA decreases the expression of the mesenchymal markers vimentin and fibronectin and induces the expression of the epithelial marker E-cadherin along with inhibition of Snail and the induction of RKIP. de Nigris et al38 reported that overexpression of the YY1 protein was associated with malignancy and deletion of YY1 by siRNA metastatic growth. These findings suggest that NO-mediated inhibition of Snail expression and activity is a result of both the inhibition of its transcription by NO-mediated inhibition of NF-κB and its direct inhibition of activity by S-nitrosylation (Fig. 4).
FIGURE 4.
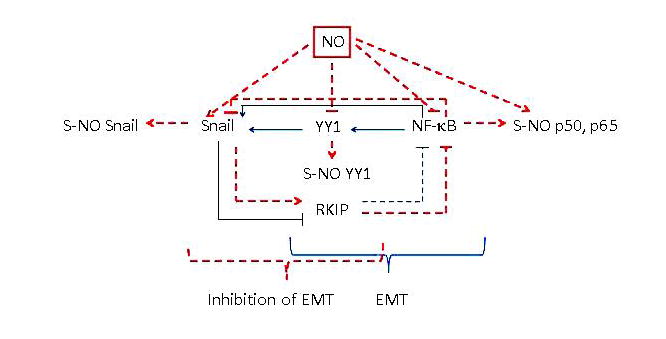
The direct role of nitric oxide (NO)–induced inhibition of Yin Yang (YY) 1 in the inhibition of the epithelial-to-mesenchymal transition (EMT). Treatment with NO inhibits YY1 activity via S-nitrosylation and its expression via inhibition of nuclear factor (NF)-κB. NO-mediated inhibition of YY1, Snail, and NF-κB results in the induction of RKIP. Overexpression of YY1 induces EMT and its knockdown inhibits EMT. Together, NO-induced inhibition of YY1 results in the inhibition of EMT.
II. D. NO-Mediated Induction of RKIP and Its Role in the Inhibition of EMT
In contrast to NO-mediated inhibition of NF-κB, Snail, and YY1, NO treatment has an inducing effect on RKIP expression. RKIP is a member of the phosphatidyl-ethanolamine protein family present in cytoplasm.39 Yeung et al40,41 have reported that RKIP inhibits the activity of both the Raf-1/MEK/extracellular signal-regulated kinase and NF-κB pathways. The expression of RKIP is downregulated significantly in several malignancies and is almost absent in several metastatic tumors. Overexpression of RKIP inhibits metastasis in experimental animal model systems.42,43 Hence, RKIP was identified as a metastatic suppressor gene product.42 Snail represses RKIP transcription.30 The direct role of RKIP in the regulation of EMT was corroborated in an experiment whereby ectopic expression of RKIP inhibited expression of Snail and the mesenchymal gene markers vimentin and fibronectin and reversed the EMT phenotype and inhibited cell migration and invasion (Fig. 5).26
FIGURE 5.
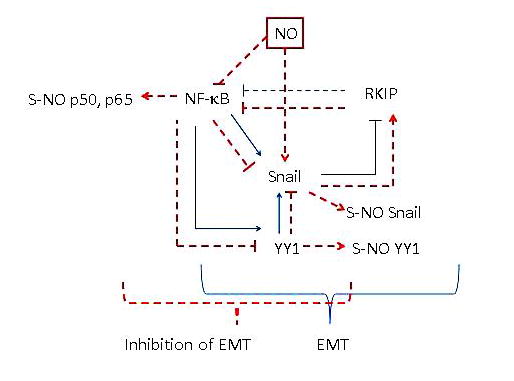
Nitric oxide (NO)–induced upregulation of RKIP in the inhibition of the epithelial-to-mesenchymal transition (EMT). Treatment with NO results in the inhibition of the transcription repressor Snail, which results in the induction of RKIP expression, a metastasis suppressor gene product. Upregulation of RKIP amplifies the inhibition of the nuclear factor (NF)-κB/Snail/Yin Yang (YY) 1 loop. Overexpression of RKIP results in the inhibition of EMT and its knockdown results in the induction of EMT. Together, NO-induced upregulation of RKIP results in the inhibition of EMT.
III. IN VIVO NO-MEDIATED INHIBITION OF THE EMT PHENOTYPE IN A METASTATIC HUMAN TUMOR XENOGRAFT
We have investigated the effect of in vivo treatment of mice bearing EMT+ PC3 human prostate carcinoma cells on the EMT phenotype. Tumor-bearing mice were treated intratumorally with DETANONOate, and samples then were analyzed by immunohistochemistry for several EMT biomarkers. The findings revealed that after treatment with DETANONOate, the expression of the mesenchymal markers fibronectin and vimentin were along with the expression of Snail. In contrast, the expression of both E-cadherin and RKIP were significantly induced (Huerta-Yepez et al, unpublished data). These findings corroborate the in vitro findings and validate the role of NO in the inhibition of the EMT phenotype in tumor-bearing mice.
IV. CONCLUSIONS
The findings reported here show that treatment of EMT+ human cancer cell lines with the NO donor DETANONOate inhibits EMT via modification of the dysregulated NF-κB/Snail/YY1/ RKIP loop, which has been shown to regulate EMT. Each of the gene products in the loop directly regulates EMT by either silencing it or following ectopic expression in EMT+ and EMT− tumor cells, respectively. The NO-mediated inhibition of EMT is a result, in part, of the S-nitrosylation of the transcription factors NF-κB, Snail, and YY1. The S-nitrosylation of these factors inhibits their DNA-binding activities. The inhibition of these factors resulted in the induction of RKIP expression, which is under its transcriptional regulation, in part, by Snail. The in vitro findings of NO-mediated inhibition of EMT were corroborated in vivo in mice bearing EMT+ tumor xenografts. Treatment with the NO donor DETANONOate inhibited in vivo the EMT phenotype. Together, these findings suggest potential therapeutic efficacy of NO donors in the inhibition of EMT and metastasis as well as their roles in sensitizing drug-resistant tumor cells to apoptosis by cytotoxic drugs.
Acknowledgments
The authors acknowledge the contributions reported by their collaborators, namely, Drs. Hermes Garban, Fumiya Hongo, Sara Huerta-Yepez, and Kam Yeung. The authors also acknowledge Daphne Liang, Melissa Cao, and Suzie Vardanyan for their assistance in the preparation of this manuscript. The work was supported, in part, by the National Cancer Institute (R01CA107023-02SI) and by the Jonsson Comprehensive Cancer Center at UCLA.
ABBREVIATIONS
- DETANONOate
(Z)-1-[2-(2-aminoethyl)-N-(2-ammonio-ethyl) amino] diazen-1-ium-1,2-diolate
- NF-κB
nuclear factor κB
- NO
nitric oxide
- RKIP
Raf kinase inhibitor protein
- YY1
Yin Yang 1
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 3.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17:2941–2953. doi: 10.1200/JCO.1999.17.9.2941. [DOI] [PubMed] [Google Scholar]
- 5.new treatments and novel insights into an old problem. Br J Cancer. 2008;99:387–391. doi: 10.1038/sj.bjc.6604510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeClerck K, Elble RC. The role of hypoxia and acidosis in promoting metastasis and resistance to chemotherapy. Front Biosci. 2010;15:213–225. doi: 10.2741/3616. [DOI] [PubMed] [Google Scholar]
- 7.Acloque H, Thiery JP, Nieto MA. The physiology and pathology of the EMT Meeting on the epithelial–mesenchymal transition. EMBO Rep. 2008;9:322–326. doi: 10.1038/embor.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Bonavida B, Baritaki S. Dual role of NO donors in the reversal of tumor cell resistance and EMT: downregulation of the NF-kB/Snail/YY1/ RKIP circuitry. Nitric Oxide. 2011;24:1–7. doi: 10.1016/j.niox.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol. 2001;153:1049–1060. doi: 10.1083/jcb.153.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 12.LaBonne C, Bronner-Fraser M. Snail-related transcriptional repressors are required in Xenopus for both the induction of the neural crest and its subsequent migration. Dev Biol. 2000;221:195–205. doi: 10.1006/dbio.2000.9609. [DOI] [PubMed] [Google Scholar]
- 13.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 14.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin K, Baritaki S, Militello L, Malaponte G, Bevelacqua Y, Bonavida B. The role of B-RAF mutations in melanoma and the induction of EMT via dysregulation of the NF-κB/ Snail/RKIP/PTEN circuit. Genes Cancer. 2010;1:409–420. doi: 10.1177/1947601910373795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu K, Bonavida B. The activated NF-kappaB-Snail-RKIP circuitry in cancer regulates both the metastatic cascade and resistance to apoptosis by cytotoxic drugs. Crit Rev Immunol. 2009;29:241–254. doi: 10.1615/critrevimmunol.v29.i3.40. [DOI] [PubMed] [Google Scholar]
- 17.Bartitaki S, Bonavida B. Nitric oxide inhibits tumor cell metastasis via dysregulation of the NF-kB/Snail/RKIP loop. In: Bonavida B, editor. Nitric Oxide and Cancer: Prognosis, Prevention, and Therapy (Cancer Drug Discovery and Development) New York: Springer; 2010. pp. 209–236. [Google Scholar]
- 18.Wink DA, Vodovotz Y, Cook JA, Krishna MC, Kim S, Coffin D, DeGraff W, Deluca AM, Liebmann J, Mitchell JB. The role of nitric oxide chemistry in cancer treatment. Biochemistry (Mosc) 1998;63:802–809. [PubMed] [Google Scholar]
- 19.Xie K, Huang S. Contribution of nitric oxide-mediated apoptosis to cancer metastasis inefficiency. Free Radic Biol Med. 2003;34:969–986. doi: 10.1016/s0891-5849(02)01364-3. [DOI] [PubMed] [Google Scholar]
- 20.Williams EL, Djamgoz MB. Nitric oxide and metastatic cell behavior. Bioessays. 2005;27:1228–1238. doi: 10.1002/bies.20324. [DOI] [PubMed] [Google Scholar]
- 21.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumor progression. Nat Rev Cancer. 2006;6:521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 22.Wink DA, Ridnour LA, Hussain SP, Harris CC. The reemergence of nitric oxide and cancer. Nitric Oxide. 2008;19:65–67. doi: 10.1016/j.niox.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104:733–744. doi: 10.1002/jcb.21695. [DOI] [PubMed] [Google Scholar]
- 24.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial–mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin SR, Sanchez-Velar N, Sherr DH, Sonenshein GE. 7,12-dimethylbenz(a)anthracene treatment of a c-rel mouse mammary tumor cell line induces epithelial to mesenchymal transition via activation of nuclear factor-kappaB. Cancer Res. 2006;66:2570–2575. doi: 10.1158/0008-5472.CAN-05-3056. [DOI] [PubMed] [Google Scholar]
- 26.Baritaki S, Huerta-Yepez S, Sahakyan A, Karagiannides I, Bakirtzi K, Jazirehi A, Bonavida B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle. 2010;9:4931–4940. doi: 10.4161/cc.9.24.14229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hongo F, Huerta-Yepez S, Vega M, Garban H, Jazirehi A, Mizutani Y, Miki T, Bonavida B. Inhibition of the transcription factor Yin Yang 1 (YY1) activity by S-nitrosylation. Biochem Biophys Res Commun. 2005;336:692–701. doi: 10.1016/j.bbrc.2005.08.150. [DOI] [PubMed] [Google Scholar]
- 28.Wu Y, Zhou BP. Snail: more than EMT. Cell Adh Migr. 2010;4:199–203. doi: 10.4161/cam.4.2.10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 30.Beach S, Tang H, Park S, Dhillon AS, Keller ET, Kolch W, Yeung KC. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene. 2008;27:2243–2248. doi: 10.1038/sj.onc.1210860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, Del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 32.Baritaki S, Yeung K, Palladino M, Berenson J, Bonavida B. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res. 2009;69:8376–8385. doi: 10.1158/0008-5472.CAN-09-1069. [DOI] [PubMed] [Google Scholar]
- 33.Baritaki S, Chapman A, Yeung K, Spandidos DA, Palladino M, Bonavida B. Inhibition of epithelial to mesenchymal transition in metastatic prostate cancer cells by the novel proteasome inhibitor, NPI-0052: pivotal roles of Snail repression and RKIP induction. Oncogene. 2009;28:3573–3585. doi: 10.1038/onc.2009.214. [DOI] [PubMed] [Google Scholar]
- 34.Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- 35.Baritaki S, Katsman A, Chatterjee D, Yeung KC, Spandidos DA, Bonavida B. Regulation of tumor cell sensitivity to TRAIL-induced apoptosis by the metastatic suppressor Raf kinase inhibitor protein via Yin Yang 1 inhibition and death receptor 5 upregulation. J Immunol. 2007;179:5441–5453. doi: 10.4049/jimmunol.179.8.5441. [DOI] [PubMed] [Google Scholar]
- 36.Palmer MB, Majumder P, Cooper JC, Yoon H, Wade PA, Boss JM. Yin yang 1 regulates the expression of snail through a distal enhancer. Mol Cancer Res. 2009;7:221–229. doi: 10.1158/1541-7786.MCR-08-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Notarbartolo M, Giannitrapani L, Vivona N, Poma P, Labbozzetta M, Florena AM, Porcasi R, Muggeo VM, Sandonato L, Cervello M, Montalto G, D’Alessandro N. Frequent alteration of the Yin Yang 1/Raf-1 kinase inhibitory protein ratio in hepatocellular carcinoma. OMICS. 2011;15:267–272. doi: 10.1089/omi.2010.0096. [DOI] [PubMed] [Google Scholar]
- 38.de Nigris F, Rossiello R, Schiano C, Arra C, Williams-Ignarro S, Barbieri A, Lanza A, Balestrieri A, Giuliano MT, Ignarro LJ, Napoli C. Deletion of Yin Yang 1 protein in osteosarcoma cells on cell invasion and CXCR4/angiogenesis and metastasis. Cancer Res. 2008;68:1797–1808. doi: 10.1158/0008-5472.CAN-07-5582. [DOI] [PubMed] [Google Scholar]
- 39.Odabaei G, Chatterjee D, Jazirehi AR, Goodglick L, Yeung K, Bonavida B. Raf-1 kinase inhibitor protein: structure, function, regulation of cell signaling, and pivotal role in apoptosis. Adv Cancer Res. 2004;91:169–200. doi: 10.1016/S0065-230X(04)91005-6. [DOI] [PubMed] [Google Scholar]
- 40.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 41.Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol. 2001;21:7207–7217. doi: 10.1128/MCB.21.21.7207-7217.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Z, Smith PC, Zhang L, Rubin MA, Dunn RL, Yao Z, Keller ET. Effects of raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst. 2003;95:878–889. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- 43.Fu Z, Kitagawa Y, Shen R, Shah R, Mehra R, Rhodes D, Keller PJ, Mizokami A, Dunn R, Chinnaiyan AM, Yao Z, Keller ET. Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate. 2006;66:248–256. doi: 10.1002/pros.20319. [DOI] [PubMed] [Google Scholar]