

Abstract
Recent studies have suggested a potentially important role for a family of tiny regulatory RNAs, known as microRNAs (miRNAs or miRs), in the control of diverse aspects of cardiac function in health and disease. Although the field of miRNA biology is relatively new, there is emerging evidence that miRNAs may play an important role in the pathogenesis of heart failure through their ability to regulate the expression levels of genes that govern the process of adaptive and maladaptive cardiac remodeling. Here we review the biology of miRNAs in relation to their role in modulating various aspects of the process of cardiac remodeling, as well as discuss the potential application of miRNA biology to the field of heart failure.
Keywords: heart failure, cardiac remodeling, microRNAs, neurohormonal activation, arrhythmia
The search for the basic mechanisms that are responsible for the development and progression of heart failure has been exhaustive; nonetheless, no single unifying mechanism has been uncovered that explains the development and progression of heart failure. Recent studies have uncovered a potentially important role for a family of tiny regulatory RNAs, known as microRNAs (miRNAs or miRs), in the control of diverse aspects of cardiac function (reviewed in 1). Although the field of miRNA biology is relatively nascent, there is emerging evidence that miRNAs may play an important role in the pathogenesis of heart failure through their ability to regulate the expression levels of genes that govern the process of adaptive and maladaptive cardiac remodeling. Herein we review the biology of miRNAs in relation to their role in modulating various aspects of the process of cardiac remodeling, as well as discuss the potential application of miRNA biology to the field of heart failure.
Biology of MicroRNAs
As reviewed elsewhere (see references1, 2 for recent reviews), miRNAs are noncoding RNAs that pair with specific “target” mRNAs and negatively regulate their expression through translational repression or mRNA degradation (gene silencing). The binding specificity of miRNAs depends upon complementary base pairing of ~7 nucleotide (nt) region at the 5′ end of the microRNA with the 3′ untranslated region (UTR) of the corresponding mRNA target. However, since miRNAs are conserved over the whole length of the mature miRNA, it is likely that the remainder of the miRNA (i.e., outside of the 7 nucleotide region that pairs with the 3′ UTR of the mRNA) may also play a role in determining the target mRNA. As shown in Figure 1 binding of miRNAs to their cognate target mRNAs commonly leads to decreased expression of target genes through translation repression or mRNA degradation. Increased expression levels of miRNAs can also result in the “paradoxical” up-regulation of previously suppressed target genes either by directly decreasing the expression of inhibitory proteins and/or transcription factors, or indirectly by inhibiting the expression levels of inhibitory miRNAs. Alternatively, decreased expression levels of inhibitory miRNAs can lead directly to increased target gene expression.3
Figure 1.
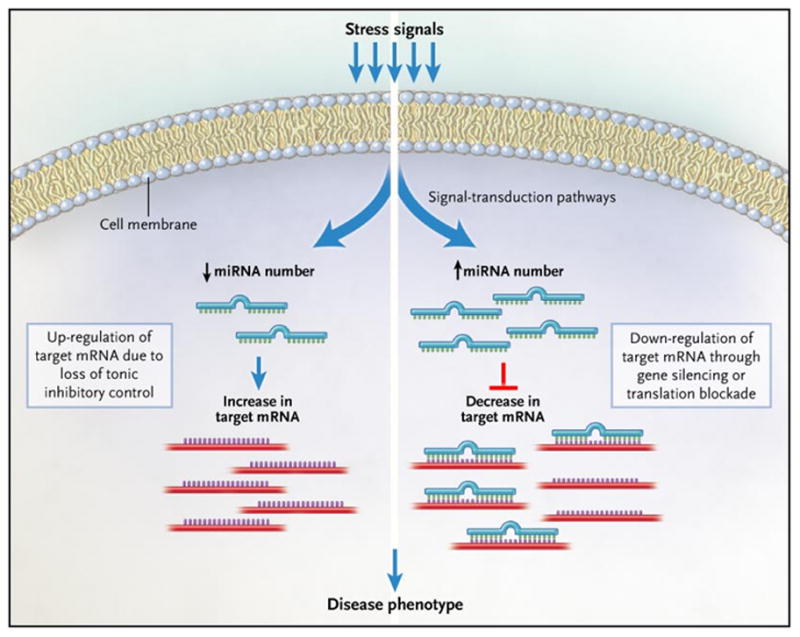
Mechanism for miRNA regulation of target mRNA levels. Stress signals (such as hemodynamic overload) activate signal transduction pathways that lead to either the up-regulation or down-regulation of specific microRNAs (miRNAs). Stress-signals that increase the expression levels of miRNAs can result in the down regulation of several target mRNAs through gene silencing, or more commonly translational blockade of the target mRNA. Alternatively, a stress induced decrease in the expression levels of inhibitory miRNAs can lead to up-regulation of previously suppressed target genes. Ultimately, it is the miRNA-induced pattern of change in gene expression that contributes to the resultant disease phenotype. (Reproduced with permission from Mann DL, MicroRNAs and the failing Heart, N Engl J Med. 2007 Jun 21;356(25):2644–5)
According to miRBase (http://microrna.sanger.ac.uk), the online reference repository of information on miRs, there are currently over 6200 published mature miRNA sequences, including over 700 miRNAs in humans. Moreover, additional miRNAs are continually being identified.4 The most common approach that has been used to study miRNAs thus far takes into account two characteristic features of miRs; that is, their tissue specificity and their disease specificity. With respect to tissue-specificity, profiling miRNAs across various tissues reveals that miR-1, let-7, miR-26a, miR-30c, miR-126-3p and miR-133 are highly expressed in the murine heart (18.5 weeks).5 However, at present we have very little information with respect to the temporal and spatial expression profiles of miRNAs in the heart. Disease-specific changes in miRNA expression have been studied using microarrays to determine which miRNAs are up- or down-regulated in response to various forms of tissue injury. By identifying the signature miRNAs that are regulated in the heart, investigators have been able to use sophisticated computational approaches to predict the potential array of mRNA targets that miRNAs can bind to, which has led to the identification of previously unrecognized targets within disease pathways of interest. That said, target prediction with the existing prediction algorithms remains challenging and requires careful experimental validation.21
Expression Profiles of MicroRNAs in Experimental Models and Human Heart Failure
At the time of this writing our understanding of the role that miRNAs play in heart failure is limited, and is based largely upon studies that have examined the miRNA expression profiles obtained from explanted hearts from patients with heart failure, or studies that have examined miRNA microarrays in experimental models that lead to pathological remodeling of the heart, such as transaortic banding (TAB) of the aorta, cardiac restricted overexpression of calcineurin, or cardiac restricted overexpression of constitutively active mutant of Akt kinase.6–8 Another approach that has been used is to perform miRNA microarrays on cultured cardiac myocytes that have been stimulated with peptides that mimic neurohormonal activation and/or that have been shown to provoke myocyte hypertrophy in vitro, such as angiotensin II (AngII), endothelin-1 (ET-1), and phenylephrine (PE).7–10
Human studies of miRNA expression profiles in heart failure are limited by the lack of standardized protocols, the small numbers of patients in the studies, and the high degree of variability in expression levels between patients. Two independent groups have performed microarrays on RNA isolated from non-failing and failing hearts. Ikeda et al. have obtained miRNA expression profiles in human heart failure using left ventricular (LV) samples from non-failing hearts, hearts from patients with ischemic or dilated cardiomyopathy, or aortic stenosis.11 Interestingly, these authors were able to use a miRNA based classifier to correctly classify the miRNA profiles into one of the above four disease specific categories in 60% of the samples, suggesting that miRNA expression profiles are sufficiently distinct to predict clinical etiology with modest accuracy. Thum et al. compared microarray data of miRNA expression profiles from non-failing, failing, and fetal human hearts, and observed that, when compared to non-failing hearts, 87% of the miRs that were up-regulated and 84% of the miRs that were down-regulated in the failing hearts were regulated concordantly in fetal heart tissue, suggesting that alterations in miRNAs levels may contribute to activation of the “fetal gene” program, which is one of the signatures of hypertrophied and failing myocardium. 12
To better compare and contrast the altered expression profiles of miRNAs in various animal models of heart disease and in human heart failure we have generated a two-dimensional graphical map (Figure 2) that displays the relevant human and experimental studies in columns with the accompanying changes in miRNA expression levels depicted in rows. Analogous to conventional heat maps that have been used for gene and miRNA arrays, we have used red to indicate changes in miRNAs that are reported to be significantly up-regulated, and green to indicate miRNAs that were reported to be significantly down-regulated. miRNA expression that was reported as unchanged is depicted in black, whereas miRNAs that were either not reported, or whose change in expression was not statistically interpretable were left uncolored. However, unlike a conventional heat-map, the color intensity of the miRNAs depicted in Figure 2 has not been adjusted to reflect the magnitude of change in miRNA expression. Using this approach, we obtained miRNA expression profiles on 172 miRs from a total of ten different experimental and clinical models, of which 5 were in human tissue, 4 were in-vivo studies in animals, and 1 was an in vitro study in cultured cardiac myocytes. For the sake of brevity and clarity, we did not graphically display 101 miRNAs whose expression levels were changed in only a single study (see data supplement for the entire analysis). The remaining 71 candidate miRNAs whose expression levels changed significantly in two or more studies are illustrated in Figure 2. Inspection of this map reveals that for miRNAs with increased levels of expression levels there was good concordance between human heart failure and experimental models of pathological remodeling. Indeed, 25 miRNAs (7b, 7c, 10b, 15b, 21, 23a, 23b, 24, 27a, 27b, 29a, 103, 125b, 140*, 195, 199a, 199a*, 199b, 208, 210, 211, 214, 330, 341, 424) were up-regulated in one or more myocardial samples from failing human hearts and experimental models, suggesting that changes in miRNA expression patterns in experimental models may provide further insight into our understanding LV remodeling in human heart failure. Interestingly, the expression profile of miRNAs with decreased expression levels were far less concordant in experimental models and human heart failure samples. Indeed, there were only 10 miRNA species (1, 10a, 26b, 30a_5p, 30b, 30c,150, 218, 451, 499) that were down-regulated in one or more myocardial samples from failing human hearts and experimental models. It should be emphasized that different microarray platforms were used in the above studies, which may account for some of the observed differences in miRNA regulation.
Figure 2.
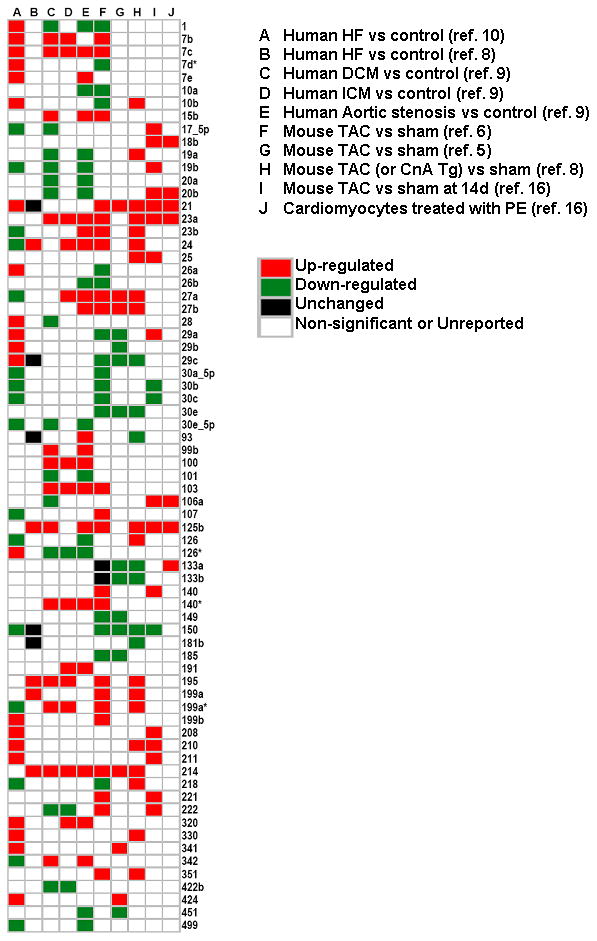
miRNA expression profiles in experimental models and human heart failure. A Pubmed search (May-June 2008) was conducted using the MeSH titles: ‘microRNA’, ‘heart disease’ and/or ‘heart failure.’ A total of 2314 articles were identified of which 614 review articles were excluded. The content of the 1696 original articles were reviewed for relevance with respect to the role of miRNAs in cardiac remodeling. Six studies reported global miRNA expression data (miRNA profiling) using micro-RNA micro-arrays, of which 3 evaluated miR expression in two or more experimental model systems.6, 7, 9–12 Differentially expressed miR candidates that were observed in at least two studies are graphically displayed with the relevant studies in columns and the accompanying changes in miRNA expression illustrated in rows. Red indicates miRs that were significantly up-regulated; green indicates miRs that were significantly down-regulated; black indicated no change in miR expression levels; white indicates miRs that were either unreported, equally expressed or not significantly different (p>0.05) between disease phenotype and controls.
In order to better understand how miRNAs may play a role in cardiac remodeling and heart failure it is useful to recognize that cardiac remodeling represents the anatomic summation of a series of changes in the biology of the cardiac myocyte and the myocardium, including the volume of cardiac myocytes and the volume/composition of the extracellular matrix (ECM) (see Table 1).13 As will be discussed below, there is emerging evidence that miRNAs play an important role both in the changes in the biology of the cardiac myocyte, as well as changes in the myocardium.
TABLE 1.
OVERVIEW OF LEFT VENTRICULAR REMODELING
| Alterations in Myocyte Biology |
| Changes in excitation contraction coupling |
| Beta-myosin heavy chain (fetal) gene expression |
| Beta-adrenergic receptor desensitization |
| Hypertrophy with loss of myofilaments |
| Loss of cytoskeletal proteins |
| Myocardial Changes |
| Myocyte loss |
| Necrosis |
| Apoptosis |
| Autophagy |
| Alterations in Extracellular Matrix |
| Matrix degradation |
| Replacement Fibrosis |
| Impaired Angiogenesis |
| Alterations in Left Ventricular Chamber Geometry |
| Left ventricular dilation |
| Increased LV wall stress |
| Mitral valve incompetence |
| Wall thinning with afterload mismatch |
MicroRNAs Involved in Cardiac Myocyte Hypertrophy
In both animal models and the human heart it is generally held that changes in the biology of the cardiac myocyte (see Table 1)14 are the primary initiating event that leads to cardiac remodeling, although it should be noted that cardiac remodeling can occur in the absence of myocyte dysfunction in some experimental models.15, 16 One of the principal changes that occurs in the biology of the failing cardiac myocyte is an increase in cell size (hypertrophy). Based on the extant literature, there is evidence that various microRNAs control and/or modulate key components of the hypertrophic process in cardiac myocytes, including reactivation of the so-called fetal gene program. Indeed, the extant experimental literature suggests that miR-1, miR-18b, miR-21, miR-133, miR-195, and miR-208 play important roles in modulating cardiac hypertrophic growth.
miR-1 and miR-133
miR-1 and miR-133 are preferentially expressed in cardiac and skeletal muscle and have been shown to regulate differentiation and proliferation of these cells. Although miR-1 and miR-133 form part of the same bicistronic unit, they are expressed as separate transcripts. Both miR-1 and miR-133 appear to play an important role in the remodeling of the heart that occurs during cardiogenesis. During cardiac development miR-1 levels are low, whereas its cognate target, Hand2 (a transcription factor that controls cardiac myocyte proliferation) is expressed at high levels, thereby promoting cardiomyocyte proliferation.17 As the heart develops, miR-1 levels increase and Hand2 protein levels decrease as cardiac myocytes exit the cell cycle and differentiate in mature myocytes. Germane to the present discussion, levels of miR-1 and miR-133 are decreased in heart failure models (Figure 2). Studies by Sayed et al. demonstrated that levels of miR-1 were suppressed as early as 1 day after aortic banding and remained depressed 7 days after TAB.6 Independently, Caré et al demonstrated reduced expression of miR-1 and miR-133 in three models of cardiac hypertrophy, namely, TAB, mice with cardiac restricted overexpression of constitutively active Akt kinase, and rats subjected to adaptive cardiac hypertrophy following endurance training.8 These same investigators confirmed that the expression level of miR-1 and miR-133 were decreased in the hearts of patients with hypertrophic cardiomyopathy or atrial dilation. Unfortunately, this study did not examine miR-1 or miR-133 expression levels in myocardial samples from human heart failure.
The functional significance of the findings with respect to miR-1 has been demonstrated in gain of function studies in vitro and in vivo. For example, overexpression of miR-1 in cultured neonatal myocytes partially inhibited phosphorylation of ribosomal S6 protein, and inhibited the computationally predicted in silico targets of miR-1 that were related to cardiac growth, including Ras GTPase-activating protein, cyclin dependent kinase 9, fibronectin, and Ras homolog expressed in brain.6 miR-1 overexpression also suppressed myocyte spreading induced by serum and endothelin-1 and expression of atrial natriuretic factor (ANF).6
Overexpression of miR-133 resulted in suppression of protein synthesis and inhibition of hypertrophic growth in PE or ET-1 treated neonatal mouse cardiac myocytes, and up-regulation of fetal genes, including those encoded by ANF, skeletal and cardiac alpha-actin and alpha- and beta-myosin heavy chain.8 Loss of function studies showed that sequestering endogenous miR-133 using a targeted 3′UTR decoy sequence resulted in marked cell hypertrophy and increased protein synthesis, increased fetal gene expression, and perinuclear localization of ANF, consistent with a potential role for miR-133 in suppressing hypertrophic growth.8 Further, treatment of mice using an antisense RNA oligonucleotide (termed an antagomir) targeted to miR-133 resulted in cardiac hypertrophy and re-induction of the fetal gene program.8 Taken together these studies suggest that down-regulation of miR-1 and miR-133 allows for the increased expression (release) of growth related genes that are responsible for cardiac hypertrophy. However, it is not clear whether these miRNAs contribute to the adverse cardiac remodeling that occurs in heart failure, insofar as the majority of studies in human tissue, save for one,11 have not show that miR-1 and miR-133 are down-regulated. Of note, the one study that did show that miR-1 and miR-133 were down-regulated in human tissue, examined dilated atria and myomectomy samples from patients with hypertrophic cardiomyopathy, rather than LV myocardial specimens from patients with heart failure.8
miR-21
The role of miR-21 in cardiac myocyte hypertrophy is controversial at present. Two studies have shown that miR-21 is highly up-regulated in mouse models of pressure-overload hypertrophy post-TAB.6, 9 Moreover, angiotensin-II (Ang-II) and phenylephrine (PE)-induced cardiac myocyte hypertrophy is accompanied by a 4 to 5-fold increase in miR-21 expression in isolated cardiac myocytes. Anti-sense mediated knockdown of miR-21 partially inhibited PE- or AngII- induced cell growth and protein synthesis in neonatal rat ventricular myocytes.9 In contrast, a different group of investigators has shown that anti-sense knockdown of miR-21 provokes hypertrophy and increased expression of fetal genes in cultured neonatal cardiac myocytes.10 The reason(s) for the discrepancy between these studies is unclear, but may be that modulation of hypertrophic growth by miR-21 in myocytes is through an indirect mechanism, rather than a direct targeting effect of miR-21 on hypertrophy related genes. And indeed, miR-21 has no validated gene targets that are related to cardiac myocyte hypertrophy. Unfortunately, further investigations into the role of miR-21 in myocyte hypertrophy have been hampered, insofar as exogenously administered pre-miR-21 fails to be processed in cardiac myocytes, resulting in the inability to overexpress mature miR-21 in these cells.7
Other candidate miRNAs involved in cardiac myocyte hypertrophy
Based on microarray data from aortic banding studies, calcineurin-transgenic mouse models and human heart failure samples, van Rooij and colleagues identified seven miRNAs that were up-regulated and 4 miRNAs that were coordinately down-regulated.7 Five of the miRNAs that were up-regulated, miR-23a, -23b, -24, -195 and -214, provoked cardiac myocyte hypertrophy when transfected into neonatal cardiac myocytes (Figure 3), whereas transfection of miR-199a resulted in elongated spindle shapes myocytes that were reminiscent of the elongated cardiac myocytes observed in dilated cardiomyopathy. Of the miRNAs that were down-regulated, miR-150 and-181b caused a reduction in cardiac myocyte size when transfected into cells.7 Cardiac-restricted overexpression of miR-195 resulted in a dilated cardiac hypertrophic phenotype at 6 weeks of age that was accompanied by exaggerated myocyte hypertrophy and activation of the fetal gene ensemble. In contrast, cardiac restricted overexpression of miR-214 had no effect on cardiac phenotype. Of interest to the present discussion, miR-195 was significantly up-regulated in 5 of the 10 studies illustrated in Figure 2, including human heart failure. Viewed together these studies suggest that miR-195 may play an important role in adverse cardiac remodeling. Thum et al. expressed three miRNAs that were up-regulated in human heart failure (miR-21, miR-129 and miR-212), and showed that although transfection of a single miRNA had little effect on cell morphology, the simultaneous overexpression of all 3 miRNAs resulted in myocyte hypertrophy and reinduction of the fetal gene program.12
Figure 3.
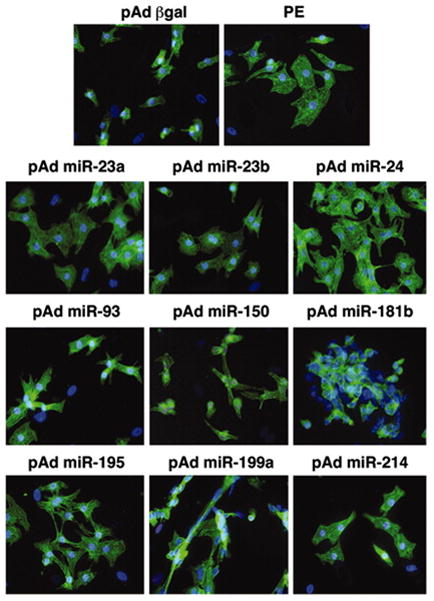
Overexpression of miRNAs in cultured cardiac myocytes. Rat cardiac myocytes in serum-free medium were infected with adenoviruses expressing individual miRNAs that were either up- or down-regulated in heart failure models. Actin staining of cardiac myocytes shows that adenoviral overexpression of several miRNAs induces morphological changes characteristic of hypertrophy. Cells were infected with pAd-gal were used as a negative control. As a positive control, cells were stimulated with phenylephrine (PE), a potent inducer of hypertrophy. (Modified with permission from van Rooij et al, PNAS, 2006;103:18255–18260).
MicroRNAs Involved in Regulating Excitation Contraction Coupling
Excitation-contraction (EC) coupling refers to the cascade of biological events that begins with cardiac action potential and ends with myocyte contraction and relaxation. Classic studies from explanted failing hearts have shown that patients with end-stage heart failure exhibit decreased contractility and impaired relaxation, which is believed to be secondary to changes in the abundance and/or phosphorylation state of critical calcium (Ca2+) regulatory proteins that are thought to play an important role in cross-bridge activation and relaxation. An additional defect in myocyte function in hypertrophy and heart failure is thought to occur secondary to changes in the actin and myosin myofibrillar cross-bridges. Indeed, early studies showed that myofibrillar ATPase was reduced in the hearts of patients who died of heart failure, and that these abnormalities in ATPase activity could be explained by an isoform switch from alpha-myosin heavy (MHC), which hydrolyzes ATP rapidly and is expressed in the adult heart, to beta-MHC which hydrolyzes more slowly and is expressed in the fetal heart. Whereas alpha-MHC accounts for approximately 33 percent of MHC mRNA in normal human myocardium, the abundance of alpha-MHC mRNA decreases to about 2 percent in the failing heart.18 Given the aforementioned link between expression levels of miRNAs and the development of cardiac myocyte hypertrophy, it is logical to consider that miRNAs might also regulate the Ca2+ regulatory proteins involved in E-C coupling that are down-regulated in cardiac hypertrophy. However, at the time of this writing there is no information on how, or whether miRNAs target genes that are related to Ca2+ handling in the heart. This statement notwithstanding, there are interesting data which suggest that miRNAs affect cross-bridge cycling.19
miR-208
Studies have shown that miR-208 is up-regulated in response to a hemodynamic pressure overload, as well as in human heart failure.10, 12 Olson and colleagues showed that miR-208, a cardiac specific microRNA that is encoded within an intron of the alpha-MHC gene, is coordinately regulated with alpha-MHC. Mice deficient for (miR-208−/−) appeared phenotypically normal at baseline; however, these mice had a blunted hypertrophic response following TAB, as well as decreased myocardial fibrosis (Figure 6). Of note, miR-208−/− mice did not express beta-MHC in response to pressure overload. Although mice with cardiac restricted overexpression of miR-208 had increased expression of beta-MHC, they did not develop pathological remodeling. Taken, together these observations suggest that miR-208 is required for the development of cardiac hypertrophy and myocardial fibrosis, and that miR-208 is a positive regulator of beta-MHC gene expression.19
Figure 6.
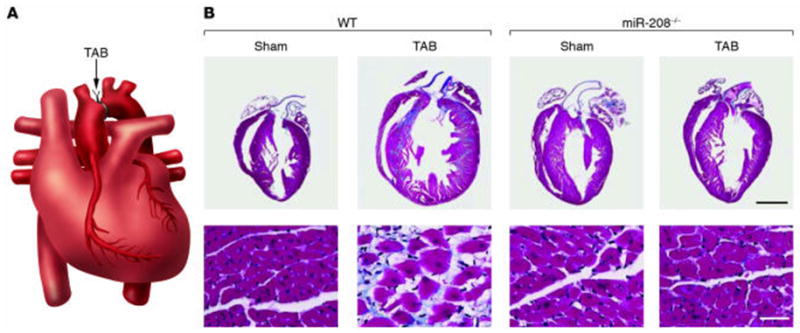
Requirement of miR-208 for cardiomyocyte hypertrophy and fibrosis. (A) Schematic diagram of a heart following thoracic aortic banding (TAB). (B) Sections of hearts of approximately 3-month-old wild-type and miR-208−/− mice are shown following sham operation or TAB for 21 days. High-magnification views of the ventricular wall are shown at the bottom. Trichrome staining identifies fibrosis in blue. Note that hypertrophy and fibrosis are diminished in miR208−/− mice compared with wild-type following TAB. (Reproduced with permission from van Rooij and Olson, J. Clin. Invest. 2007; 117:2369–2376).
miR-21
Alterations in the expression or activity, or both, of myofilament regulatory proteins has also been proposed as a potential mechanism for the decrease in cardiac contractile function in heart failure, including changes in myosin light chains and the troponin-tropomyosin complex. As noted above some,6, 9 but not all 10 studies have implicated a role for miR-21 in regulating hypertrophic growth in cardiac myocytes. Although not yet studied in human heart failure, miR-21 targets the 3′ UTR of tropomyosin and inhibits its translation in tumor cells.20 Further studies will be necessary to delineate the role of miR-21 in cross-bridge cycling in heart failure.
MicroRNAs involved in Regulating the Cytoskeleton
The cytoskeleton of cardiac myocytes consists of actin, the intermediate filament desmin, the sarcomeric protein titin and alpha- and beta-tubulin that form the microtubules by polymerization. Vinculin, talin, dystrophin and spectrin represent a separate group of membrane-associated cytoskeletal proteins. Disruption of cytoskeletal and/or membrane-associated proteins has been implicated in the pathogenesis of heart failure in numerous studies. Indeed, the loss of integrity of the cytoskeleton with a resultant loss of linkage of the sarcomere to the sarcolemma and extracellular matrix would be expected to lead to contractile dysfunction at the myocyte level, as well as at the myocardial level. At present there is limited evidence that miRNAs are involved in regulating the cytoskeleton, insofar are relatively few studies have examined these targets.
miR-1 and miR-133
As noted above, the decreased expression of miR-1 and miR-133 allows for the increased expression of growth related genes that are responsible for cardiac hypertrophy. Adenoviral mediated overexpression of miR-1 suppressed sarcomeric alpha-actin organization that is normally observed in serum-deprived neonatal cardiac myocytes. The effects of miR-1 on cytoskeletal reorganization were also thought to be responsible for the inhibition of endothelin induced cell-spreading in neonatal myocytes.6 Similarly, adenoviral transfection with miR-133 inhibited the reorganization of the actin myofilaments observed in hypertrophic growth of cardiac myocytes (Figure 4).8 Two validated gene targets of miR-133 are RhoA and Cdc42, which is of interest in that members of the Rho- family of GTP binding proteins are involved in myofibrillar rearrangement, and are therefore important in the cell motility and contractility. miR-1 also targets a number of cytoskeletal related proteins including microtubule related proteins, kinectins, actin binding proteins and cadherins. Insofar as miR-1 and miR-133 do not appear to be down-regulated in human heart failure, the significance of the aforementioned miR-induced changes in the cytoskeleton vis-a-vis the biology of the failing myocyte remains to be determined.
Figure 4.
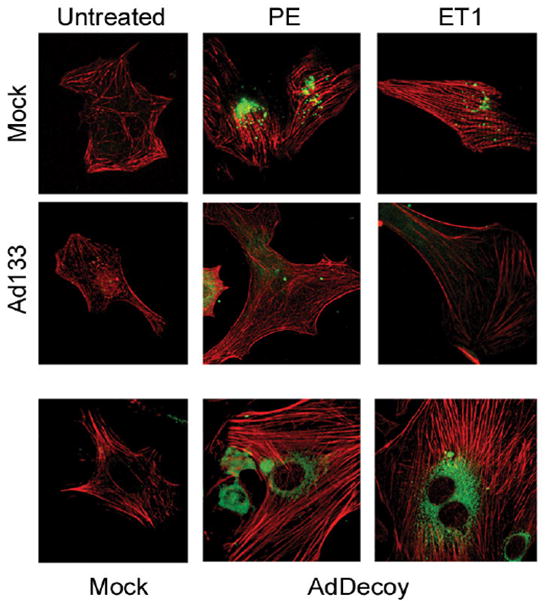
Cytoskeletal changes in myocyte induced by miRNAs. Immunofluorescence images of diluent, phenylephrine (PE) and endothelin-1 (ET1) treated adult cardiac myocytes that were transfected with mock adenovirus, adenovirus-miR-133 (Ad133) and a decoy 3′UTR transcript to miR-133 (AdDecoy). The myocyte cultures were immunostained for actin (red) and ANF (green) proteins. (Modified from Care et al, Nature Medicine, 2007; 13: 613–618 [data supplement])
MicroRNAs Involved in Myocardial Alterations in the Failing Heart
The alterations that occur in failing myocardium may be categorized broadly into those that affect the number of cardiac myocytes, as well as those changes that occur in the volume and composition of the extracellular matrix (Table 1). With respect to the changes that occur in cardiac myocyte component of the myocardium there is increasing evidence which suggests that progressive myocyte loss, through necrotic, apoptotic or autophagic cell death pathways, may contribute to progressive cardiac dysfunction and LV remodeling.
MicroRNAs involved in Regulating Cell Fate
Progressive myocyte loss secondary to apoptosis occurs in failing hearts, and contributes to progressive cardiac dysfunction and LV remodeling. Apoptosis requires activation of a a phylogenetically conserved ensemble of proteins belonging to the extrinsic (death receptor mediated) or intrinsic (mitochondrial) pathways that ultimately lead to activation of executioner caspases and myocyte loss. The extensive synergistic regulation of pro- and anti-apoptotic proteins in response to stress signals raises the possibility that miRs may be involved in regulating programmed cell death.
miR-21
miR-21 is up-regulated in various cancers (malignant gliobastoma, colorectal carcinoma, cervical adenocarcinoma) and, based on its in-silico predicted pro-apoptotic gene targets, has been proposed as a potential anti-apoptotic miRNA. Knockdown of miR-21 in glioblastoma cells leads to caspase activation and apoptotic cell death,21 and depletion of miR-21 in vascular smooth muscle cells lead to a dose-dependent increase in apoptosis and decrease in cell proliferation.22 As noted above, several independent groups have shown that the expression levels of miR-21 are increased 2–4 fold in the heart post-TAB.6, 9 Although miR-21 levels were not examined in relation to myocyte apoptosis in these studies, the upregulation of miR-21 may represent a pro-survival stress response in response to hemodynamic pressure overload. In this regard, it is interesting to note that the anti-apoptotic protein Bcl-2 is an indirect target of miR-21.22
miR-1 and miR-133
Although miR-1 and miR-133 act synergistically in controlling myocyte proliferation and differentiation (see above), the extant literature suggests that these miRNAs have opposite roles with respect to regulating cell fate. Xu et al. demonstrated that overexpression of miR-1 in H9c2 rat myoblasts provoked apoptotic cell death, which was partially rescued by treatment with miR-133.23 Similar effects on oxidative-stress induced apoptosis were observed in H2O2 treated H9c2 cells, wherein co-transfection with miR-1 led to 60% reduction in the IC50 value necessary for oxidative stress induced DNA fragmentation, and co-transfection with miR-133 resulted in a 40% increase in the IC50 value required for oxidative stress induced DNA fragmentation. Qualitatively similar results were obtained when rat neonatal ventricular myocytes were exposed to H2O2.23 Subsequent computational analyses predicted that HSP60 and HSP70 were targets for miR-1, and that caspase-9 was a target for miR-133. And indeed, experimentally miR-1 suppressed HSP60 and HSP70 protein levels by ~70 and ~60%, respectively, whereas miR-133 decreased protein levels of caspase-9 and caspase-9 activity levels in rat H9c2 cells. The authors postulated that the relative levels of miR-1 and miR-133 may determine cell fate.23 In this regard, it is noteworthy that miR-1 levels are overexpressed in coronary disease, and are highly up-regulated in ischemic myocardium,24 wherein apoptosis is an important mode of cell death.
MicroRNAs involved in Regulating Angiogenesis
Adequate myocardial perfusion is essential for maintaining cardiac myocyte viability, particularly in the failing heart, wherein left ventricular filling pressures are elevated and subendocardial perfusion is compromised. Prior studies have suggested that fewer capillaries are present in end-stage dilated cardiomyopathy25, 26 and post-partum cardiomyopathy.27 The failure to appropriately regulate angiogenesis and vasculogenesis in the failing heart can result in myocardial ischemia with subsequent loss of cardiac myocytes and/or interstitial fibrosis, and may thus contribute to progressive myocardial dysfunction in heart failure. There is increasing evidence that miRNAs may regulate vascular integrity, angiogenesis, and wound repair. Indeed, a number of pro- and anti-angiogenic microRNAs have been identified that target key proteins involved in endothelial tube formation, as well as endothelial cell proliferation, migration and apoptosis.28 However, it should be emphasized that many of the miRNAs that have been studied thus far have been identified in neoplastic tissue, and may therefore not be relevant to cardiovascular physiology. Nonetheless, two recent studies have implicated miR-126 in angiogenesis and regulation of vascular integrity.29, 30
miR-126
Srivastava and colleagues found that miR-126 was enriched in endothelial cells derived from mouse embryonic stem cells and in developing mouse embryos. Subsequent studies in zebrafish showed that miR-126 knockdown resulted in loss of vascular integrity, as well as hemorrhage during embryonic development. These authors found that miR-126 directly repressed two negative regulators of the vascular endothelial growth factor (VEGF) pathway, namely Sprouty-related protein (Spred1) and the phosphoinositol-3 kinase regulatory subunit 2 (PIK3R2/p85-beta). Increased expression of Spred1 or inhibition of VEGF signaling in zebrafish resulted in defects similar to miR-126 knockdown.29 Analogous studies in mice revealed that endothelial cell-restricted expression of miR-126 mediated developmental angiogenesis, whereas targeted deletion of miR-126 resulted in leaky vessels, hemorrhaging, and partial embryonic lethality, due to a loss of vascular integrity and defects in endothelial cell proliferation, migration, and angiogenesis. Relevant to the present discussion, the subset of mutant mice that were not embryonic lethal had impaired myocardial neovascularization following myocardial infarction. It was further shown that miR-126 enhanced the pro-angiogenic actions of VEGF and fibroblast growth factor and promoted formation of new blood vessels by repressing the expression of Spred1.30 Taken together, these findings illustrate that a miR-126 is sufficient to regulate vascular integrity and angiogenesis. However, the relevance of these experimental findings with respect to human heart failure is unclear, insofar as miR-126 is up-regulated in failing human hearts.
MicroRNAs involved in Regulating The Extracellular Matrix
Changes within the extracellular matrix (ECM) constitute the second important myocardial adaptation that occurs during cardiac remodeling. The myocardial ECM consists of a basement membrane, a fibrillar collagen network that surrounds the myocytes, proteoglycans and glycosaminoglycans, and biologically active signaling molecules. Important changes occur in the ECM during cardiac remodeling, including changes in fibrillar collagen synthesis and degradation, as well as changes in the degree of collagen cross-linking. One of the histological signatures of advancing heart failure is the progressive increase in collagen content of the heart (myocardial fibrosis). The increased fibrous tissue would be expected to lead to increased myocardial stiffness, which would theoretically result in decreased myocardial shortening for a given degree of afterload. In addition, myocardial fibrosis may provide the structural substrate for atrial and ventricular arrhythmias, thus potentially contributing to sudden death. At time of this writing, the role of miRNAs in regulating the extracellular matrix is relatively unexplored.
miR-208
As noted above, Olson and colleagues have shown that miR-208, a cardiac specific micro RNA encoded within an intron of the alpha-MHC gene, is required for cardiac fibrosis in response to hemodynamic pressure overload. Indeed, miR-208 deficient mice (mir-208−/−) were resistant to developing fibrosis following TAB, as well as cardiac restricted overexpression of calcineurin, a calcium and calmodulin-dependent phosphatase that provokes pathological remodeling of the heart.19 Thus, miR-208 appears to be required for the development of cardiac fibrosis. Further studies will be needed to explore the role of miRs in the development of cardiac fibrosis.
mirR-29
The miR-29 family is comprised of three members, mirR-29a-c, that are expressed from two bicistronic miRNA clusters: miR-29b-1 is coexpressed along with miR-29a, whereas the second copy of miR-29b (miR-29b-2) is coexpressed with miR-29c. In an effort to identify miRNAs that were dysregulated in acute myocardial infarction van Rooij and colleagues observed that miR-29 was down-regulated in the border zone mouse hearts following acute occlusion of the left anterior descending artery and in the border zone of hearts from patients who were receiving cardiac transplant.31 A Targetscan prediction algorithm (www.targetscan.org) identified a high number of fibrosis related mRNAs for miR-29. Down-regulation of miR-29 with anti-miRs induced collagen expression both in vitro and in vivo, whereas overexpression of a miR-29 mimic in isolated cardiac fibroblasts reduced collagen transcripts. Moreover, stimulation of isolated cardiac fibroblasts with transforming growth factor-beta, a pro-fibrotic peptide, also resulted in decreased miR-29 expression. Taken together, these results suggest that the dynamic regulation of miR-29 in the heart may contribute to the development of myocardial fibrosis.
MicroRNAs Involved in Neurohormonal Activation
Extensive clinical and experimental data support the point of view that heart failure progresses as a result of the overexpression of biologically active molecules (collectively referred to as neurohormones) that are capable of exerting deleterious effects on the heart and the peripheral circulation.13 The portfolio of biologically active molecules that have been described thus far include those that belong to the adrenergic nervous system (norepinephrine) and renin angiotensin system (RAS [angiotensin II and aldosterone]), which are responsible for maintaining cardiac output through increased retention of salt and water, peripheral arterial vasconstriction and increased contractility, as well as inflammatory mediators (tumor necrosis factor, interleukin-1 and interleukin-6) that are responsible for cardiac repair and remodeling. Although the role of miRNAs in regulating the components of RAS and the adrenergic system is not well understood, several recent observations are worth noting. As discussed above, treating isolated neonatal cardiac myocytes with ligands that mimick neurohormonal activation in vitro, such as phenylephrine, endothelin-1, or angiotensin II is sufficient to up-regulate miRs that are linked to hypertrophic growth, including miR 21, 23a and miR-133.7, 9, 10 As discussed below, miRNAs may also contribute to “neurohormonal activation” through enhanced RAS signaling.
miR-155
miR-155 is sufficient to suppress the levels of the type I angiotensin receptor (AT1R) in primary human lung fibroblasts by binding directly to the 3′-untranslated region of AT1R mRNA. Functional studies demonstrated that transfection of miR-155 into human primary lung fibroblasts resulted in a significant reduction in Ang II-induced activation of the extracellular signal-related kinase ½ (ERK1/2). Furthermore, when fibroblasts were transfected with an antisense inhibitor to miR-155, endogenous AT1R expression and angiotensin II-induced ERK1/2 activation were increased significantly.32 Interestingly, a silent polymorphism (+1166 A/C) in the human AT1R gene that is associated with cardiovascular disease (possibly due to enhanced AT1R activity) is a target of miR-155. When the +1166 C-allele is present, base-pairing complementarity to the 3′-untranslated region of the AT1R is interrupted, thereby decreasing the ability of miR-155 to interact with the cis-regulatory site. The result is that miR-155 is no longer able efficiently attenuate the translation of the AT1R gene, leading to increased endogenous levels of AT1R and increased angiotensin II-induced activation of ERK1/2.33 Whether these observations have any relevance in the setting of heart failure is unknown, and will require further study.
mir-21
The role of miR-21 in cardiac hypertrophy and cell apoptosis has been discussed above. Of note, angiotensin II has been shown to up-regulate miR-21 expression in human adrenal cells.34 Moreover, overexpression of miR-21 resulted in increased aldosterone secretion and cell proliferation in these cells. Although performed in a cell type that resides outside the heart, this study raises the interesting possibility that increased miRNA-21 expression levels outside of the heart may contribute to adverse cardiac remodeling through dysregulation of angiotensin-II mediated signaling and enhanced aldosterone secretion.
MicroRNAs Involved in Electrophysiological Alterations
In addition to cardiac remodeling and neurohormonal activation, there are important changes in ion channel function and expression in the failing heart that lead to alterations in the electrical phenotype of both atrial and ventricular myocytes. This “electrophysiological remodeling” renders the heart more vulnerable to ventricular arrhythmias that underlie sudden cardiac death.35 Although the exact role of microRNAs in the development of atrial and ventricular arrhythmias in the setting of clinical heart failure is unknown, the experimental literature suggests that cardiac specific microRNAs are sufficient to provoke cardiac arrhythmias.
miR-1 and miR-133
The pacemaker function of sino-atrial (SA), atrioventricular (AV and cells in the ventricular conducting system depends upon their ability to undergo diastolic depolarizations. The ionic mechanism underlying pacemaker depolarizations is governed by activation of the hyperpolarization-activated current (If).36 The HCN ion channel subunit gene family encodes hyperpolarization-activated cation channels that are permeable to Na+ and K+. It has been suggested that upregulation of HCN channels may contribute to enhanced automaticity and arrhythmias in heart failure.37 Relevant to the present discussion, down-regulation of miR-1 and miR-133 have been associated with increased protein levels of HCN2/HCN4 in hypertrophic hearts, whereas transfection of miR-1 and miR-133 into angiotensin II stimulated neonatal cardiac myocytes prevented overexpression of HCN2/HCN4.38 Given that miR-1 and miR-133 (Figure 2) are down-regulated in heart failure models, the significance of these studies with respect to IK1 and IKs and the development of cardiac arrhythmia in heart failure is uncertain. Changes in the inward rectifier K+ current (IK1) have been suggested to play an important role in the genesis of both atrial and ventricular arrhythmia.39, 40 In cardiac myocytes, IK1 establishes the resting membrane potential and modulates the terminal phase of repolarization. Transfection of miR-1 into healthy or infarcted rat hearts resulted in significant widening of the QRS complex, prolonged the QT interval and exacerbated arrhythmia, whereas knock-down of miR-1 in infarcted rat hearts using an antisense resulted in decreased arrhythmia.41 Importantly, miR-1 targets genes that encode for connexin 43 (GJA1) and the Kir2.1 potassium channel subunit (KCNJ2), which may partially explain the proarrhythmogenic potential of miR-1. miR-1 and miR-133 have been shown experimentally to target KCNE1 and KCNQ1, which are the genes that comprise the channels that regulates the slow delayed rectifier current (IKs). However, given that miR-1 and miR-133 are down-regulated in heart failure models, the significance of these studies with respect to IK1 and IKs and the development of cardiac arrhythmia in heart failure is uncertain.
CONCLUSION
In the foregoing discussion, we have reviewed the emerging importance of miRNAs in the failing heart. Although our understanding of miRNA biology in heart failure is embryonic at present, the experimental literature reviewed herein suggests that miRNAs contribute to adverse/pathological remodeling, and hence are sufficient to contribute to disease progression in heart failure. As shown in Figure 7, miRNAs regulate key components of the remodeling process, including cardiac myocyte biology, cell fate, extracellular matrix remodeling, and neurohormonal activation. Given that miRNAs are coordinately regulated in response to stress signals, and given that miRNAs regulate the expression levels of gene networks that determine the so-called “heart failure phenotype,” it is tempting to speculate that miRNAs, acting singly or in combination, may be responsible for modulating the transition from adaptive to pathological cardiac remodeling. Although predicting the success of future therapeutic strategies in the field of heart failure is fraught with difficulty, one can envision that miRNA biology may contribute to our understanding of heart failure in at least three important areas. First, as modern computational methodologies continue to improve, it is likely that studying the expression patterns of miRNAs will lead to the identification of interesting new disease targets that are not obvious with classic “pathway driven (e.g., neurohormonal)” approaches. Moreover, this type of strategy would, for the first time, engender therapeutic strategies that directly address the fundamental biology of heart failure, insofar as they would be directed at the targets that are responsible for cardiac remodeling. Second, expression profiling of miRNAs in circulating leukocytes and/or myocardial tissue may one day be used to help to identify personalized strategies for heart failure patients, as is currently being done in the field of oncology.42, 43 For example expression profiling of miRNAs might also be used to help in the earlier implantation and/or removal of circulatory assist devices if investigators are able to reliably determine miRNA signatures for worsening heart failure and/or myocardial recovery. Third, it is possible that certain miRNAs may themselves become may become therapeutic targets in heart failure. As discussed in depth elsewhere, 44–46 the use of chemically modified oligonucleotides to target specific miRNAs and/or to disrupt the binding between a specific miRNA and a specific mRNA target is now possible. Although this type of a therapeutic approach will be subject to many of the same challenges that have plagued gene therapy in terms of modes of delivery and toxicity, it is nonetheless completely feasible. Targeted miRNA approaches may also prove challenging because of the potential for a myriad of “off-target” effects with this approach. As with all things in heart failure, progress in this field will require the collaborative efforts between basic scientists and clinical scientists in order to perform the requisite target validation and the careful clinical phenotyping that are required in order to move from the bench to the bedside. Given how little we know about miRNAs in heart failure at present, and given that there are seemingly an infinite number of interactions between miRNAs and their cognate target genes, some of which have yet to be characterized, it is almost certain that our learning curve will be extremely steep now and for the foreseeable future.
Figure 7.
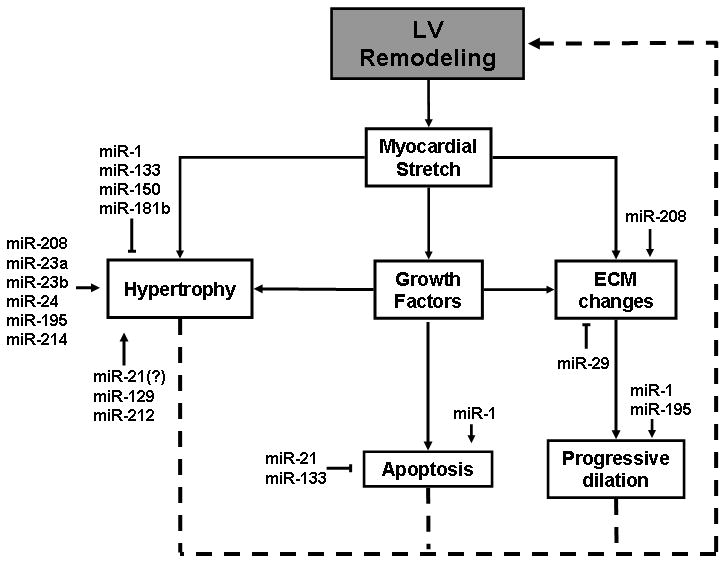
Role of miRNAs in LV remodeling. Candidate miRNAs that have either been reported or have suggested roles in cardiac remodeling are depicted in relation to the components of the remodeling process that they impact. Postulated mechanisms with limited evidence are represented by question marks.
Supplementary Material
Figure 5.
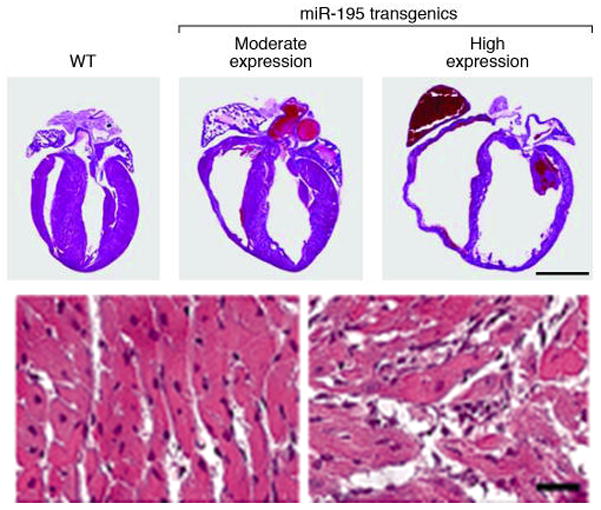
Cardiac-specific overexpression of miR-195 is sufficient to drive cardiomyopathy. Hematoxylin and eosin stained sections show that overexpression of miR-195 induces cardiac hypertrophic growth at 2 weeks of age that progresses to a dilated phenotype within 6 weeks. Moderate and high expression indicate transgenic mouse lines with 26- and 29-fold expression of miR-195 resulting in cardiac hypertrophy and dilated cardiomyopathy, respectively. The bottom two panels show a high magnification of the hematoxylin and eosin section, indicating severe myocyte disorganization in the miR-195 transgenic (TG) animals compared with wild-type (WT). (Modified with permission from van Rooij et al, PNAS, 2006;103:18255–60).
Acknowledgments
SOURCES OF FUNDING
This research was supported by research funds from the N.I.H. (UO1 HL084890-01 and RO1 HL58081, RO1 HL61543, HL-42250).
Footnotes
DISCLOSURES
Dr. Mann is a consultant for Miragen, Boulder Colorado.
References
- 1.van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117:2369–76. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism and function. Cell. 2008;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths-Jones S, Grocock RJ, van DS, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34 (Database issue):D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–9. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 6.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–24. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 7.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255–60. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–8. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 9.Cheng YH, Ji RR, Yue JM, Yang J, Liu XJ, Chen H, Dean DB, Zhang CX. MicroRNAs are aberrantly expressed in hypertrophic heart - Do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831–40. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137–41. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiological Genomics. 2007;31:367–73. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 12.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart - A clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–67. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 13.Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005;111:2837–49. doi: 10.1161/CIRCULATIONAHA.104.500546. [DOI] [PubMed] [Google Scholar]
- 14.Dash R, Kadambi V, Schmidt AG, Tepe NM, Biniakiewicz D, Gerst MJ, Canning AM, Abraham WT, Hoit BD, Liggett SB, Lorenz JN, Dorn GW, Kranias EG. Interactions between phospholamban and beta-adrenergic drive may lead to cardiomyopathy and early mortality. Circulation. 2001;103:889–96. doi: 10.1161/01.cir.103.6.889. [DOI] [PubMed] [Google Scholar]
- 15.Urabe Y, Hamada Y, Spinale FG, Carabello BA, Kent RL, Cooper G, IV, Mann DL. Cardiocyte contractile performance in experimental biventricular volume-overload hypertrophy. Am J Physiol. 1993;264:H1615–H1623. doi: 10.1152/ajpheart.1993.264.5.H1615. [DOI] [PubMed] [Google Scholar]
- 16.Anand IS, Liu D, Chugh SS, Prahash AJ, Gupta S, John R, Popescu F, Chandrashekhar Y. Isolated myocyte contractile function is normal in postinfarct remodeled rat heart with systolic dysfunction. Circulation. 1997;96:3974–84. doi: 10.1161/01.cir.96.11.3974. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–20. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 18.Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Invest. 1997;100:2362–70. doi: 10.1172/JCI119776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 20.Zhu SM, Si ML, Wu HL, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J Biol Chem. 2007;282:14328–36. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 21.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 22.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–88. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 23.Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H, Xiao J, Shan H, Wang Z, Yang B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci. 2007;120:3045–52. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- 24.Yang BF, Lin HX, Xiao JN, Lu YJ, Luo XB, Li BX, Zhang Y, Xu CQ, Bai YL, Wang HZ, Chen GH, Wang ZG. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nature Med. 2007;13:486–91. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 25.Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V, Perriard JC, Dewerchin M, Flameng W, Nagy A, Lupu F, Moons L, Collen D, D’amore PA, Shima DT. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med. 1999;5:495–502. doi: 10.1038/8379. [DOI] [PubMed] [Google Scholar]
- 26.de Boer RA, Pinto YM, van Veldhuisen DJ. The imbalance between oxygen demand and supply as a potential mechanism in the pathophysiology of heart failure: the role of microvascular growth and abnormalities. Microcirculation. 2003;10:113–26. doi: 10.1038/sj.mn.7800188. [DOI] [PubMed] [Google Scholar]
- 27.Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, Forster O, Quint A, Landmesser U, Doerries C, Luchtefeld M, Poli V, Schneider MD, Balligand JL, Desjardins F, Ansari A, Struman I, Nguyen NQ, Zschemisch NH, Klein G, Heusch G, Schulz R, Hilfiker A, Drexler H. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. 2007;128:589–600. doi: 10.1016/j.cell.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 28.Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79:581–8. doi: 10.1093/cvr/cvn156. [DOI] [PubMed] [Google Scholar]
- 29.Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–84. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–71. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Rooij E, Sutherland LB, Thatcher JE, DiMaio M, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–32. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin MM, Lee EJ, Buckenberger JA, Schmittgen TD, Elton TS. MicroRNA-155 regulates human angiotensin II type 1 receptor expression in fibroblasts. J Biol Chem. 2006;28:18277–84. doi: 10.1074/jbc.M601496200. [DOI] [PubMed] [Google Scholar]
- 33.Martin MM, Buckenberger JA, Jiang J, Malana GE, Nuovo GJ, Chotani M, Feldman DS, Schmittgen TD, Elton TS. The human angiotensin II type 1 receptor +1166 A/C polymorphism attenuates microrna-155 binding. J Biol Chem. 2007;282:24262–9. doi: 10.1074/jbc.M701050200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Romero DG, Plonczynski MW, Carvajal CA, Gomez-Sanchez EP, Gomez-Sanchez CE. Microribonucleic acid-21 increases aldosterone secretion and proliferation in H295R human adrenocortical cells. Endocrinology. 2008;149:2477–83. doi: 10.1210/en.2007-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nass RD, Aiba T, Tomaselli GF, Akar FG. Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat Clin Pract Cardiovasc Med. 2008;5:196–207. doi: 10.1038/ncpcardio1130. [DOI] [PubMed] [Google Scholar]
- 36.Accili EA, Proenza C, Baruscotti M, DiFrancesco D. From funny current to HCN channels: 20 years of excitation. News Physiol Sci. 2002;17:32–7. doi: 10.1152/physiologyonline.2002.17.1.32. [DOI] [PubMed] [Google Scholar]
- 37.Zicha S, Fernandez-Velasco M, Lonardo G, L’Heureux N, Nattel S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res. 2005;66:472–81. doi: 10.1016/j.cardiores.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Luo X, Lin H, Pan Z, Xiao J, Zhang Y, Lu Y, Yang B, Wang Z. Downregulation of MIRNA-1/MIRNA-133 contributes to re-expression of pacemaker channel genes HCN2 and HCN4 in hypertrophic heart. J Biol Chem. 2008;284:20045–20052. doi: 10.1074/jbc.M801035200. [DOI] [PubMed] [Google Scholar]
- 39.Samie FH, Berenfeld O, Anumonwo J, Mironov SF, Udassi S, Beaumont J, Taffet S, Pertsov AM, Jalife J. Rectification of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ Res. 2001;89:1216–23. doi: 10.1161/hh2401.100818. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Garratt CJ, Zhu J, Holden AV. Role of up-regulation of IK1 in action potential shortening associated with atrial fibrillation in humans. Cardiovasc Res. 2005;66:493–502. doi: 10.1016/j.cardiores.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 41.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486–91. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 42.Blower PE, Verducci JS, LIN S, Zhou J, Chung JH, Dai Z, Liu CG, Reinhold W, Lorenzi PL, Kaldjian EP, Croce CM, Weinstein JN, Sadee W. MicroRNA expression profiles for the NCI-60 cancer cell panel. Mol Cancer Ther. 2007;6:1483–91. doi: 10.1158/1535-7163.MCT-07-0009. [DOI] [PubMed] [Google Scholar]
- 43.Salter KH, Acharya CR, Walters KS, Redman R, Anguiano A, Garman KS, Anders CK, Mukherjee S, Dressman HK, Barry WT, Marcom KP, Olson J, Nevins JR, Potti A. An integrated approach to the prediction of chemotherapeutic response in patients with breast cancer. PLoS ONE. 2008;3:e1908. doi: 10.1371/journal.pone.0001908. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Krutzfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–92. doi: 10.1093/nar/gkm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 46.Xiao JN, Yang BF, Lin HX, Lu YJ, Luo XB, Wang ZG. Novel approaches for gene-specific interference manipulating actions of via microRNAS: Examination on the pacemaker channel genes HCN2 and HCN4. J Cell Physiol. 2007;212:285–92. doi: 10.1002/jcp.21062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.