

Abstract
The treatment of patients with invasive breast cancer remains a major issue because of the acquisition of drug resistance to conventional chemotherapy. Here we propose a new therapeutic strategy by combining DNA methyltransferase inhibitors (DMTIs) with suramin. Cytotoxic effects of suramin or combination treatment with DMTIs were determined in highly invasive breast cancer cell lines MDA-MB-231, BT-20 and HCC1954, or control cells. In addition, effects on cell invasion were determined in 3-dimensional cell culture assays. DMTI-mediated upregulation of Protein Kinase D1 (PKD1) expression was shown by Western blotting. Effects of suramin on PKD1 activity was determined in vitro and in cells. The importance of PKD1 in mediating the effects of such combination treatment in cell invasion was demonstrated using 3D cell culture assays. A proof of principal animal experiment was performed showing that PKD1 is critical for breast cancer growth. We show that when used in combination, suramin and DMTIs impair the invasive phenotype of breast cancer cells. We show that PKD1, a kinase that previously has been described as a suppressor of tumor cell invasion, is an interface for both FDA-approved drugs, since the additive effects observed are due to DMTI-mediated re-expression and suramin-induced activation of PKD1. Our data reveal a mechanism of how a combination treatment with non-toxic doses of suramin and DMTIs may be of therapeutic benefit for patients with aggressive, multi-drug resistant breast cancer.
Keywords: Breast cancer, Suramin, Methyltransferase inhibitors, Invasive phenotype, MDA-MB-231
Introduction
According to recent statistics, about 200,000 women in the United States will be diagnosed this year with invasive breast cancer which will be lethal for nearly 40,000 of them. Most of clinical treatments, including chemotherapy or hormonal therapy, are not very successful in targeting metastatic cells, and therefore, invasive breast cancer still remains highly lethal. Another reason for an increased death rate is acquisition of drug resistance to these therapies which leaves patient with advanced disease with limited treatment options. Therefore, a main focus for developing novel treatment strategies may be to target mechanisms that promote invasion and mediate drug resistance in combination with conventional chemotherapy.
Epigenetic events such as aberrant de novo methylation of gene promoters have recently been defined as a hallmark of human cancers and as such therapeutic strategies targeting these mechanisms currently are tested in several clinical trials [1, 2]. Among the compounds being tested, DNA methyltransferase inhibitors (DMTIs) such as decitabine (5-aza-2′-deoxycytidine) are able to reverse the silencing of tumor-suppressor genes such as TP53, ESR1, and PRKD1 and upregulate their expression [3–5]. In addition to decreasing promoter methylation in tumors cells, DMTIs can also act as cytotoxic agents by inducing cell cycle arrest and apoptosis, i.e., through the upregulation of p21 [3].
Chemoresistance of tumor cells can be mediated by many factors. For example, high expression of growth factors (GFs) such as aFGF and bFGF is observed in most cancer [6–11], and was associated with resistance to several chemotherapeutic agents [12–14]. Interestingly, suramin, a polysulfonyl naphtylurea, which was originally used for the treatment of sleeping sickness or other parasitic disease [15], is also able to block the binding of several GFs, including aFGF and bFGF, to their receptors [16–19]. Later it was shown that suramin can decrease tumor growth, by inducing tumor cell differentiation [20–22] and inhibiting cell proliferation [23, 24] and angiogenesis [12–14]. The different mechanisms mediating these anti-tumor effects of suramin highlighted its potential as a promising agent for tumor therapy and led to a phase I/II trial, in which suramin was combined with paclitaxel in metastatic breast cancer.
Protein kinase D1 (PKD1) is a serine/threonine kinase expressed in ductal epithelial cells of the normal breast where it prevents epithelial-to-mesenchymal transition and maintains the epithelial phenotype [4, 25–27]. PKD1 also has been shown to be a negative regulator of actin reorganization processes necessary for cell migration and invasion [28]. Consequently, PKD1 expression is lost during breast tumor progression to an aggressive metastatic phenotype [4], and this is mediated by hypermethylation and inactivation of its promoter [5]. A key function for PKD1 in regulating breast tumor cell invasiveness was demonstrated by comparing MCF-7 and MDA-MB-231 cells. Both represent bona fide cell lines for either non-invasive cells that endogenously express PKD1 (MCF-7) or highly invasive cells that do not express PKD1 due to PKD1 promoter methylation (MDA-MB-231) [5]. Moreover, a knockdown of PKD1 in MCF-7 cells led to an acquisition of invasiveness, whereas a re-expression of active PKD1 decreased the invasiveness of MDA-MB-231 cells [4], clearly showing the dependence of cell invasion on the absence of PKD1.
Using the highly invasive breast cancer cell lines MDA-MB-231 (TN, claudin low), BT-20 (TN) and HCC1954 (Her2+), we here show that PKD1 is the interface for both DMTIs and suramin. We found that DMTIs induced the re-expression of PKD1 but its activation status remained modest. When used in combination with suramin which induced an additional strong activation of PKD1 in vitro as well as in vivo, we observed a dramatic impact on the invasive phenotype. Our data predict that drug combinations leading to re-expression and increased activation of tumor suppressors such as PKD1 in highly invasive breast cancer cells (BC) represent new strategies for therapy.
Materials and methods
Cell lines, antibodies, and reagents
HeLa, MCF-10A, MCF-7, BT-20, HCC1954, and MDA-MB-231 were obtained from American Type Culture Collection ATCC (Manassas, VA), and HuMEC cells were from Invitrogen (Carlsbad, CA). HeLa, MCF-7, and MDA-MB-231 were maintained in DMEM with 10 % FBS. BT-20 were maintained in EMEM with 10 % FBS, 2 mM L-glutamine, 1.5 g/l sodium bicarbonate, 0.1 mM NEAA, and 1 mM sodium pyruvate. HCC1954 were maintained in RPMI with 10 % FBS. MCF-10A were maintained in DMEM/Ham F10 (50:50, v/v) with 5 % horse serum, 20 ng/ml EGF, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 10 μg/ml insulin, and 1 % penicillin/streptomycin. HuMEC cells were maintained in HMEC Culture System from Invitrogen. EGF was from Peprotech (Rocky Hill, NJ) and insulin and hydrocortisone from Sigma Aldrich (Saint Louis, MO). MDA-MB-231 cell lines stably expressing PKD1 or control were generated by transfection with pcDNA3 or pcDNA3-GFP-PKD1 plasmids (wildtype PKD1 or PKD1.KD (kinase-dead (KD) version; PKD1.K612W mutation)). Cell pools were selected for 14 days using G418 (100 μg/ml). MDA-MB-231 cells were transduced with scr-shRNA or PKD1-shRNA lentivirus to generate stable cell lines. After infection cell pools were selected with Puromycin (1 μg/ml) for 14 days, Decitabine (5-aza-2′-deoxycytidine) and RG108 (2-1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-3-(1H-indol-3-yl)propionic acid) were purchased from Millipore (Billerica, MA). Suramin, Caetocin, and Curcumin were from Sigma Aldrich. Puromycin was from InvivoGen (San Diego, CA) and G418 from Millipore. The monoclonal antibody specific for PKD1 was described in detail previously [5]. Anti-β-actin and anti-HA antibodies were from Sigma Aldrich, anti-pS910-PKD from Cell Signaling Technology (Danvers, MA), and anti-pS738/742-PKD from Abcam (Cambridge, MA). Secondary HRP-linked antibodies were from Roche (Indianapolis, IN).
DNA constructs, shRNA constructs, and production of lentivirus
The expression plasmids for HA-tagged PKD1 and a KD PKD1 version (PKD1.K612W mutant) were described before [29, 30]. The specific lentiviral expression construct for shRNA targeting human PKD1 is commercially available from Sigma (NM_002742.x -2498s1c1, SHDNA MISSION® shRNAPlasmid DNA; St Louis, MO). Lentivirus was produced in Hek293FT cells using the ViraPower Lentiviral Expression System (Life technologies, Carlsbad, CA).
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Cellular RNA was isolated using RNA-Bee (TEL-TEST, Friendswood, TX) according to the manufacturer’s instructions, and mRNA was transcribed into cDNA using Superscript II (Invitrogen). For the transcription reaction, 1 μg Oligo dT(18) primer (New England Biolabs, Beverly, MA) and 1 μg RNA were incubated in a total volume of 10 μl H2O at 70 °C for 10 min. 5× buffer, 40 U RNAsin (Roche, Mannheim, Germany), 200 μM dNTP (NEB), 10 mM DTT, and 300 U Superscript II reverse transcriptase were added to a total volume of 20 μl. The reaction was carried out at 45 °C for 60 min and then heat inactivated at 95 °C for 5 min. The resulting cDNA pool was subjected to PCR analysis using specific primer sets. Primers for human PKD1 were TTCTTCCACCTCAGGTCATC and TGCCAGAGCACATAACGAAG; PKD2 primers were CAACCCACACTGCTTTGAGA and CACACAGCTTCACCTGAGGA; PKD3 primers were TCATTGACAAACTGCGCTTC and GTACATGATCACGCCCACTG. Primers for actin were CCTCGCCTTTGCCGATCC and GGATCTTCATGAGGTAGTCAGTC. Reaction conditions for the PCR reaction were 1 min annealing at 55 °C and 1 min amplification at 72 °C with 20, 35, and 40 cycles.
Protein extraction from formalin-fixed paraffin-embedded (FFPE) tissue
Protein extraction from FFPE tissue was performed as previously described by Guo et al. [31]. In brief, eight 10 μm sections each FFPE tissue sample were deparaffinized in an eppendorf tube using 500 μl xylene for 10 min at RT. The samples were centrifuged at 14,000×g for 3 min, and the xylene deparaffinization step was repeated a second time. Rehydration was performed stepwise using different percentages of ethanol at 100, 95, 90, and 75 %, each for 10 min. Each step was performed in a volume of 500 μl at RT. Centrifugation to pellet the tissue after each step was performed at 14,000×g at RT for 3 min. After the last centrifugation, the ethanol was removed and the tissue re-suspended in 20 mM Tris–HCl buffer pH 9 plus 2 % SDS. Samples then were incubated on ice for 5 min, vortexed, boiled at 100 °C for 20 min, and then incubated at 80 °C for 2 h. The insolubilized material was pelleted at 14,000×g for 20 min at RT and discarded. Lysates then were normalized to 1 mg/ml protein and analyzed by immunostaining.
Cell lysates, immunoprecipitation, and immunostaining
Cells were lysed in lysis buffer (50 mM Tris/HCl pH 7.4, 1 % TritonX-100, 150 mM NaCl, and 5 mM EDTA) plus Protease Inhibitor Cocktail (Sigma), and either lysates were used for Western Blot analysis or proteins of interest were immunoprecipitated (1 h incubation with the respective antibody (2 μg) followed by a 30 min incubation with protein A/G-agarose (Santa Cruz), all at 4 °C). Immune-complexes were washed five times with TBS (50 mM Tris/HCl pH 7.4, 150 mM NaCl), resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunostaining.
In vitro kinase assay
In vitro kinase assays were performed using 250 ng of purified PKD1 (Millipore) per sample combined with the indicated amount of suramin in a total volume of 30 μl FJ kinase buffer (50 mM Tris pH 7.4, 10 mM MgCl2, and 2 mM DTT) supplemented with 100 μM ATP. The kinase reaction (30 min, RT) was stopped by adding 2× Laemmli buffer.
Cell viability assay (MTT)
Cells were seeded in 96-well plates and stimulated the next day as indicated. To measure cell viability, 10 μl of MTT in PBS (5 mg/ml) were added per well. Cells were incubated at 37 °C for 4 h and then 100 μl solubilization solution (10 % SDS in 0.001 M HCl) was added over night. Plates were analyzed by reading at 600 nm using a SynergyHT plate reader (Bio Tek Instruments, Winooski, VT).
Multicellular spheroids/3D cell culture assay
3D analysis of morphology was performed as described previously [4]. In brief, cell culture dishes (24 well plates) were pre-coated with undiluted phenol red-free Matrigel (10 mg/ml). 104 cells (per well of a 24 well plate) were suspended in a volume of 200 μl PBS and mixed with 100 μl of cold Matrigel (10 mg/ml). The cell suspension was added dropwise over the bottom layer to cover it. After the cell layer was set complete, culture media was added over the top. Media was changed every 2 days, without disturbing the cell/matrix layer. Photos were taken after indicated days using a 10× magnification for an overview and 40× to document 3D phenotype.
Orthotopic animal model
All animal experiments were performed under protocols (A15207 and A14810) approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC). Seven mice (female, nu/nu) per group were anesthetized, and breast carcinoma cell clones were injected subcutaneously into the mammary gland fat pad in the upper thorax region between the clearly visible nipples of the 2nd and the 3rd mammary glands on the right side of the animal. For each injection, a total of 106 cells, washed three times with PBS and mixed with 30 μl of extracellular matrix (complete Matrigel without phenol red, BD Biosciences) were used. Tumor growth was continuously monitored (starting from week 2) using a caliper. At week 5 (end point), primary tumors were removed, and tumor weight and volume were determined. Tumor volume was determined using a caliper and the formula length × width × height × π/6. Tumors then were fixed with formalin and embedded in paraffin for further immunohistochemical analysis.
Immunohistochemistry
Slides were deparaffinized (1 h, 60 °C), de-waxed in xylene (five times for 4 min), and gradually re-hydrated with ethanol (100, 95, 75 %, and each two times for 3 min). The re-hydrated samples were rinsed in water and subjected to antigen retrieval in 10 mM sodium citrate buffer (pH 6.0) as described by the manufacturer (DAKO, Carpinteria, CA). Slides were treated with 3 % H2O2 (5 min) to reduce endogenous peroxidase activity, washed with PBS containing 0.5 % Tween 20, and blocked with protein block serum-free solution (DAKO) for 5 min at room temperature. Samples were stained with H&E or anti-PKD1 antibody (dilution: 1:2,500) in Antibody Diluent Background Reducing Solution (DAKO) and visualized using the Envision Plus Dual Labeled Polymer Kit (DAKO) according to the manufacturer’s instructions. Images were captured using the ScanScope XT scanner and ImageScope software (Aperio, Vista, CA).
Statistical analysis
GraphPad Prism version 4.0c (GraphPad Prism Software, Inc.) was used for all statistical analyses. Statistical significance was determined using an unpaired t test. For all analyses, P ≤ 0.05 was considered statistically significant.
Results
A combination treatment with decitabine and suramin is non-toxic to cells, but decreases the invasive potential of breast cancer cells
DMTIs such as decitabine can have inhibitory effects on breast cancer cell invasion by upregulating tumor-suppressor genes including TP53, ESR1, and PRKD1 [3–5]. Suramin previously had been shown to decrease proliferation of breast cancer cells when applied at high doses (i.e., EC50 = 50 μM for MCF-7) [32]. However, when applied at high doses, suramin is too toxic for patient treatment. On the other hand low-dose suramin treatment, although not effective on its own, had been shown to improve the activity of paclitaxel in animal models [33]. Based on this, we tested a combination of suramin and DMTIs on breast cancer cell viability and invasiveness. We first tested if suramin affects the viability of normal (HuMEC and MCF-10A) or breast cancer cells (MCF-7, MDA-MB-231, BT-20, and HCC1954). At a dosage up to 30 μM, suramin had neither growth inhibitory effects nor did it induce cell death (Fig. 1a, b, c). We also tested combination treatment with suramin and the DMTIs decitabine or RG108 in the highly invasive breast cancer cell lines (MDA-MB-231, BT-20, and HCC1954), but also did not find any growth inhibitory or toxic effects (Fig. 1b, c).
Fig. 1.

A combination treatment with decitabine and suramin decreases the invasive potential of MDA-MB-231 cells, but is non-toxic to cells. a HuMEC, MCF-10A, or MCF-7 cells were treated with suramin. After 48 h, a MTT assay was performed. b, c MDA-MB-231 (b) and BT-20 or HCC1954 (c) cells were treated with suramin at indicated concentrations, either alone or in combination with the DNA methyltransferase inhibitors Decitabine (25 μM) or RG108 (300 nM). After 48 h, a MTT assay was performed. d BT-20 or HCC1954 cells were seeded in Matrigel 3D culture and treated with control vehicle or Decitabine (5 μM). After 48 h, cells were additionally treated with suramin (1 μM) as indicated. Treatment as indicated was repeated every other day over a time period of 9 days. Representative cell clusters are shown. The bar indicates 25 μm. e MDA-MB-231 cells were seeded in Matrigel 3D culture and treated with control vehicle or Decitabine (5 μM). After 48 h, cells were additionally treated with suramin (1 μM) as indicated. Treatment as indicated was repeated every other day over a time period of 12 days. Representative cell clusters are shown. The bar indicates 25 μm. All experiments shown in a–c were independently performed three times with identical results
Eventually, we determined the effects of combination treatment on the invasiveness of cells (MDA-MB-231, BT-20, and HCC1954) in 3D cell culture. While decitabine or suramin alone in this setting had no significant inhibitory effects, a combination of both compounds effectively blocked cell invasion into the surrounding extracellular matrix (Fig. 1d, e). Similar as observed in the cytotox studies, tumor cell proliferation was not affected.
Combination treatment induces PKD1 re-expression and activation in MDA-MB-231 cells
Our next focus was on identifying a molecular mechanism underlying the decrease in cell invasion induced by combination treatment. Recently, we have shown that loss of PKD1 expression correlates with invasiveness of breast cancer cells and human breast cancer tissue and have identified promoter hypermethylation as the cause for decreased PKD1 expression [4, 5]. Accordingly, highly invasive MDA-MB-231, BT-20, and HCC1954 cells do not express PKD1, whereas non-motile MCF-7 cells express PKD1 (Fig. 2a, b). Consequently, treatment of cells with the DMTIs decitabine or RG108 restored PKD1 expression (Fig. 2c, d). We also determined the activity of PKD1 re-expressed with this strategy using a phospho-site specific antibody for the phosphorylation status at the activation loop serines S738 and S742 (Fig. 2d). The phosphorylation of PKD1 in these serine residues in the activation loop is mediated by PKC and has been shown to be prerequisite for its activation [34]. However, although DMTIs can induce PKD1 expression, the activity of the re-expressed kinase was only moderate after such treatment (Fig. 2d).
Fig. 2.

DNA methyltransferase inhibitors induce PKD1 re-expression in MDA-MB-231 cells. a Total RNA from BT-20, HCC1954, MCF-7, and MDA-MB-231 cells was analyzed for PKD1, PKD2, and PKD3 mRNA expression using RT-PCR. b MCF-7 and MDA-MB-231 cells were analyzed for PKD1 expression by immunoprecipitation for endogenous PKD1 (anti-PKD1 antibody). Samples were resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunoblot for PKD1 using an anti-PKD1 antibody. c MDA-MB-231 cells were treated with decitabine (10 μM) for indicated times. Endogenous PKD1 was immunoprecipitated (anti-PKD1 antibody). Samples were resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunoblot for PKD1 using an anti-PKD1 antibody. Lysates also were analyzed by Western blot for expression of β-actin. d MDA-MB-231 cells were treated with RG108 (250 nM, 24 h). Endogenous PKD1 was immunoprecipitated (anti-PKD1 antibody). Samples were resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunoblot for PKD1 activation loop phosphorylation using the pS738/742-PKD antibody. Blots were stripped and re-probed for total PKD1 using an anti-PKD1 antibody. All experiments shown in a–d were independently performed three times with identical results
Suramin, a symmetric molecule that contains 8 benzene rings (Fig. 3a), had been shown to activate PKC isoforms in vitro by unknown mechanisms [35]. In an in vitro kinase assay, in which we used recombinant PKD1 and measured PKD1 autophosphorylation at residue S910, we found that suramin at approximately 5–10 μM is also an activator of PKD1 (Fig. 3b). Phosphorylation at this residue has been shown to serve as a measure of PKD1 activity [36]. In such an in vitro setting, suramin-induced maximum PKD1 activity at 5 μM. However, activity dropped at high concentrations of suramin. This somewhat unexpected effect may be explained by the negative charged nature of suramin which may interfere with the in vitro kinase assay. We next tested if a more significant and prolonged activation can be obtained in cells in which suramin is expected to activate both, PKC and PKD1. Therefore, we first ectopically expressed PKD1 in cells and determined an optimal dose for PKD1 activation (Fig. 4a). The PKD1 activation status was determined by phosphorylation of its activation loop serines S738/S742, which is mediated by PKC [34]. The dose of suramin needed for maximal activation of PKD1 was below the dose needed for PKD1 activation in vitro. This is not surprising since suramin in cells can also act as an activator of PKC enzymes, which are upstream kinases for PKD activation. Using suramin at 1 μM, we then performed a time kinetic over a period of 30 min and found maximum PKD1 activity at 5–10 min of treatment (Fig. 4b). Using MCF-7, we found that suramin also increases activation loop phosphorylation and activity of endogenous PKD1 (Fig. 4c).
Fig. 3.
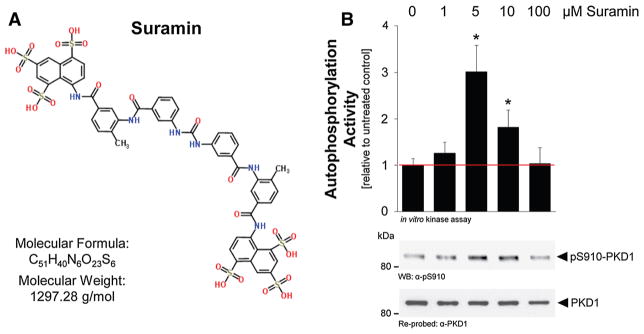
Suramin is a direct activator of PKD1 in vitro. a Structure, molecular formula, and molecular weight of suramin. b Purified PKD1 was incubated with suramin at indicated concentrations, and an in vitro kinase assay was performed. PKD1 activity was measured using the pS910-PKD antibody (directed against the PKD1 autophosphorylation site) in Western blots. Blots were stripped and re-probed for total PKD1 using an anti-PKD1 antibody. P values were acquired with the student’s t test using Prism v5 software. Asterisks indicate extreme statistical significance, P ≤ 0.005. This experiment was independently performed three times with identical results
Fig. 4.

Suramin is an activator of PKD1 in cells. a, b HeLa cells, were transfected with wildtype, HA-tagged PKD1 and then treated with suramin at indicated concentrations (1, 10 μM), or a time course was performed (1 μM, 0–30 min). Ectopically expressed PKD1 was immunoprecipitated (anti-HA). Samples were resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunoblot for PKD1 activation loop phosphorylation using the pS738/742-PKD antibody. Blots were stripped and re-probed for total PKD1 using an anti-PKD1 antibody. c MCF-7 cells were treated with suramin as indicated. Endogenous PKD1 was immunoprecipitated (anti-PKD1 antibody). Samples were resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunoblot for PKD1 activation loop phosphorylation using the pS738/742-PKD antibody. Blots were stripped and re-probed for total PKD1 using an anti-PKD1 antibody. d MDA-MB-231 cells were treated as indicated with vehicle control or decitabine (10 μM, 48 h) and then additionally stimulated with suramin (1 μM, 10 min). Endogenous PKD1 was immunoprecipitated (anti-PKD1 antibody). Samples were resolved by SDS-PAGE, transferred to nitrocellulose, and analyzed by immunoblot for PKD1 activation loop phosphorylation using the pS738/742-PKD antibody. Blots were stripped and re-probed for total PKD1 using an anti-PKD1 antibody. All experiments shown in a–d were independently performed three times with identical results
Finally, we tested that if combination treatment with decitabine and suramin can lead to the upregulation of PKD1 expression (decitabine effect) as well as increased activity (suramin effect) in MDA-MB-231 cells. MDA-MB-231 cells re-expressed PKD1 to physiological levels when treated with decitabine alone. Although such re-expression already resulted in minimal PKD1 activity (measured by activation loop phosphorylation by its upstream kinases), additional treatment with suramin further increased PKD1 activity (Fig. 4d). Taken together, a possible explanation for the significant effects of the decitabine/suramin combination treatment on cell invasion could be that decitabine upregulates PKD1 expression and suramin leads to increased activity. This also would explain why treatment with the individual drugs is relatively ineffective.
Combination treatment affects invasiveness of cells through PKD1
We next determined if the anti-invasive effects observed after combination treatment in 3D culture are mediated through upregulation and re-activation of PKD1. To test this, we stably infected MDA-MB-231 cells with PKD1-shRNA. Although MDA-MB-231 cells due to promoter hypermethylation do not express PKD1 mRNA [5], the presence of PKD1-shRNA prevents PKD1 mRNA re-expression when the PKD1 gene is reactivated by inhibition of DNA methyltransferases. By comparing PKD1-shRNA to control cells (scr-shRNA), PKD1-specific effects on cell fate can be determined. In this experiment, instead of using decitabine, we used RG108 since, unlike decitabine, it has been shown to directly interact with DNA methyltransferases. RG108 was used in combination with suramin at two different doses (1 and 10 μM), and effects on cell growth in 3D Matrigel culture were determined. In control cells, as predicted, combination treatment with both compounds blocked cell invasion (Fig. 5, top row). In cells where PKD1 re-expression is prevented by the presence of PKD1-shRNA, the combination of both drugs was ineffective on cell invasion (Fig. 5, bottom row). This suggests that effects observed on tumor cell invasiveness are indeed due to re-activation of PKD1.
Fig. 5.
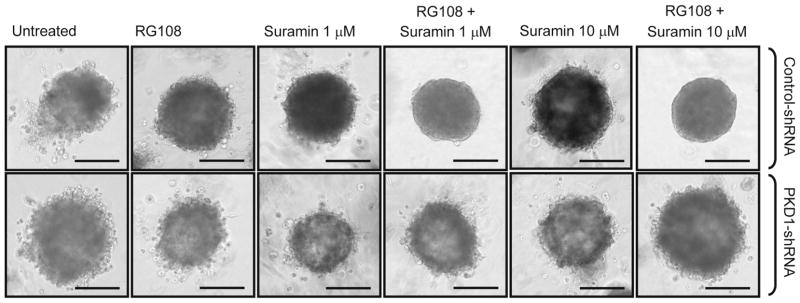
Combination treatment blocks invasiveness of cells through PKD1. MDA-MB-231 cells stably infected with control shRNA (scr-shRNA) or PKD1-shRNA (combination of two different previously validates shRNAs specific for PKD1) were seeded in Matrigel 3D culture and then treated with control vehicle or RG108 (5 μM). 48 h after initial treatment, cells were additionally treated with suramin (1 or 10 μM) as indicated. Treatment was performed every other day over a time period of 4 weeks. Representative cell clusters are shown. The bars indicate 200 μm. This experiment was independently performed three times with identical results
Expression of PKD1 in MDA-MB-231 cells decreases their invasiveness and inhibits orthotopic tumor growth
As a proof of principle, we next tested if MDA-MB-231 cells expressing PKD1 indeed show a similar phenotype in 3D culture than cells obtaining the combination treatment of DNA methyltransferase inhibitor and suramin. Therefore, we generated MDA-MB-231 cells stably expressing PKD1 or vector control. When analyzing these cell lines in 3D Matrigel culture assays, we observed a similar non-invasive phenotype, when PKD1 was expressed as obtained by the combination treatment (Fig. 6a, compare to Figs. 1e, 5). Over the time period of the assay, stable re-expression of PKD1 in MDA-MB-231 cells had no effect on their proliferation (not shown). Previously, we and others have shown that effects obtained in such 3D culture reflect effects obtained for orthotopic tumor growth in animal models [37]. Indeed, in accordance with the phenotype observed in 3D culture, in mouse orthotopic xenografts, PKD1-expressing MDA-MB-231 cells developed smaller, more encapsulated primary tumors with significant decreases in tumor volume and weight than vector control cells or cells expressing a PKD1.KD version of PKD1 (Fig. 6b, c). This most likely is due to decreased ability for local invasion. Immunohistochemical analysis indicated that PKD1 and mutant were similarly expressed (Fig. 6d, e). The significant effects observed in absence of an additional PKD1 activator are most likely due to activation of overexpressed PKD1 by upstream kinases. Since antibodies currently available to detect PKD1 activity cannot be used for IHC, because they also recognize other active PKD isoforms (anti-S738/742 also cross-reacts with PKD2 and PKD3; anti-S910 cross-reacts with PKD2), we were not able to proof this in formalin-fixed tumor samples. Therefore, we performed a protein extraction from FFPE tissue and performed Western blotting analysis for total PKD1. We found that overexpressed wildtype PKD1 as compared to the PKD1.KD version showed a shift in migration in the gel (see arrows in Fig. 6e), indicating phosphorylation-mediated activity. However, this does not necessarily mean that restoring PKD1 expression to physiological levels with DMTI will lead to full activation of PKD1 in vivo, and therefore, an additional activator such as suramin may be very well needed.
Fig. 6.

PKD1 expressing breast cancer cells show no invasive behavior and develop smaller primary tumors. a MDA-MB-231 cells stably expressing vector control or wildtype PKD1 were seeded in Matrigel 3D culture. Cell morphology was analyzed at day 20. Representative cell clusters are shown. The bars indicate 250 μm. Western blots for total PKD1 were performed using an anti-PKD1 antibody. Equal loading of the gel was controlled by staining for β-actin (anti-β-actin antibody). Experiments shown were independently performed three times with identical results. b–e MDA-MB-231 cells stably expressing vector control (labeled: Control), wildtype PKD1 (labeled: PKD1) or kinase-dead (K612W mutation) PKD1 (labeled: PKD1.KD) were orthotopically injected into the mfp of seven mice per experimental group (n = 7). At week 5 primary tumors were removed, photographed (B), and volume and weight (C) determined as described in the Materials and methods section. Asterisks indicate extreme statistical significance between Control and PKD1-expressing tumors (P = 0.0,002 for tumor volume; P<0.0,001 for tumor weight). d Immunohistological analysis. Primary tumors were fixed with formalin and either H&E stained or analyzed by IHC for expression of PKD1 and PKD1.KD using anti-PKD1 antibody. e Western blot (anti-PKD1 antibody) for ectopically expressed PKD1 after protein extraction from FFPE tumor tissue. The shifting of wildtype PKD1 (as compared to PKD1.KD) indicates activity. The animal experiments were performed twice with identical results
In summary, our data show that a combination treatment of DMTIs and suramin can be effective in inhibiting breast cancer cell invasion. A potential effector of such treatment is PKD1, since we clearly demonstrate an inhibitory function of PKD1 on tumor formation and invasiveness in these cells. Our data also implicate that chemically induced re-expression and re-activation of PKD1 could be a strategy to decrease the invasiveness of tumor cells.
Discussion
Suramin is a FDA-approved drug used for the treatment of sleeping sickness and other parasitic diseases. Although also showing anti-tumorigenic effects, initial clinical studies prevented the use of suramin for the treatment of cancer. Phase I/II clinical studies indicated high toxicity [38–40], as well as lack of significant therapeutic benefits when used in combination with conventional chemotherapy [41–44]. However, recent data suggests that the potential use of suramin for tumor therapy needs to be revisited since several studies have established that a non-cytotoxic dose of suramin not only prevents growth factor-induced drug resistance but also increases anti-tumor effects induced by chemotherapy in vitro in tumor cells [45]. Anti-tumor effects of suramin were also observed in xenografts of prostate, lung, and breast cancer cells, when used in combination with cytotoxic agents or chemotherapy [12, 33, 46, 47].
Here, we propose a new approach to improve the therapeutic benefits of suramin by using it in combination with DMTIs such as decitabine or RG108. Epigenetic modifications including promoter-specific DNA methylation have been shown to play a key role in tumor progression, and the interest in using epigenetic modulators as potential therapeutic agents for the treatment of cancer has increased greatly. At low doses, DMTIs induce the re-expression of tumor-suppressor genes such as TP53, CDKN1A, or ESR1 [48, 49] and can promote differentiation of tumor cells in vitro [50, 51]. We recently have shown that DMTIs also lead to re-expression of PKD1, a kinase that negatively affects directed cell migration and invasion in breast cancer cells [4, 5].
Here, we show that low dose of suramin used alone or in combination with DMTIs is not toxic to cells. However, combination treatment of suramin and RG108 impairs the invasive potential of MDA-MB-231, BT-20, and HCC1954 in 3D culture, whereas suramin or RG108 alone have no visible effect (Figs. 1, 5). These results are in accordance with a previous study showing that suramin used as a single agent does not affect tumor growth of breast tumor xenografts [33]. With identifying PKD1 as an interface between both compounds, we define one possible molecular mechanism underlying the anti-tumor effect of suramin in combination with DMTIs. Previous studies have shown that PKD1 is expressed in ductal epithelial cells of the normal breast where it maintains the epithelial phenotype by upregulating the expression of the adhesion protein E-cadherin [25]. Other functions are inhibiting the expression of several matrix metalloproteinases [4], as well as directed cell migration [52–56]. Consequently, loss of PKD1 was observed in osteosarcoma, advanced gastric, prostate, and breast cancers and has been associated with increased invasiveness and metastasis [4, 26, 27, 57, 58]. Silencing of PKD1 in invasive gastric and breast cancer is mediated by hypermethylation of its gene promoter [5, 57]. MDA-MB-231 as well as BT-20 and HCC1954 represent bona fide cell models for invasive breast cancer. These cells also do not express PKD1 due to promoter methylation, and treatment with DMTIs (decitabine, RG108) lead to PKD1 re-expression (Fig. 2). Although re-expressed at physiological levels, PKD1 activity remained rather modest (Fig. 2d). Suramin on the other hand has been shown to be an activator of PKD and PKC enzymes [35], and suramin in an in vitro kinase assay directly activated PKD1 (Fig. 3). In MDA-MB-231 cells, when combined with decitabine, suramin further increased PKD1 activity (Fig. 4), as did other PKD activators such as curcumin (not shown [59, 60]). Combination treatment with RG108 and suramin effectively decreased the invasiveness of MDA-MB-231, BT-20, and HCC1954 cells growing in 3D cell culture, whereas both drugs alone were ineffective (Figs. 1d, e, 5). Moreover, these effects were dependent on the presence of PKD1, since cells expressing PKD1-shRNA were affected in their invasiveness (Fig. 5, bottom row). In these cells, the presence of PKD1-shRNA prevents DMTI-induced upregulation of PKD1 mRNA and protein. Due to their decreased invasiveness when orthotopically implanted in the mammary fat pad in mice, MDA-MB-231 cells expressing PKD1 develop smaller tumors (Fig. 6). A next step now, beyond the scope of this study, is to decipher the optimal conditions of DMTI/suramin combination therapy in mice by further assessing the tolerability as well as the anti-tumor efficiency; and then to translate this for clinical trials.
Taken together, the results of this study suggest a potential therapeutic benefit of a combinatorial therapy using FDA-approved DMTIs with suramin. Such treatment may revert the invasive phenotype of breast cancer cells—similar as shown here for MDA-MB-231, BT-20, and HCC1954 cells.
Acknowledgments
This work was supported by grants from the NIH (GM086435) and the Bankhead-Coley Program of the Florida Department of Health (1BG11). Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under award number P50CA116201. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We also thank the Luther and Susie Harrison Foundation for their support, Irene K. Yan for technical assistance, and Alicia Fleming for help with the manuscript.
Abbreviations
- BC
Breast cancer
- DMTI
DNA methyltransferase inhibitor
- EMT
Epithelial-to-mesenchymal transition
- FFPE
Formalin-fixed paraffin-embedded
- GF
Growth factor
- IHC
Immunohistochemistry
- Mfp
Mammary fat pad
- MMP
Matrix metalloproteinase
- PKC
Protein kinase C
- PKD
Protein kinase D
- RT
Room temperature
- TN
Triple negative
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenet. 2013;5:3. doi: 10.1186/1868-7083-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh V, Sharma P, Capalash N. DNA methyltransferase inhibitors as epigenetic therapy for cancer. Curr Cancer Drug Targets. 2013;13:379–399. doi: 10.2174/15680096113139990077. [DOI] [PubMed] [Google Scholar]
- 3.Karpf AR, Moore BC, Ririe TO, Jones DA. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2′-deoxycytidine. Mol Pharmacol. 2001;59:751–757. [PubMed] [Google Scholar]
- 4.Eiseler T, Doppler H, Yan IK, Goodison S, Storz P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009;11:R13. doi: 10.1186/bcr2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borges S, Doppler H, Perez EA, Andorfer CA, Sun Z, et al. Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res. 2013;15:R66. doi: 10.1186/bcr3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chopin DK, Caruelle JP, Colombel M, Palcy S, Ravery V, et al. Increased immunodetection of acidic fibroblast growth factor in bladder cancer, detectable in urine. J Urol. 1993;150:1126–1130. doi: 10.1016/s0022-5347(17)35705-1. [DOI] [PubMed] [Google Scholar]
- 7.Cronauer MV, Hittmair A, Eder IE, Hobisch A, Culig Z, et al. Basic fibroblast growth factor levels in cancer cells and in sera of patients suffering from proliferative disorders of the prostate. Prostate. 1997;31:223–233. doi: 10.1002/(sici)1097-0045(19970601)31:4<223::aid-pros3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 8.Ravery V, Jouanneau J, Gil Diez S, Abbou CC, Caruelle JP, et al. Immunohistochemical detection of acidic fibroblast growth factor in bladder transitional cell carcinoma. Urol Res. 1992;20:211–214. doi: 10.1007/BF00299719. [DOI] [PubMed] [Google Scholar]
- 9.Ropiquet F, Giri D, Kwabi-Addo B, Mansukhani A, Ittmann M. Increased expression of fibroblast growth factor 6 in human prostatic intraepithelial neoplasia and prostate cancer. Cancer Res. 2000;60:4245–4250. [PubMed] [Google Scholar]
- 10.Singh RK, Bucana CD, Gutman M, Fan D, Wilson MR, et al. Organ site-dependent expression of basic fibroblast growth factor in human renal cell carcinoma cells. Am J Pathol. 1994;145:365–374. [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki K, Tokue A, Kamiakito T, Kuriki K, Saito K, et al. Predominant expression of fibroblast growth factor (FGF) 8, FGF4, and FGF receptor 1 in nonseminomatous and highly proliferative components of testicular germ cell tumors. Virchows Arch. 2001;439:616–621. doi: 10.1007/s004280100437. [DOI] [PubMed] [Google Scholar]
- 12.Bernsen HJ, Rijken PF, Peters JP, Bakker JH, Boerman RH, et al. Suramin treatment of human glioma xenografts; effects on tumor vasculature and oxygenation status. J Neurooncol. 1999;44:129–136. doi: 10.1023/a:1006363215260. [DOI] [PubMed] [Google Scholar]
- 13.Bhargava S, Hotz B, Hines OJ, Reber HA, Buhr HJ, et al. Suramin inhibits not only tumor growth and metastasis but also angiogenesis in experimental pancreatic cancer. J Gastrointest Surg. 2007;11:171–178. doi: 10.1007/s11605-006-0081-z. [DOI] [PubMed] [Google Scholar]
- 14.Danesi R, Del Bianchi S, Soldani P, Campagni A, La Rocca RV, et al. Suramin inhibits bFGF-induced endothelial cell proliferation and angiogenesis in the chick chorioallantoic membrane. Br J Cancer. 1993;68:932–938. doi: 10.1038/bjc.1993.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawking F. Suramin: with special reference to onchocerciasis. Adv Pharmacol Chemother. 1978;15:289–322. doi: 10.1016/s1054-3589(08)60486-x. [DOI] [PubMed] [Google Scholar]
- 16.Coffey RJ, Jr, Goustin AS, Soderquist AM, Shipley GD, Wolfshohl J, et al. Transforming growth factor alpha and beta expression in human colon cancer lines: implications for an autocrine model. Cancer Res. 1987;47:4590–4594. [PubMed] [Google Scholar]
- 17.Hosang M. Suramin binds to platelet-derived growth factor and inhibits its biological activity. J Cell Biochem. 1985;29:265–273. doi: 10.1002/jcb.240290310. [DOI] [PubMed] [Google Scholar]
- 18.Pollak M, Richard M. Suramin blockade of insulinlike growth factor I-stimulated proliferation of human osteosarcoma cells. J Natl Cancer Inst. 1990;82:1349–1352. doi: 10.1093/jnci/82.16.1349. [DOI] [PubMed] [Google Scholar]
- 19.Williams LT, Tremble PM, Lavin MF, Sunday ME. Platelet-derived growth factor receptors form a high affinity state in membrane preparations. Kinetics and affinity cross-linking studies. J Biol Chem. 1984;259:5287–5294. [PubMed] [Google Scholar]
- 20.Hensey CE, Boscoboinik D, Azzi A. Suramin, an anti-cancer drug, inhibits protein kinase C and induces differentiation in neuroblastoma cell clone NB2A. FEBS Lett. 1989;258:156–158. doi: 10.1016/0014-5793(89)81639-4. [DOI] [PubMed] [Google Scholar]
- 21.Buchinger B, Spitzer S, Karlic H, Klaushofer K, Varga F. Lysyl oxidase (LOX) mRNA expression and genes of the differentiated osteoblastic phenotype are upregulated in human osteosarcoma cells by suramin. Cancer Lett. 2008;265:45–54. doi: 10.1016/j.canlet.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Wiese C, Nikolova T, Zahanich I, Sulzbacher S, Fuchs J, et al. Differentiation induction of mouse embryonic stem cells into sinus node-like cells by suramin. Int J Cardiol. 2011;147:95–111. doi: 10.1016/j.ijcard.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang S, Chen X, Li C, Zhang X, Zhang T, et al. Suramin inhibits the growth of nasopharyngeal carcinoma cells via the downregulation of osteopontin. Mol Med Rep. 2012;6:1351–1354. doi: 10.3892/mmr.2012.1074. [DOI] [PubMed] [Google Scholar]
- 24.Stein CA. Suramin: a novel antineoplastic agent with multiple potential mechanisms of action. Cancer Res. 1993;53:2239–2248. [PubMed] [Google Scholar]
- 25.Bastea LI, Doppler H, Balogun B, Storz P. Protein kinase D1 maintains the epithelial phenotype by inducing a DNA-bound, inactive SNAI1 transcriptional repressor complex. PLoS ONE. 2012;7:e30459. doi: 10.1371/journal.pone.0030459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du C, Zhang C, Hassan S, Biswas MH, Balaji KC. Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res. 2010;70:7810–7819. doi: 10.1158/0008-5472.CAN-09-4481. [DOI] [PubMed] [Google Scholar]
- 27.Jaggi M, Rao PS, Smith DJ, Hemstreet GP, Balaji KC. Protein kinase C mu is down-regulated in androgen-independent prostate cancer. Biochem Biophys Res Commun. 2003;307:254–260. doi: 10.1016/s0006-291x(03)01161-6. [DOI] [PubMed] [Google Scholar]
- 28.Olayioye MA, Barisic S, Hausser A. Multi-level control of actin dynamics by protein kinase D. Cell Signal. 2013;25:1739–1747. doi: 10.1016/j.cellsig.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 29.Storz P, Doppler H, Johannes FJ, Toker A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J Biol Chem. 2003;278:17969–17976. doi: 10.1074/jbc.M213224200. [DOI] [PubMed] [Google Scholar]
- 30.Cowell CF, Doppler H, Yan IK, Hausser A, Umezawa Y, et al. Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J Cell Sci. 2009;122:919–928. doi: 10.1242/jcs.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo H, Liu W, Ju Z, Tamboli P, Jonasch E, et al. An efficient procedure for protein extraction from formalin-fixed, paraffin-embedded tissues for reverse phase protein arrays. Proteome Sci. 2012;10:56. doi: 10.1186/1477-5956-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vignon F, Prebois C, Rochefort H. Inhibition of breast cancer growth by suramin. J Natl Cancer Inst. 1992;84:38–42. doi: 10.1093/jnci/84.1.38. [DOI] [PubMed] [Google Scholar]
- 33.Song S, Yu B, Wei Y, Wientjes MG, Au JL. Low-dose suramin enhanced paclitaxel activity in chemotherapy-naive and paclitaxel-pretreated human breast xenograft tumors. Clin Cancer Res. 2004;10:6058–6065. doi: 10.1158/1078-0432.CCR-04-0595. [DOI] [PubMed] [Google Scholar]
- 34.Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem. 2003;278:154–163. doi: 10.1074/jbc.M208075200. [DOI] [PubMed] [Google Scholar]
- 35.Gschwendt M, Kittstein W, Johannes FJ. Differential effects of suramin on protein kinase C isoenzymes. A novel tool for discriminating protein kinase C activities. FEBS Lett. 1998;421:165–168. doi: 10.1016/s0014-5793(97)01530-5. [DOI] [PubMed] [Google Scholar]
- 36.Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem. 1999;274:26543–26549. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- 37.Storz P, Doppler H, Copland JA, Simpson KJ, Toker A. FOXO3a promotes tumor cell invasion through the induction of matrix metalloproteinases. Mol Cell Biol. 2009;29:4906–4917. doi: 10.1128/MCB.00077-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowden CJ, Figg WD, Dawson NA, Sartor O, Bitton RJ, et al. A phase I/II study of continuous infusion suramin in patients with hormone-refractory prostate cancer: toxicity and response. Cancer Chemother Pharmacol. 1996;39:1–8. doi: 10.1007/s002800050531. [DOI] [PubMed] [Google Scholar]
- 39.Dreicer R, Smith DC, Williams RD, See WA. Phase II trial of suramin in patients with metastatic renal cell carcinoma. Invest New Drugs. 1999;17:183–186. doi: 10.1023/a:1006331518952. [DOI] [PubMed] [Google Scholar]
- 40.Falcone A, Pfanner E, Cianci C, Danesi R, Brunetti I, et al. Suramin in patients with metastatic colorectal cancer pretreated with fluoropyrimidine-based chemotherapy. A phase II study. Cancer. 1995;75:440–443. doi: 10.1002/1097-0142(19950115)75:2<440::aid-cncr2820750205>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 41.George S, Dreicer R, Au JJ, Shen T, Rini BI, et al. Phase I/II trial of 5-fluorouracil and a noncytotoxic dose level of suramin in patients with metastatic renal cell carcinoma. Clin Genitourin Cancer. 2008;6:79–85. doi: 10.3816/CGC.2008.n.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lustberg MB, Pant S, Ruppert AS, Shen T, Wei Y, et al. Phase I/II trial of non-cytotoxic suramin in combination with weekly paclitaxel in metastatic breast cancer treated with prior taxanes. Cancer Chemother Pharmacol. 2012;70:49–56. doi: 10.1007/s00280-012-1887-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villalona-Calero MA, Otterson GA, Wientjes MG, Weber F, Bekaii-Saab T, et al. Noncytotoxic suramin as a chemosensitizer in patients with advanced non-small-cell lung cancer: a phase II study. Ann Oncol. 2008;19:1903–1909. doi: 10.1093/annonc/mdn412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villalona-Calero MA, Wientjes MG, Otterson GA, Kanter S, Young D, et al. Phase I study of low-dose suramin as a chemosensitizer in patients with advanced non-small cell lung cancer. Clin Cancer Res. 2003;9:3303–3311. [PubMed] [Google Scholar]
- 45.Song S, Wientjes MG, Gan Y, Au JL. Fibroblast growth factors: an epigenetic mechanism of broad spectrum resistance to anticancer drugs. Proc Natl Acad Sci U S A. 2000;97:8658–8663. doi: 10.1073/pnas.140210697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song S, Wientjes MG, Walsh C, Au JL. Nontoxic doses of suramin enhance activity of paclitaxel against lung metastases. Cancer Res. 2001;61:6145–6150. [PubMed] [Google Scholar]
- 47.Zhang Y, Song S, Yang F, Au JL, Wientjes MG. Nontoxic doses of suramin enhance activity of doxorubicin in prostate tumors. J Pharmacol Exp Ther. 2001;299:426–433. [PubMed] [Google Scholar]
- 48.Skliris GP, Munot K, Bell SM, Carder PJ, Lane S, et al. Reduced expression of oestrogen receptor beta in invasive breast cancer and its re-expression using DNA methyl transferase inhibitors in a cell line model. J Pathol. 2003;201:213–220. doi: 10.1002/path.1436. [DOI] [PubMed] [Google Scholar]
- 49.Zhu WG, Hileman T, Ke Y, Wang P, Lu S, et al. 5-Aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J Biol Chem. 2004;279:15161–15166. doi: 10.1074/jbc.M311703200. [DOI] [PubMed] [Google Scholar]
- 50.Constantinides PG, Jones PA, Gevers W. Functional striated muscle cells from non-myoblast precursors following 5-azacytidine treatment. Nature. 1977;267:364–366. doi: 10.1038/267364a0. [DOI] [PubMed] [Google Scholar]
- 51.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 52.Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, et al. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol. 2009;11:545–556. doi: 10.1038/ncb1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eiseler T, Hausser A, De Kimpe L, Van Lint J, Pfizenmaier K. Protein kinase D controls actin polymerization and cell motility through phosphorylation of cortactin. J Biol Chem. 2010;285:18672–18683. doi: 10.1074/jbc.M109.093880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eiseler T, Schmid MA, Topbas F, Pfizenmaier K, Hausser A. PKD is recruited to sites of actin remodelling at the leading edge and negatively regulates cell migration. FEBS Lett. 2007;581:4279–4287. doi: 10.1016/j.febslet.2007.07.079. [DOI] [PubMed] [Google Scholar]
- 55.Peterburs P, Heering J, Link G, Pfizenmaier K, Olayioye MA, et al. Protein kinase D regulates cell migration by direct phosphorylation of the cofilin phosphatase slingshot 1 like. Cancer Res. 2009;69:5634–5638. doi: 10.1158/0008-5472.CAN-09-0718. [DOI] [PubMed] [Google Scholar]
- 56.Spratley SJ, Bastea LI, Doppler H, Mizuno K, Storz P. Protein kinase D regulates cofilin activity through p21-activated kinase 4. J Biol Chem. 2011;286:34254–34261. doi: 10.1074/jbc.M111.259424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim M, Jang HR, Kim JH, Noh SM, Song KS, et al. Epigenetic inactivation of protein kinase D1 in gastric cancer and its role in gastric cancer cell migration and invasion. Carcinogenesis. 2008;29:629–637. doi: 10.1093/carcin/bgm291. [DOI] [PubMed] [Google Scholar]
- 58.Onishi Y, Kawamoto T, Kishimoto K, Hara H, Fukase N, et al. PKD1 negatively regulates cell invasion, migration and proliferation ability of human osteosarcoma. Int J Oncol. 2012;40:1839–1848. doi: 10.3892/ijo.2012.1400. [DOI] [PubMed] [Google Scholar]
- 59.Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, et al. Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood. 2007;109:2579–2588. doi: 10.1182/blood-2006-07-027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sundram V, Chauhan SC, Ebeling M, Jaggi M. Curcumin attenuates beta-catenin signaling in prostate cancer cells through activation of protein kinase D1. PLoS ONE. 2012;7:e35368. doi: 10.1371/journal.pone.0035368. [DOI] [PMC free article] [PubMed] [Google Scholar]