

Abstract
Thioredoxin reductases (TrxRs) are flavin-containing dithioloxidoreductases that couple reduction equivalents from the soluble NAD(P)H pool to the soluble protein thioredoxin (Trx). Previous crystallographic studies of the Escherichia coli enzyme (ecTrxR) have shown that low molecular weight TrxRs can adopt two distinct conformations: the first (FO) is required for the oxidation of the flavin cofactor and the generation of reduced Trx; the second (FR) is adopted for the reduction of the flavin by NAD(P)H. Here, protein electrochemistry has been used to interrogate the equilibrium between the oxidized and reduced conformations of the ecTrxR and a novel, low molecular weight TrxR from the thermophilic archaeon Thermoplasma acidophilum (taTrxR) that is characterized structurally and biochemically in the accompanying paper [Hernandez et al. (2008) Biochemistry 47, 9728−9737]. A reversible electrochemical response is observed that reveals a dynamic behavior dependent upon the temperature of the experiment. At low temperatures (283 K) a broad, quasi-reversible electrochemical envelope is observed centered at a value of ∼−300 mV and displaying a peak width of over 150 mV. The voltammetric response sharpens dramatically as the temperature increases, becoming much more reversible (as determined by peak separation and peak width). The overall potential and shape of the voltammetric data indicate that the flavin (FAD/FADH2) and disulfide/dithiol couples are very close in thermodynamic potentials, and the data are interpreted in terms of the model of two-state conformational change between flavin reducing (FR) and flavin oxidizing (FO) states, where the difference in potential for the flavin and disulfide cofactors must be within 40 mV of one another. In this model, the low temperature peak broadening is interpreted as an indication of a heterogeneous population of TrxR conformations that exist at low temperature; at higher temperatures, FO and FR conformers can rapidly interconvert, and voltammetry reports upon an average potential of the conformations.
The ubiquitous protein thioredoxin (Trx)1 functions as a reductant for many key cellular processes, including DNA synthesis, oxidative stress response, and transcriptional activation (1). The reduced form of Trx is generated by thioredoxin reductase (TrxR), and the pair maintains the reducing conditions of the cytoplasmic environment, as well as being responsible for the reduction of ribonucleotide reductase and peroxiredoxins and the reductive activation of several transcription factors (2). The maintenance of redox environment is achieved through the coupling of reduction equivalents available from NAD(P)H to the TrxR active site, which includes a flavin moiety and a redox-active disulfide bond (3−5). TrxRs are homodimeric enzymes that are differentiated by complexity and molecular mass: the prokaryotic enzymes (exemplified by the enzyme from Escherichia coli) are of relatively low Mr (∼35000 Da per monomer) when compared to their higher Mr mammalian homologues (∼55000 Da per monomer) which contain an additional redox active unit, such as a selenocysteine-cysteine pair (6). To date the E. coli TrxR (ecTrxR) has proven to be a robust model system for studies of low Mr TrxR structural dynamics and catalytic chemistry. Preliminary structural analyses by Kuriyan revealed that NADPH binding occurs far from the flavin domain of the protein, while the redox-active disulfide pair (Cys135 and Cys138) are on the re face of the flavin, in a buried state, forbidding direct interaction with Trx (7). Further analysis of this conformer, the flavin oxidizing (FO) state, by Waksman suggested that flavin reduction could be achieved by a 67° ball-in-socket rotation of the NADPH binding and flavin domains (8). Additional kinetic and spectroscopic studies of wild-type and mutant forms of ecTrxR have indicated that not only is a large conformation change required in the course of ecTrxR catalysis for the generation of the flavin reducing (FR) form of the enzyme but that the two forms, FO and FR, are in equilibrium in solution (9−12). By crystallographically characterizing a site-directed mutant of TrxR cross-linked to a Trx “doorstop”, Ludwig and co-workers successfully provided the first structural snapshot of the FR conformation of ecTrxR, as shown in Figure 1(13,14).
Figure 1.
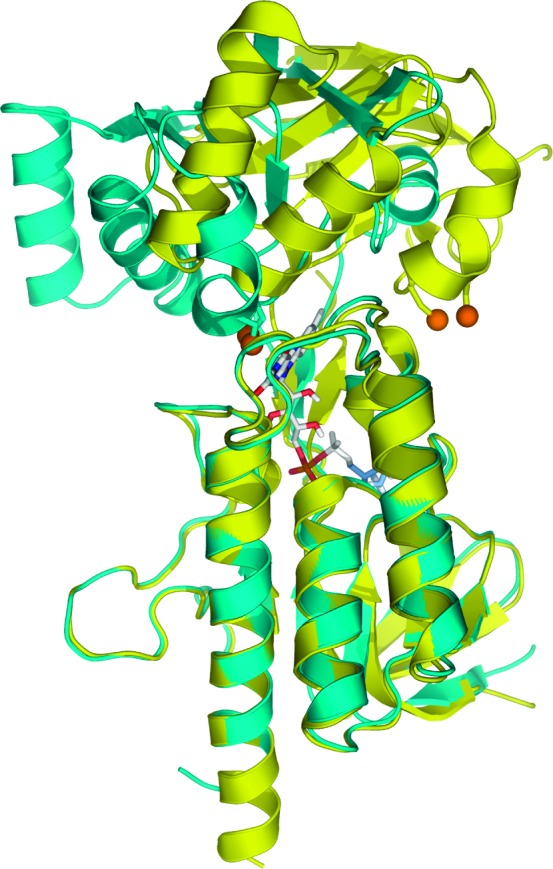
The FO and FR conformations of ecTrxR, superimposed in cyan and yellow, respectively. Orange spheres are used to represent the disulfide-forming cysteines in the FO conformation, and the cysteine/serine pair is used in the crystallographic trapping of the ecTrxR FR conformation. The image was constructed from the FO and FR PDB files 1TDF and 1F6M, respectively.
The picture of the ecTrxR mechanism that emerges requires a ternary complex (15) that redox cycles between the two- and four-electron reduced states (16), where redox transformations must be coupled to potential conformational changes. However, it has proven challenging to develop a detailed picture of the impact of conformational dynamics upon the redox properties of the FAD cofactor and the Cys135-Cys138 disulfide. Previous potentiometric titrations suggest that the thermodynamic potentials of the ecTrxR redox cofactors (FADH2/FAD and the Cys135-Cys138 disulfide) are very close, with Em varying by only 7 mV at pH 7.6 (16,17). Specifically, the enzyme FAD/FADH2 potential has been calculated as −263 and −280 mV for the active site disulfide and dithiol forms, respectively, while the enzyme active site disulfide/dithiol potential has been calculated as −270 and −287 mV for the flavin oxidized and flavin reduced forms, respectively (16,17). However, such calculations have been previously based upon data that cannot discriminate between the FR and FO conformers, which rapidly interconvert at room temperature (18). Thus, we hypothesize that the former measurements represent potentials that are best considered as an average between the populations of each conformation.
Here we have looked to direct electrochemical methods in order to parse the relationship between conformational dynamics and redox chemistry. We have used protein film voltammetry (PFV) as a method to investigate the redox characteristics of the low Mr TrxR enzymes from E. coli, as well as the enzyme for the archaeon Thermoplasma acidophilum, an enzyme that has been biochemically and structurally characterized in the accompanying report (19). PFV has the ability to interrogate redox equilibria that are coupled to additional chemical processes, particularly when chemical steps result in modulations in the midpoint potential of the system. For example, studies of proton-coupled steps in redox reactions of bacterial ferredoxin (20,21) have resolved proton transfer reactions of a single side chain. No electrochemical studies of TrxR (of any type) have been reported to date, and the reports of potentials have been limited to techniques that will average away any contribution due to distinct conformational changes, which are rapid on the time scale of potentiometry. However, typical electrochemical investigations of flavoenzymes are at times hindered by the stability of the flavin-bound form of the protein: protein binding of the flavin cofactor can be compromised upon exposure to a typical graphite working electrode frequently used in PFV experiments, as has been reported for flavodoxin and P450BM322,23. Given this, we have opted to study both the low MrecTrxR and a novel TrxR from the thermophilic organism T. acidophilum, whose crystal structure and preliminary biochemical characterization are reported in the accompanying paper (19). Using this strategy, it is our hope that the greater protein stability of the thermophilic enzyme not only will yield better electrochemical results but will enable a dissection of the contribution of FO and FR conformations to the control of flavin and disulfide reduction potentials. As described in the preceding paper, the taTrxR has been cloned, purified, and subjected to structural and biochemical analyses, demonstrating that this new TrxR is an excellent homologue of the ecTrxR, with the caveat that it cannot use NADPH as an electron donor (19).
Experimental Procedures
Protein Purification
The taTrxR was purified as a His-tagged construct following cloning from genomic DNA from T. acidophilum. The details of protein purification and characterization are described in the accompanying paper (19). The ecTrxR was produced from the system described by Mulrooney (24), with the following modifications: cells were streaked on 2×YT agar plates with 100 μg/mL ampicillin and 25 μg/mL kanamycin and incubated at 310 K overnight. One colony was used for a 600 mL culture that grew with gentle shaking overnight at 310 K in 2×YT media with 50 mM phosphate (pH 7), 20 mM glucose, 100 μg/mL ampicillin, and 25 μg/mL kanamycin. The cell mixture was cooled on ice and centrifuged; then the pellets were collected and frozen at 253 K overnight. Frozen cells were thawed and resuspended by adding 20 mL of 10 mM phosphate (pH 7.6), 0.3 mM EDTA, and 0.5 mM phenylmethanesulfonyl fluoride. Cells were then sonicated 5 times for 10 s, allowing 20 s between sonications to cool on ice. After sonication, 100 mg of streptomycin sulfate was added, and the mixture was centrifuged, supernatant was saved, and ammonium sulfate was added to the final concentration of 0.56 g/mL. The precipitate was collected by centrifuging the mixture, and the pellet was resuspended in 10 mL of 10 mM phosphate (pH 7.6) and 0.3 mM EDTA and dialyzed overnight in 1 L of 10 mM phosphate (pH 7.6) and 0.3 mM EDTA; the solution was refreshed three times. The protein solution was then passed through a 45 μm syringe filter and then loaded onto a 3 cm × 5 cm 2′,5′-ADP Sepharose 4B column. Protein was eluted using a 120 mL 10 mM phosphate (pH 7.6), 0.3 mM EDTA buffer with a 0.15 mM to 1 M NaCl linear gradient. Fractions were combined based on 280 nm/456 nm ratio and then precipitated by adding ammonium sulfate to the final concentration of 0.56 g/mL. The protein precipitate was collected by centrifugation, and the pellet was resuspended in 10 mM phosphate (pH 7.6), 0.3 mM EDTA, and 100 μM FAD, incubated for several hours at 4 °C, and then dialyzed overnight in 1 L of 10 mM phosphate (pH 7.6) and 0.3 mM EDTA; the solution was refreshed three times. Protein was refridgerated for immediate use. The plasmid pTrR301 was the generous donation of Dr. Scott B. Mulrooney.
Protein Electrochemistry
Protein film voltammetry was conducted using a PGSTAT 12 electrochemical analyzer (EcoChemie), in a three electrode cell configuration, using a pyrolytic graphite edge (PGE) working electrode, a calomel reference electrode, and a platinum wire counterelectrode. All experiments were performed in a water-jacketed all-glass electrochemical cell that was thermostated using a circulating water bath. The reference electrode was maintained at room temperature in the course of the experiments, and all potentials described here are reported versus the hydrogen potential, correcting for the discrepancy between SHE and the calomel electrode as a function of temperature.
Electroactive films of taTrxR were generated by first polishing the PGE surface with an aqueous slurry of 1.0 μm alumina (Buhler), followed by sonication. The polished electrode was washed thoroughly with distilled water, and 1.0 μL of TrxR (typically a 100 μM solution in pH 8.0 HEPES buffer with 150 mM NaCl) was deposited on the graphite surface. Excess protein was removed with washing, and the resulting protein film was placed immediately in the electrochemical cell compartment, in which 2.0 mL of a working cell buffer was purged under a continuous nitrogen blanket. Electroactive films of ecTrxR were similarly obtained by first poising a freshly polished PGE at −510 mV in a protein-free cell solution for 5 min. Then, 1.0 μL of ecTrxR (260 μM) was directly added to the electrode tip, and excess protein was removed by washing with a small amount of ice-cold buffer.
Electrochemistry was conducted as staircase cyclic voltammetry. The resulting voltammograms were subjected to baseline subtraction of a polynomial background, representing the nonfaradaic (charging) component of the current. The entropic component of the redox reaction of TrxR was determined using the slope of a plot of Em vs T (ΔS °rc = nF(dEm/dT)) for data collected using a cell in which the reference calomel electrode was maintained at a constant temperature (293 ± 0.5 K), while the working electrode-containing cell temperature was varied. The enthalpic component was similarly determined by a Gibbs−Helmholtz plot (Em/T vs 1/T).
Redox Potentiometry
Additional determinations of the thermodynamic potentials of taTrxR and ecTrxR were conducted using the xanthine/xanthine oxidase system described by Massey (25). Specifically, potentials were determined under anaerobic conditions in a glovebox using a S. I. Photonics 400 series spectrophotometer. The solution contained 100 mM HEPES (pH 7.0), 1 μM benzyl viologen, 1 μM methyl viologen, 250 μM xanthine, xanthine oxidase, 18 μM enzyme, and 5 μM safranine O as a reference dye (−289 mV).
Results
Protein Electrochemistry
The taTrxR was investigated at pyrolytic graphite edge electrodes using cyclic voltammetry, at 283 K, as shown in Figure 2 (solid line). The electrochemical response reveals a single pair of highly broadened voltammetric peaks, centered at a midpoint potential of −315 mV. On initial analysis, the data do not seem to be in accord with theoretical predictions of redox-active molecules adsorbed at an electrode: ideal peak shapes would display zero separation of cathodic and anodic peaks, while the data shown in Figure 2A display a peak separation of nearly 110 mV. However, the relationship of cathodic and anodic peak heights (ic and ia) with respect to the scan rate (v) shows a linear relationship, indicating an adsorbed species. Further, the ratio of ic/ia is invariant with respect to scan rate, indicative of a simple redox process with no further preceding, or following, chemical reactions (Figure 2A, inset). Like the unusual increase of peak separation at 283 K, the peak widths (δ) of both the cathodic and anodic peaks (Figure 2B) also deviate from a simple, ideal system. By theory, δ ≈ 3.53(RT/nF), so at 283 K, a δ value of 86/n mV is predicted. Thus, even if the voltammogram represents the sum of two distinct, overlapping electrochemical responses, each corresponding to an n = 2 redox reaction (for the FAD/FADH2 and active site disulfide/dithiol couples), the observed data appear to be unusually broad. Plotting the peak width at half-height versus temperature (Figure 2B) shows that the peak width decreases substantially as the temperature increases, becoming closer to the predicted value for a n = 2 process at temperatures greater 313 K. Notably, theory states that, for a routine redox process, the decrease should not occur (δ should increase as a function of temperature), indicating that other processes must be at work.
Figure 2.

PFV response of taTrxR upon PGE electrodes. (A) Raw PFV data of taTrxR at low temperature (solid line) superimposed upon the baseline response of the PGE electrode used for taTrxR immobilization (dashed line). Experimental conditions were as follows: scan rate, 100 mV/s; pH 7.0; 100 mM NaCl electrolyte; 283 K. Inset: Dependence of peak height (ip) as a function of scan rate. (B) The average peak width at half-height for oxidative and reductive scans, as a function of temperature (open squares), compared to computed values of peak width for a n = 1e− (solid) or n = 2e− (dashed) feature. The solid square represents the observed average peak width after returning a heated taTrxR protein film to 293 K.
The observed voltammetric response is strongly affected by changes in temperature. The dependence of the voltammetric peak shape upon temperature is shown in Figure 3A. By warming the cell solution using an externally controlled circulator, the peak separation, peak width, and midpoint potential of the taTrxR PFV response were altered. As mentioned above, the peak width decreases as a function of temperature (Figure 2B), and as Figure 3A indicates, no additional features are observed in the voltammetry. At low temperature, a broad envelope-like signal appears, while at elevated temperatures the voltammetric response sharpens considerably. In a similar way the peak separation also decreases as a function of temperature, which is also unexpected on the basis of electrochemical theory for a simple reversible redox equilibrium. Figure 3B shows that as the temperature increases to the upward limit of the stability of the electroactive protein film, 333 K, the peak separation decreases, though the ideal value of 0 mV is never attained. While perfect reversibility is not observed at higher temperatures, the peak separation (typically ∼30−50 mV) is close to values observed for other systems analyzed with PFV. As discussed below, the nonzero peak separation may be due to heterogeneity upon the electrode, including distinct global conformations of taTrxR at the electrode surface, but also the FR and FO conformations unique to low Mr TrxR enzymes. The intensity, or area, of the electrochemical response is also strongly affected by temperature; as Figure 3A indicates, the area of both the oxidative and the reductive signals is reduced as the temperature is elevated. In the higher temperature regime (e.g., 313 K), the taTrxR electrochemistry still corresponds to an immobilized species, as determined by a linear relation of ia or ic with v (data not shown). In the minutes required to attain and stabilize the protein at a specific temperature, desorption of the protein film did occur, though as all experiments were conducted in fresh, protein-free solutions, the desorption was not significant in the time scale of a single cyclic voltammogram, collected at 150 mV/s. Figure 3A clearly indicates that cooled protein films initially yield pronounced surface coverage (heavy, solid line). Heating not only leads to peak narrowing and a decrease in peak separation but, when a protein film at elevated temperature is returned to the cool temperature (283 K), broadening and peak separation occurs again, and although the signal intensity is depleted (Figure 3A, dotted line) the peak width (Figure 2A, open square) and peak separation (Figure 3B, closed square) are nearly identical upon the return to 283 K, as compared to the initial experiments. We interpret the dramatic changes in the PFV response as a demonstration of the conformational dynamics of the FR and FO states as the temperature approaches the optimal growth temperature of T. acidophilum.
Figure 3.
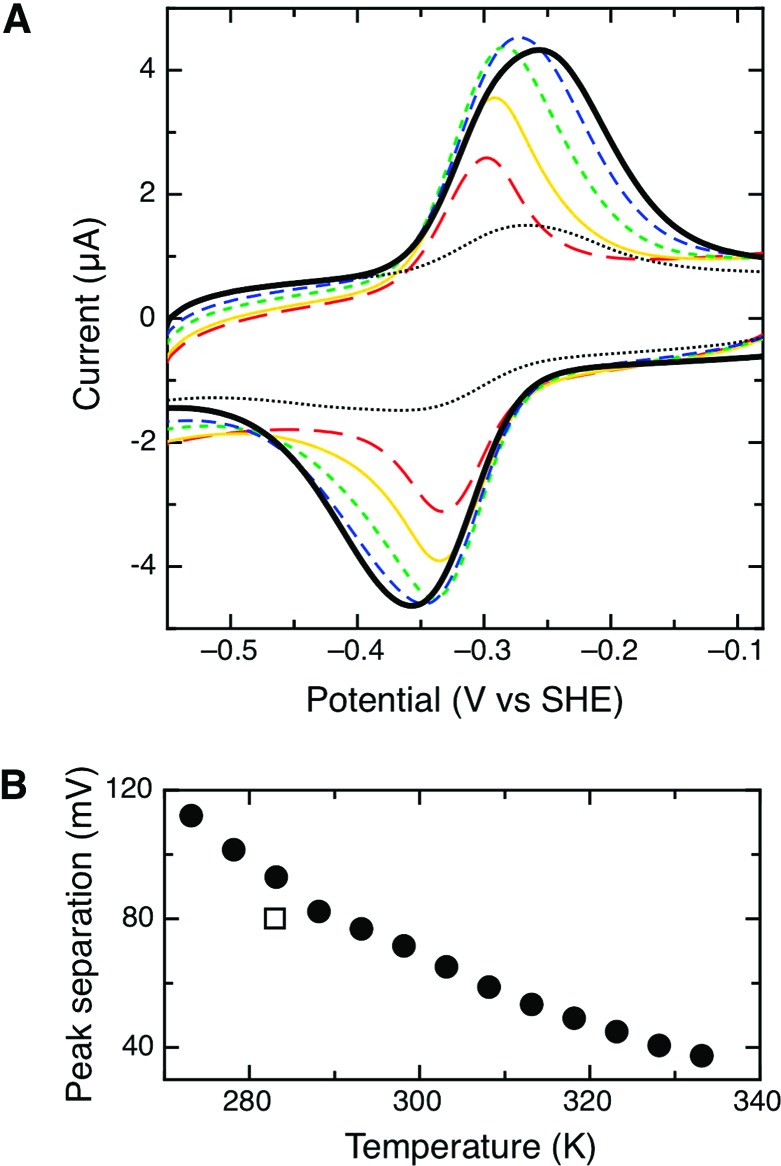
Variable temperature, PFV response of the taTrxR. (A) The taTrxR PFV response as a function of temperature. Protein films generated at 283 K (solid line, heavy) were then subsequently warmed to 293 (blue dashed), 303 (green dash), 313 (yellow), and then 323 K (red dash). Finally, the experimental temperature was lowered to 283 K again (dotted line), indicating the loss of the protein surface coverage during the course of the experiment. All data were collected at pH 7.0, 100 mM NaCl electrolyte, and with a scan rate of 150 mV/s. (B) Peak separation as a function of temperature, for a single experiment, heating the protein film in increasing steps of five degrees; the open square represents the peak separation observed when returning the taTrxR film to 293 K.
The redox thermodynamics for the overall taTrxR electrochemical response is illustrated in Figure 4, which depicts the measured midpoint potential of the overall voltammetric response (taking as Em the average of the cathodic and anodic peak) as function of temperature. The dependence of the observed midpoint potential displays a clear downward trend, as in the case of the redox thermodynamics of flavins measured in nonaqueous solvents (26), as well as in flavoproteins (23,27). If the midpoint potentials are taken as a superposition of the two redox cofactors (i.e., the FAD/FADH2 and the disulfide/dithiol couples), and the behavior of the two couples is assumed to be similar, the slope of the linear fit shown in Figure 4A can be related to the entropy change for the redox reaction, ΔS °rc/nF, yielding a value of −78.1 J/(K·mol). As the individual redox couples cannot be observed directly, this value can serve as an estimate of ΔS °rc only. (Indeed, the redox thermodynamics of the two cofactors may be different, due to temperature-dependent fluctuations of the midpoint potential itself, as well as distinct pKa values.) A similar interpretation of a Gibbs−Helmholtz plot (Figure 4B) conveys ΔH°rc for the process at the electrode, giving a value of −19.2 kJ/mol.
Figure 4.

Redox thermodynamics of taTrxR protein films, modeling the data to two overlapping 2e− features. (A) ΔS °rc is determined from the slope of the Em vs T plot. Data are fit to a linear regression analysis, and the slope of which is interpreted as slope = ΔS °rc/nF, where n is assumed to be 2e−. (B) Gibbs−Helmholtz plot of the same data.
Possible Role of Adsorbed Flavin
The propensity for several flavoproteins and enzymes to lose flavin cofactors at graphite electrodes forced us to consider the possibility that the apparent electrochemical reponse of taTrxR could be due to FAD adsorbed upon the electrode. To investigate this possibility, we have conducted several experiments:
First, we have compared adsorbed FAD at a graphite electrode with the taTrxR response. A microliter of a buffered FAD solution (50 μM) was deposited on a graphite electrode, in a similar fashion to the taTrxR PFV. The resulting electrochemically active species displayed a midpoint potential ∼50 mV more positive than the taTrxR response.
Second, in order to ensure that our taTrxR data did not correspond to denatured protein in the presence of extra FAD in a non-native conformation, we have supplemented taTrxR solutions used to generate protein films with additional “free” flavin. Free FAD can be added to the electrochemical cell, with taTrxR already adsorbed onto the working electrode. In this experiment, the typical taTrxR PFV signal (Figure 5A, light solid line) in the absence of exogenous flavin transforms into a voltammogram that is a convolution of overlapping “free FAD” and “taTrxR-based” signals as a function of the added FAD (Figure 5A). Identical results are achieved with FMN or riboflavin addition, and it is clear that the FAD-based signal is discernible from the TrxR-based signal, at all pH values in this study (Figure 5B). In the case of both FAD and taTrxR, a pH dependence of −60 mV/pH is observed (Figure 5B), indicating a 1:1 ratio of protons and electrons involved in the redox couples.
Figure 5.

Demonstration that the PFV response of taTrxR is not due to flavin desorbed from the protein. (A) Addition of exogenous FAD to a taTrxR protein film. Buildup of the voltammetric response of the FAD/FADH2 couple is observed by starting with a taTrxR-bound electrode (light solid line) and adding FAD to the electrochemical cell at final concentrations of 2, 4, and 10 μM (dotted, dashed, and heavy solid line). The buildup of the free FAD species is observable at ∼−220 mV. Other experimental features: pH 7.0, 10 °C, v = 150 mV/s. (B) Free FAD (●) and taTrxR (open square) midpoint potentials as a function of pH.
Third, the E. coli TrxR (ecTrxR) can be interrogated using PFV (Figure 6) and reveals a significant loss of “free flavin” at the EPG electrode, indicating that a simple difference between the ec and ta enzymes is the enhanced stability of the TrxR from the thermophile T. acidophilum in retaining flavin, when bound to the electrode. Similar to the taTrxR findings, the ecTrxR shows a “free flavin” based electrochemical signature that grows upon the addition of exogenous flavin into the cell solution (data not shown). In general, the ecTrxR behaves very much like the taTrxR although the overall electroactive surface coverage (Γ) is significantly lower for the ecTrxR as compared to the taTrxR. Yet, the average peak width of the ecTrxR does decrease with increasing temperature (Figure 6B), as observed for the T. acidophilum protein (Figure 2B). As well, the peak widths are similar when the temperature is near growth optimum, 65 mV for taTrxR at 333 K (Figure 2B) and 67 mV for ecTrxR at 303 K (Figure 6B). Thus, we believe that the essential redox characteristics of the TrxR cofactors are nearly identical.
Figure 6.

PFV response of ecTrxR. (A) Raw PFV and baseline-subtracted voltammetric features for ecTrxR. Parameters for the experiment: pH 7.0, 10 °C, and v = 150 mV/s. (B) Average peak width at half-height as a function of temperature for the ecTrxR component of the PFV response shown in (A).
Finally, we have noted that electrodes modified with taTrxR behave very much like other protein film electrodes, in contrast with FAD-modified electrodes. Electroactive films generated from taTrxR solutions are easily removed by repolishing the electrode surface with an alumina slurry, and electrodes are themselves sensitive to changes in buffer conditions. Neither of these parameters is true for a graphite electrode modified with FAD, which is quite difficult to remove from the graphite surface, even with multiple cycles of mechanical abrasion and alumina polishing. Thus, due to these four lines of inquiry, we are convinced that the taTrxR-based electrodes genuinely represent the redox chemistry of the taTrxR and are not grossly compromised due to the loss of the flavin cofactor.
Verification of Potentials via Redox Potentiometry
In order to probe further the similarity between the taTrxR and ecTrxR, we measured the observable flavin potential for taTrxR using the xanthine/xanthine oxidase-coupled potentiometric assay described by Massey (25). Figure 7 depicts the result of this experiment, using the redox indicator safranine O (SafO). The solution-based method cleanly reduces the taTrxR flavin from the oxidized to reduced forms, without the generation of the flavin semiquinone, yielding a value of the flavin potential of −305 ± 5 mV, at 303 K, in reasonable agreement with measurements made by PFV (−322 mV for the entire taTrxR envelop, Figure 4). Control experiments were conducted using the ecTrxR, which we analyzed using an identical protocol, resulting in a potential of −291 ± 5 mV, agreeing reasonably well with with previous measurements of the ecTrxR flavin potential, −252 mV, as determined by O’Donnell and Williams, using a potentiometric method involving NADPH- and Trx-based equilibria.
Figure 7.

Redox titration of the taTrxR flavin using the xanthine/xanthine oxidase reduction methodology of Massey (25), showing the decrease in the visible absorption spectrum, as collected every ∼5 min during the course of the reduction of the flavin cofactor. SafO was used as the reporter dye (maximum at 520 nm), as described in the Experimental Procedures. Inset: Plot of log [Ox]/[Red] for SafO vs log [Ox]/[Red] for TrxR, used to find the midpoint potential with respect to the reference value of SafO (−289 mV).
Variable Scan Rate Measurements
In order to better describe the temperature-induced alteration of the taTrxR electrochemical response, we examined the impact of scan rates (and therefore, time scale) used in PFV. By collecting voltammetry data at faster and faster time scales (increasing scan rate), we presumed that, in the higher temperature regime, fast scan rates would allow us to detect the distinct FO and FR conformations on the electrochemical time scale. Thus, if conformational dynamics were a substantial contributor to the narrow voltammetric envelope observed at higher temperatures, we could reverse the narrowing effect by using fast scan rates. Figure 8 shows the impact upon the PFV experiment achieved by increasing the scan rate over 2 orders of magnitude, while the temperature is held at a constant value of 308 K. By normalizing the baseline-subtracted data for the scan rate (and small deviations over all area due to film loss associated with the thermal desorption of the TrxR from the electrode), it clear that as v is increased from 200 mV/s (Figure 8, dotted line) to 2 V/s (dashed line) to 20 V/s (solid), the data mimic the impact of lowering the temperatures. As shown in Figure 8, as v is increased by 2 orders of magnitude, the oxidative and reductive peaks shift from the midpoint position by over 110 mV, and the shape of the peak broadens by ∼50%. At temperatures greater than 308 K identical behavior was observed when the scan rate is increased, though the electrochemical response is complicated by thermal desorption of the taTrxR which yields signals of smaller magnitudes (and for this reason, we show illustrative data at 308 K in Figure 8). The overall scan rate-dependent behavior is likely compounded with the natural increase of peak separation and peak width at half-height that can otherwise be utilized to find interfacial electron transfer kinetics. Yet, qualitatively, we have found a striking similarity between experiments conducted using faster scan rate yet higher temperatures and those at slower scan rates and lower temperatures. As discussed below, we take these findings as supportive of the model of TrxR conformational dynamics upon the electrode.
Figure 8.

Impact of increasing scan rate upon the taTrxR voltammetry at 308 K. Baseline-subtracted data are given to illustrate how, at elevated temperatures, the scan rate of the experiment can be raised from 200 mV/s (dotted line) to 2 V/s (dashed) to 20 V/s (solid), which results in broadening due to the observation of FO and FR states.
Discussion
We have shown that the direct electrochemistry can be used to interrogate the redox-active cofactors of low Mr TrxRs through the study of the newly characterized enzyme from T. acidophilum as well as the TrxR from E. coli. As described in the preceding paper, the taTrxR is a genuine thioredoxin reductase, yet it cannot be reduced by NADPH (the typical reductant of low Mr TrxRs) due to clear structural determinants that preclude binding of the cosubstrate. The structural analysis of the ta enzyme reveals the protein in a FR conformation that would enable flavin reduction by exogenous cosubsrate, which implies that a conformational change must occur to allow for the FADH2 to reduce the redox-active disulfide. Indeed, it was found that the closest cysteine S atom is 11.28 Å away from the FAD(N5) similar to the ecTrxR FR structure (11.08 Å), suggesting a similar conformational change is responsible for adopting the FO conformer and effective electron transfer from the flavin to the disulfide (14). At this time, the nature of the in vivo reduction system is unknown, though the strength of the biochemical and structural data compels the interpretation of our electrochemical data in terms of a model of taTrxR reactivity that includes interconversion between the FO and FR states.
In the most general terms, our data are in reasonable agreement with control experiments carried out with a xanthine/xanthine oxidase method for the determination of flavoprotein redox potentials. Other methods of spectrophotometrically titrating the reduction potential of the flavin would typically utilize the soluble redox agent responsible for supporting turnover in vivo, such as NADPH, an ineffective reductant of the taTrxR. Regardless, the use of the xanthine/xanthine oxidase method revealed a flavin potential for taTrxR of −305 mV at 303 K, consistent with the broad electrochemical response centered at −322 mV observed at the same temperature. We have ruled out the possibility that the PFV results are due to free flavin cofactor that has dissociated from the taTrxR during the course of our experiments, and we have found that there is a similarity between the ecTrxR electrochemistry and that observed for the ta enzyme. In both cases, the voltammetric response shows broad, yet symmetrical voltammetry that likely contains information about both the flavin cofactor and the redox-active disulfide group associated with thioredoxin reductases. However, two distinct voltammetric features are not clearly resolved, regardless of the temperature, pH, or ionic strength (not shown), each of which could be a factor that would preferentially modulate one of the two cofactors’ redox properties. Further, the redox thermodynamics of the voltammetric feature observed shows a single, linear dependence of Em as a function of temperature, indicating that PFV is reporting upon redox processes which are behaving similarly at the electrode. However, taTrxR reveals a stable electrochemical response that displays a peak shape that is very sensitive to the temperature of the electrochemical cell. While requiring significantly lower temperatures, McEvoy and Armstrong have also noted that cryotemperature-induced broadening can occur for thermophilic proteins, such as the ta ferredoxin (28). Here we hypothesize that low temperature-induced “broadening” is further enhanced by the dynamics of the TrxR enzyme. In the following points, we will attempt to resolve an electrochemical model of the data, which rationalizes how the distinct conformations associated with TrxR activity (FR and FO) result in a modest modulation in the potentials for redox-active cofactors associated with TrxR, thus yielding the apparently broadened voltammograms.
General Considerations of an Electrochemical Model
As the FO and FR conformations can interconvert regardless of redox state, a model describing the relationships between the conformational states and redox potentials must allow for each redox reaction and each interconversion between FO and FR. Scheme 1 depicts such a model, considering that PFV will report upon thermodynamic potentials resulting from four features (Em for the FAD/FADH2 and the disulfide/dithiol couples, in both the FR and FO conformations). In the case of the taTrxR, the relative populations of the FR and FO conformers are not known; for our electrochemical analyses, this should make itself manifest as a specific electroactive coverage for both the FR (ΓFR) and the FO (ΓFO) conformers. In the following analysis, we will assume that the broad electrochemical envelope observed at low temperature is due to kinetic trapping of FR and FO states upon the electrode; thus, heating the taTrxR allows for interconversion between the states, and the resulting voltammetric features represent an averaging between specific redox couples (e.g., the FAD/FADH2 couple) as experienced in the FO and FR conformers.
Scheme 1. A Simplified Model in Which Individual Microscopic Potentials Will Not Be Addressable in the PFV Experiment.
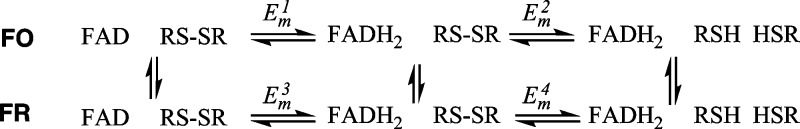
PFV Results of the taTrxR at 333 K
As observed in the raw data (Figure 3A) conducting PFV at temperatures near the growth optimum of T. acidophilum results in highly symmetric, nearly reversible voltammograms. In our model this feature should contain information regarding the FAD/FADH2 and disulfide/dithiol redox couples of the taTrxR, averaged between the interconverting FO and FR conformations. Thus, we have initially attempted to describe these data by a set of two redox couples of equal electroactive area, as the stoichiometry of the flavin cofactor to the disulfide is 1:1. Figure 9 shows the result of fitting the baseline-subtracted, oxidative scan, computing an electroactive coverage for the TrxR, and determining a two-electron midpoint potential for both the flavin and the disulfide, each of which is described by the formalism derived by Laviron, given in eq 1(29–31).
| 1 |
where
and
Here, F is the Faraday, R the ideal gas constant, T the temperature, v the scan rate, A the geometric area, and Γ the electroactive coverage. In all cases, qualitatively identical results were observed for the reductive scan of each experiment.
Figure 9.

Fitting of the baseline-subtracted, oxidative half-scan of taTrxR to two distinct Nernstian features, with an identical value of the electroactive coverage, at 333 K. For each feature the stoichiometric number of electrons was held to 2, while the separation between the EI/R and EO/R potentials was allowed to vary, as indicated by eq 1. (A) The case where the two one-electron potentials are crossed, with the difference in potential being greater than 200 mV; (B) the difference in crossing of potentials is ∼50 mV.
As Figure 9 illustrates, at 333 K the PFV response of taTrxR is modeled by just two n = 2e− features that are indicated by the dotted lines. In this way, the two voltammetric features at −295 and −330 mV are directly observed and most likely represent the cooperative FAD/FADH2 and disulfide/dithiol couples. The two features are separated in potential by approximately 35 mV, somewhat larger than the difference of 7 mV suggested by Williams and co-workers for the E. coli enzyme (9,16,17). Additionally, the higher potential feature is in good agreement with the value for the FAD cofactor determined by redox titration at 303 K, −305 mV. Figure 9A shows the fitting for a case where the cooperativity of each cofactor is perfect (by requiring a large separation in the crossed, formal one-electron potentials described by eq 1). The result is only slightly improved by allowing for the possibility that the redox behavior is somewhat nonideal. Figure 9B shows that a discernible, but modest improvement in the fit (and less than 5 mV total separation in the two detectable potentials) is achieved by allowing for breakdown of perfect n = 2e− behavior, represented here by slight broadening of the component features (Figure 9B, dotted lines). In the Laviron description of two-electron redox chemistry, a decrease in cooperativity corresponds to the two one-electron potentials becoming closer in value. The apparent loss of cooperativity could also be reflective of dispersion of the taTrxR molecules upon the electrode that is still present at the elevated temperature. From this analysis, it is clear that the averaged potentials are close in absolute value and cannot be more than ∼35 mV separated from one another, or the electrochemical data would reveal a clear indication of two peaks in the raw data.
PFV Results at 283 K
A similar approach to the analysis of data collected at lower temperatures (here, 283 K) is shown in Figure 10. Attempting to describe the data in terms of two n = 2e− components is not at all possible, given the overall broadening observed in the data. However, allowing for the loss of cooperativity is illustrated in Figure 10A, which shows the fitting of the oxidative half-scan of data collected at 283 K, where the dotted lines correspond to two n = 2e− components that both display artificially broadened peak shapes. The breadth in the case of the fitting shown in Figure 10A arises from the loss of cooperativity for the redox chemistry of both cofactors (i.e., for both features the two one-electron potentials are apparently no longer crossed). However, there are no additional indicators from solution experiments that, at 283 K, either the taTrxR flavin or the disulfide engage in one-electron chemistry. Indeed, the redox titrations of the flavin all show clean n = 2e− behavior at low temperatures (Figure 7). As such, we take the apparent loss of cooperativity to be synonymous with the effect dispersion generated by the “freezing out” of the FO and FR conformations. If we do attempt to account for the possibility of distinct conformations, by modeling the low-temperature response with four species, we can indeed obtain an excellent agreement with the data, shown in Figure 10B. In this model, four features are apparent, corresponding to both n = 2e− redox cofactors being present in the FO and FR conformations. The key result from this analysis is that we can estimate that the impact of FR and FO conformers is a modulation of 40−60 mV for both the FAD/FADH2 and disulfide/dithiol reduction potentials. However, we cannot assign specific redox couples to the voltammetric components of the envelope. Instead, PFV elucidates the similarity in the formal potentials for the cofactors and the dynamics of TrxRs at an electrode interface.
Figure 10.

Fitting of the baseline-subtracted, oxidative half-scan of taTrxR to either two noncooperative (A) or four cooperative (B) n = 2e− Nernstian features, assuming identical electroactive coverages, and a temperature of 283 K.
While the development of a more precise mechanistic model will require greater insight into the assignment of the Nernstian peaks observed in our fitting (potentially through the generation of a disulfide-free mutant of the taTrxR), an attractive hypothesis would allow for the flavin to possess a higher potential in the FR conformation, such that reduction of the flavin is efficient; and then in the FO conformation if the potential has been lowered by ∼40−60 mV with respect to the disulfide, reduction of the disulfide will again be favored. Finally, when the FR conformer is adopted again, the resulting dithiol will be able to readily react with Trx in a thermodynamically favorable manner. Thus, the TrxR scaffold may be able to achieve precise and unidirectional redox chemistry by tuning the potential of flavin and disulfide by discrete steps.
Concluding Remarks
Here we have interrogated low Mr thioredoxin reductases using direct electrochemistry for the first time. We investigated the redox properties of a classic TrxR from E. coli, as well as newly described TrxR from the archaeon T. acidophilum(19). Structural and biochemical studies have demonstrated that the taTrxR is very much like the better characterized bacterial homologue from E. coli except in its selection for its reductant. By using PFV we have been able to exploit the superlative thermostability of the taTrxR to our advantage and demonstrate that protein dynamics between the FO and FR conformations contribute to the formal potentials of the flavin and disulfide cofactors.
Acknowledgments
The authors thank Dr. Scott Mulrooney and Prof. Charles Williams, Jr., for the generous donation of the ecTrxR overexpression system.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: Trx, thioredoxin; TrxR, thioredoxin reductase; PFV, protein film voltammetry; Em, reduction potential; ec, Escherichia coli; ta, Thermoplasma acidophilum; EDTA, ethylenediaminetetraacetic acid; PGE, pyrolytic graphite edge; FAD, flavin adenine dinucleotide; FADH2, flavin adenine dinucleotide, reduced form; FO, flavin oxidizing conformation; FR, flavin reducing conformation; NADPH, nicotinamide adenine dinucleotide phosphate, reduced form; SafO, safranine O; XO, xanthine oxidase; n, the number of electrons transferred in a redox process; v, scan rate; Γ, electroactive coverage; e−, electrons; F, the Faraday; T, temperature; R, the ideal gas constant.
References
- Holmgren A. (1985) Thioredoxin. Annu. Rev. Biochem. 54, 237–271. [DOI] [PubMed] [Google Scholar]
- Arner E. S. J.; Holmgren A. (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–6109. [DOI] [PubMed] [Google Scholar]
- Moore E. C.; Reichard P.; Thelander L. (1964) Enzymatic synthesis of deoxyribonucleotiedes. V. Purification and properties of thioredoxin reductase from Escherichia coli B. J. Biol. Chem. 239, 3445–3452. [PubMed] [Google Scholar]
- Russel M.; Model P. (1988) Sequence of thioredoxin reductase from Escherichia coli—Relationship to other flavoprotein disulfide oxidoreductases. J. Biol. Chem. 263, 9015–9019. [PubMed] [Google Scholar]
- Zanetti G.; Williams C. H. (1967) Characterization of the active center of thioredoxin reductase. J. Biol. Chem. 242, 5232–5236. [PubMed] [Google Scholar]
- Williams C. H. (2000) Thioredoxin-thioredoxin reductase—a system that has come of age. Eur. J. Biochem. 267, 6101–6101. [DOI] [PubMed] [Google Scholar]
- Kuriyan J.; Wong L.; Russel M.; Model P. (1989) Crystallization and preliminary-X-ray characterization of thioredoxin reductase from Escherichia coli. J. Biol. Chem. 264, 12752–12753. [PubMed] [Google Scholar]
- Waksman G.; Krishna T. S. R.; Williams C. H.; Kuriyan J. (1994) Crystal-structure of Escherichia-coli thioredoxin reductase refined at 2-angstrom resolution—Implications for a large conformational change during catalysis. J. Mol. Biol. 236, 800–816. [PubMed] [Google Scholar]
- Lennon B. W.; Williams C. H. (1997) Reductive half-reaction of thioredoxin reductase from Escherichia coli. Biochemistry 36, 9464–9477. [DOI] [PubMed] [Google Scholar]
- Mulrooney S. B.; Williams C. H. (1997) Evidence for two conformational states of thioredoxin reductase from Escherichia coli: Use of intrinsic and extrinsic quenchers of flavin fluorescence as probes to observe domain rotation. Protein Sci. 6, 2188–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veine D. M.; Ohnishi K.; Williams C. H. (1998) Thioredoxin reductase from Escherichia coli: Evidence of restriction to a single conformation upon formation of a crosslink between engineered cysteines. Protein Sci. 7, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. F.; Veine D. M.; Ahn S. H.; Williams C. H. (1996) A stable mixed disulfide between thioredoxin reductase and its substrate, thioredoxin: Preparation and characterization. Biochemistry 35, 4812–4819. [DOI] [PubMed] [Google Scholar]
- Lennon B. W.; Williams C. H.; Ludwig M. L. (1999) Crystal structure of reduced thioredoxin reductase from Escherichia coli: Structural flexibility in the isoalloxazine ring of the flavin adenine dinucleotide cofactor. Protein Sci. 8, 2366–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon B. W.; Williams C. H.; Ludwig M. L. (2000) Twists in catalysis: Alternating conformations of Escherichia coli thioredoxin reductase. Science 289, 1190–1194. [DOI] [PubMed] [Google Scholar]
- Lennon B. W.; Williams C. H. (1995) Effect of pyridine-nucleotide on the oxidative half-reaction of Escherichia coli thioredoxin reductase. Biochemistry 34, 3670–3677. [DOI] [PubMed] [Google Scholar]
- Lennon B. W.; Williams C. H. (1996) Enzyme-monitored turnover of Escherichia coli thioredoxin reductase: Insights for catalysis. Biochemistry 35, 4704–4712. [DOI] [PubMed] [Google Scholar]
- O’Donnell M. E.; Williams C. H. (1983) Proton stoichiometry in the reduction of the Fad and disulfide of Escherichia coli thioredoxin reductase—Evidence for a base at the active-site. J. Biol. Chem. 258, 13795–13805. [PubMed] [Google Scholar]
- Van den Berg P. A. W.; Mulrooney S. B.; Gobets B.; Van Stokkum I. H. M.; Van Hoek A.; Williams C. H.; Visser A. (2001) Exploring the conformational equilibrium of E. coli thioredoxin reductase: Characterization of two catalytically important states by ultrafast flavin fluorescence spectroscopy. Protein Sci. 10, 2037–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez H. H.; Jaquez O. A.; Hamill M. J.; Elliott S. J.; Drennan C. L. (2008) Thioredoxin reductase from Thermoplasma acidophilum: A new twist on redox regulation. Biochemistry 47, 9728–9737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. S.; Bonagura C. A.; Tilley G. J.; McEvoy J. P.; Jung Y. S.; Armstrong F. A.; Stout C. D.; Burgess B. K. (2002) Crystal structures of ferredoxin variants exhibiting large changes in [Fe-S] reduction potential. Nat. Struct. Biol. 9, 188–192. [DOI] [PubMed] [Google Scholar]
- Chen K. S.; Hirst J.; Camba R.; Bonagura C. A.; Stout C. D.; Burgess B. K.; Armstrong F. A. (2000) Atomically defined mechanism for proton transfer to a buried redox centre in a protein. Nature 405, 814–817. [DOI] [PubMed] [Google Scholar]
- Fleming B. D.; Tian Y.; Bell S. G.; Wong L. L.; Urlacher V.; Hill H. A. O. (2003) Redox properties of cytochrome P450(BM3) measured by direct methods. Eur. J. Biochem. 270, 4082–4088. [DOI] [PubMed] [Google Scholar]
- Heering H. A.; Hagen W. R. (1996) Complex electrochemistry of flavodoxin at carbon-based electrodes: Results from a combination of direct electron transfer, flavin-mediated electron transfer and comproportionation. J. Electroanal. Chem. 404, 249–260. [Google Scholar]
- Mulrooney S. B. (1997) Application of a single-plasmid vector for mutagenesis and high-level expression of thioredoxin reductase and its use to examine flavin cofactor incorporation. Protein Expression Purif. 9, 372–378. [DOI] [PubMed] [Google Scholar]
- Massey V. (1991) in Flavins and Flavoproteins 1990 (Curti B., Ronchi S., and Zanetti G., Eds.) pp 59−66, Walter de Gruyter & Co., Berlin. [Google Scholar]
- Cuello A. O.; McIntosh C. M.; Rotello V. M. (2000) Model systems for flavoenzyme activity. The role of N(3)-H hydrogen bonding in flavin redox processes. J. Am. Chem. Soc. 122, 3517–3521. [Google Scholar]
- Lindgren A.; Larsson T.; Ruzgas T.; Gorton L. (2000) Direct electron transfer between the heme of cellobiose dehydrogenase and thiol modified gold electrodes. J. Electroanal. Chem. 494, 105–113. [Google Scholar]
- McEvoy J. P.; Armstrong F. A. (1999) Protein film cryovoltammetry: demonstrations with a 7Fe ([3Fe-4S]+[4Fe-4S]) ferredoxin. Chem. Commun. 1635–1636. [Google Scholar]
- Plichon V.; Laviron E. (1976) Theoretical study of a two-step reversible electrochemical reaction associated with irreversible chemical reactions in thin layer linear potential sweep voltammetry. J. Electroanal. Chem. 71, 143–156. [Google Scholar]
- Savéant J. M. (2006) Elements of molecular and biomolecular electrochemistry, John Wiley & Sons, Hoboken, NJ. [Google Scholar]
- Léger C., and Bertrand, P. (2008) Direct electrochemistry of redox enzymes as a tool for mechanistic studies. Chem. Rev. 108, 2379–2438. [DOI] [PubMed] [Google Scholar]