

Abstract
Dietary salt intake has been linked to hypertension and cardiovascular disease. Accumulating evidence has indicated that salt-sensitive individuals on high salt intake are more likely to develop renal fibrosis. Epithelial-to-mesenchymal transition (EMT) participates in the development and progression of renal fibrosis in humans and animals. The objective of this study was to investigate the impact of a high-salt diet on EMT in Dahl salt-sensitive (SS) rats. Twenty-four male SS and consomic SS-13BN rats were randomized to a normal diet or a high-salt diet. After 4 weeks, systolic blood pressure (SBP) and albuminuria were analyzed, and renal fibrosis was histopathologically evaluated. Tubular EMT was evaluated using immunohistochemistry and real-time PCR with E-cadherin and alpha smooth muscle actin (α-SMA). After 4 weeks, SBP and albuminuria were significantly increased in the SS high-salt group compared with the normal diet group. Dietary salt intake induced renal fibrosis and tubular EMT as identified by reduced expression of E-cadherin and enhanced expression of α-SMA in SS rats. Both blood pressure and renal interstitial fibrosis were negatively correlated with E-cadherin but positively correlated with α-SMA. Salt intake induced tubular EMT and renal injury in SS rats, and this relationship might depend on the increase in blood pressure.
Keywords: Dahl salt-sensitive rats, High salt intake, EMT, Renal fibrosis
Introduction
Salt has been linked to hypertension for many years (1). Previously, it was thought that volume overload induced by salt retention was the main underlying mechanism, and that this in turn was the driving factor for left ventricular hypertrophy. Recently, evidence has accumulated that dietary salt intake by itself, even without causing hypertension or volume overload, might be deleterious, resulting in cardiac remodeling, fibrosis, and left ventricular hypertrophy (2). Also, in kidneys, it has been shown that salt intake leads to enhanced glomerulosclerosis and deterioration of residual renal function (3,4).
Epidemiologic studies have demonstrated that salt-sensitive individuals had a higher incidence of death or cardiovascular events, endothelial dysfunction, and left ventricular hypertrophy than their salt-resistant counterparts (5,6). In addition, salt-sensitive subjects on a high-salt diet showed increased microalbuminuria, elevated renal vascular resistance, and decreased filtration fractions (7,8). Rapid development of renal injury is a prominent feature of salt-induced hypertension in Dahl salt-sensitive (SS) rats. Within a few weeks of high salt exposure, SS rats develop substantial injuries in preglomerular vessels, glomeruli, and the tubulointerstitial compartment (9,10). However, the mechanisms underlying the induction of renal injury, especially renal fibrosis, in salt-sensitive subjects on a high-salt diet are not fully understood.
Recently, a large body of evidence has suggested that epithelial-to-mesenchymal transition (EMT) played a crucial role in the progression of renal fibrosis in humans (11,12) and animals (13). Iwano et al. (13) reported that, in the unilateral ureteral obstruction model, about one-third of fibroblasts were derived from tubular EMT. Additionally, animal experiments have demonstrated that salt loading could induce EMT and peritoneal fibrosis (14). However, there is little information on the relationship between salt loading and renal EMT in SS rats. The aim of this study was to examine the effect of a high-salt diet on tubular EMT in SS and SS-13BN rats.
Material and Methods
Laboratory animals
Experiments were performed in 24 male Dahl SS rats and consomic SS-13BN rats (Charles River Laboratories International, USA) that received care in accordance with the national Guidelines for the Care and Use of Laboratory Animals. The consomic SS-13BN rats were 98% identical to the SS rats, differing only by a subset of genes present on chromosome 13 that were carried from the parental Brown Norway strain (9). They do not develop elevated blood pressure on a high-salt diet as do SS rats (9). The Medical College of Xi'an Jiaotong University Animal Ethics Committee approved the study protocol.
Study protocol
The SS rats were randomly divided into two groups according to their diet, which was either normal (0.3% NaCl, SN group) or high salt (8% NaCl, SH group). Similarly, the SS-13BN rats were randomly divided into two groups and given either a normal diet (0.3% NaCl, BN group) or a high-salt diet (8% NaCl, BH group). Four rats in each treatment group were placed in metabolic cages for 24-h urine collection before termination of the experiment. At the end of 4 weeks, the animals were killed with a 3 mL/kg intraperitoneal injection of 10% chloral hydrate; plasma was collected from the abdominal aorta, and the left kidney was removed and divided into two parts. One part was fixed in 10% neutral buffered formalin and embedded for histological and immunohistochemical examination. The other part was snap-frozen in liquid nitrogen and maintained at -80°C until analysis.
Measurement of biochemical parameters and blood pressure
The blood pressure of conscious rats was measured every 2 weeks using tail-cuff plethysmography (Amplifier Model 229, IITC Life Science, USA). All rats did adaptive training that was adjusted to the blood pressure measurements by tail-cuff plethysmography prior to the final measurements. A cuff was placed around the tail of each rat and inflated to 240 mmHg. Systolic blood pressure (SBP) was recorded as the pressure at the point when the first tail pulse was detected. SBP was measured four times, and the average value was calculated. Serum NaK, serum creatinine, 24-h urinary protein, and 24-h urinary NaK were analyzed with a Hitachi 7060 automatic biochemical analyzer (Hitachi, Ltd., Japan).
Morphometric analysis
After fixation in 10% neutral buffered formalin, tissue was processed with paraffin, sectioned at 5 μm, and stained with Masson's trichrome for evaluation of the collagen deposition within glomeruli and interstitium. The fractional area of collagen in glomeruli and interstitium was determined according to the method described by Moriyama et al. (15). Briefly, 10 glomerular capsules and 10 fields without vessels or glomeruli were randomly selected from each kidney section. The glomerular and tubulointerstitial collagen volume fractions were then calculated as a percentage of the stained area within traced glomerular capsules and as a percentage of the total area within a high-power field, respectively. Glomerular and tubulointerstitial fibrosis were determined separately using the Image Pro Plus 6.0 imaging software (Media Cybernetics, USA).
Immunohistochemistry
Immunohistochemistry was performed using a power vision two-step kit (ZhongShan Golden Bridge Biotechnology, China). Paraffin-embedded fixed tissue sections (5 μm) were deparaffinized, rehydrated, and pretreated for antigen retrieval in Tris/EDTA. The sections were incubated with anti-E-cadherin rabbit polyclonal antibody and anti-α-smooth muscle actin (α-SMA) mouse monoclonal antibody (Santa Cruz Biotechnology, USA) at 4°C overnight, with the secondary antibody at room temperature for 30 min. Positive immunoreaction was identified after incubation with 3,3-diaminobenzidine tetrahydrochloride (ZhongShan Golden Bridge Biotechnology) and counterstaining with hematoxylin. Negative controls without primary antibodies were carefully examined for each reaction. Sections were photographed with an upright fluorescence microscopic imaging system (BX51 Olympus, Japan). To analyze the expression of E-cadherin and α-SMA in glomeruli and interstitium, 10 nonoverlapping high-power fields (magnification 400×) were randomly selected from each kidney section. Data are reported as the mean optical density in each field area.
Quantitative real-time PCR
Renal tissues were homogenized and then RNAiso Plus (Takara Bio, Japan), chloroform and isopropanol were added to the extract tissue RNA. The tissue was then washed with ethanol and solubilized. The concentration and purity of the extracted RNA were determined using a nucleic acid analyzer NANODROP-1000 (Thermo Scientific, USA). Reverse transcription was performed using the RevertAid™ First-Strand cDNA Synthesis kit (Thermo Scientific). All cDNA samples were stored at −40°C until analysis. Quantitative real-time PCR was done using SYBR Premix Ex Taq™ II (Takara Bio) and the IQ™ 5 PCR System (Bio-Rad, USA) according to the manufacturer's protocol with the primers shown in Table 1. Thermal cycling conditions were: 95°C, 10 s predenaturation; 95°C 5 s, 58°C 15 s, 72°C 10 s, for a total of 40 cycles. The samples were run in triplicate and normalized to GAPDH levels, which were used as the endogenous control. Relative expression was determined by comparing mRNA expression of the target genes to that of the endogenous reference gene using the 2−ΔΔCt method.
Statistical analysis
Data are reported as means±SE and analysis was performed with SPSS version 16.0 (SPSS Inc., USA). Comparisons between groups were made using one-way ANOVA followed by a least significant difference post hoc test for multiple comparisons. Correlation between variables was evaluated by the Pearson correlation analysis. Differences were considered to be statistically significant at P<0.05.
Results
General data of laboratory animals
All rats had normal physiological activities and were healthy during the dietary intervention. At baseline, no significant differences in blood pressure were detected among groups. After the salt diet was switched from 0.3 to 8.0%, SBP in the SS rats increased significantly by 2 weeks of high salt when compared with the first day of the 0.3% salt diet (151±3 mmHg) and continued to rise to 184±4 mmHg by 4 weeks of the 8.0% high-salt diet. The blood pressure of SS-13BN rats fed a high-salt diet was significantly higher at 2 weeks than at baseline, and remained stable at 4 weeks (P<0.05; Figure 1). Compared with rats on the normal diet, rats on the high-salt diet showed increased 24-h urinary NaK and elevated albuminuria (P<0.01). In addition, the values of 24-h albuminuria, kidney weight, and the ratio of kidney weight to body weight were significantly higher in the SH group than those of rats in the other groups (P<0.01; Table 2).
Figure 1. Systolic blood pressure (SBP) at baseline, 2 and 4 weeks. SN: Dahl salt-sensitive (SS) rats on a normal-salt diet (0.3% NaCl); SH: Dahl SS rats on a high-salt diet (8% NaCl); BN: SS-13BN rats on a normal-salt diet (0.3% NaCl); BH: SS-13BN rats on a high-salt diet (8% NaCl). +P<0.05, ++P<0.01 compared with the SN group at the same time point; *P<0.05 compared with the SH group at the same time point; #P<0.05 compared with the SN group at the same time point (one-way ANOVA).

Renal fibrosis
As expected, after 4 weeks of salt loading, Masson staining of collagen in kidneys was significantly more pronounced in the high-salt rats than in the normal-diet rats (P<0.01). SH rats showed prominent glomerular and interstitial fibrosis compared with BH rats (P<0.05; Figure 2B and D).
Figure 2. Representative histopathology of renal cortex for all groups; A, SN group; B, SH group; C, BN group; D, BH group. a, Tubulointerstitial and glomerular collagen area fractions were determined by Masson trichrome staining. SN: Dahl salt-sensitive (SS) rats on a normal-salt diet (0.3% NaCl); SH: Dahl SS rats on a high-salt diet (8% NaCl); BN: SS-13BN rats on a normal-salt diet (0.3% NaCl); BH: SS-13BN rats on a high-salt diet (8% NaCl). One-way ANOVA was used for statistical analysis.

Tubular EMT
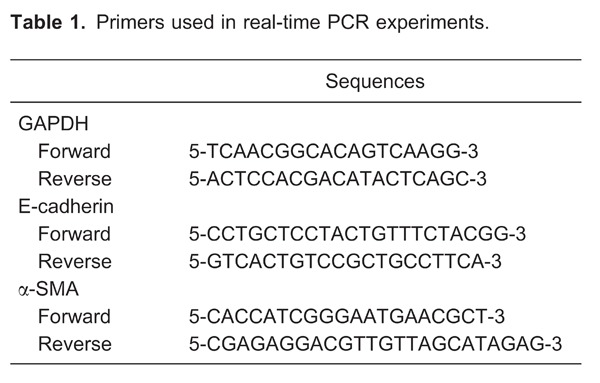
To demonstrate EMT in tubular epithelial cells, we examined the expression of E-cadherin, a phenotypic marker of epithelial cells, and α-SMA, a phenotypic marker of myofibroblast cells. As shown in Figure 3, in kidneys of rats on a normal diet, E-cadherin was strongly expressed only in renal tubular epithelial cells. The expression of E-cadherin in tubular epithelial cells of the SH group was significantly decreased compared with the SN group (P<0.01). However, immunohistochemical results and quantification of tissue for E-cadherin showed that E-cadherin expression was not statistically reduced in the BH group compared with the BN group (Figure 3A, C, and D).
Figure 3. Representative images of immunostaining for E-cadherin in the renal medulla of all groups. A, SN group; B, SH group; C, BN group; D, BH group. a, Positive immunostaining of E-cadherin was quantified in renal medulla. SN: Dahl salt-sensitive (SS) rats on a normal-salt diet (0.3% NaCl); SH: Dahl SS rats on a high-salt diet (8% NaCl); BN: SS-13BN rats on a normal-salt diet (0.3% NaCl); BH: SS-13BN rats on a high-salt diet (8% NaCl). One-way ANOVA was used for statistical analysis.
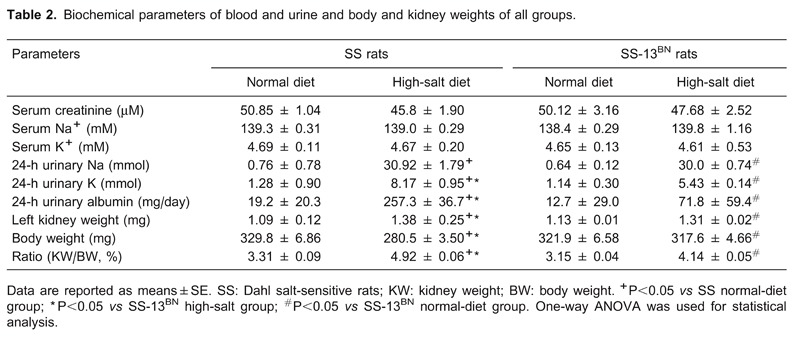
As shown in Figure 4, staining for α-SMA was limited to the muscularis of the blood vessels in all the normal-diet rats. In BH and SH rats, additional, extensive staining was observed in the glomeruli and interstitium. Quantification of α-SMA-positive areas in the glomeruli and interstitium, as summarized in Figure 4A, indicated a significantly higher expression of α-SMA in the SH group than in the normal-diet group (P<0.01).
Figure 4. Representative images of immunostaining for α-SMA in renal medulla of all groups. A, SN group; B, SH group; C, BN group; D, BH group. a, Positive immunostaining of α-SMA was quantified in both glomeruli and interstitium. SN: Dahl salt-sensitive (SS) rats on a normal-salt diet (0.3% NaCl); SH: Dahl SS rats on a high-salt diet (8% NaCl); BN: SS-13BN rats on a normal-salt diet (0.3% NaCl); BH: SS-13BN rats on a high-salt diet (8% NaCl). One-way ANOVA was used for statistical analysis.
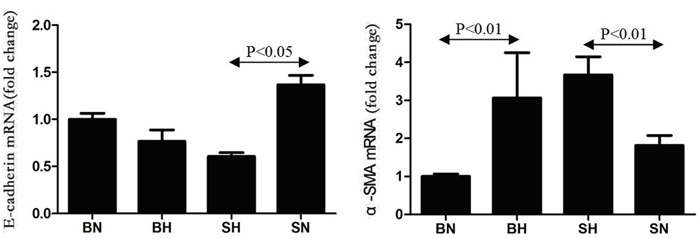
mRNA expression of E-cadherin and α-SMA
Compared with the SN group, E-cadherin mRNA expression was significantly decreased in the SH rats (P<0.01), but it was not significantly decreased in the BH rats compared with the BN group (P>0.05). Expression of α-SMA mRNA was significantly increased in BH and SH rats compared with the normal control groups (Figure 5).
Figure 5. Kidney E-cadherin and α-SMA mRNA expression in all groups. SN: Dahl salt-sensitive (SS) rats on a normal-salt diet (0.3% NaCl); SH: Dahl SS rats on a high-salt diet (8% NaCl); BN: SS-13BN rats on a normal-salt diet (0.3% NaCl); BH: SS-13BN rats on a high-salt diet (8% NaCl). One-way ANOVA was used for statistical analysis.
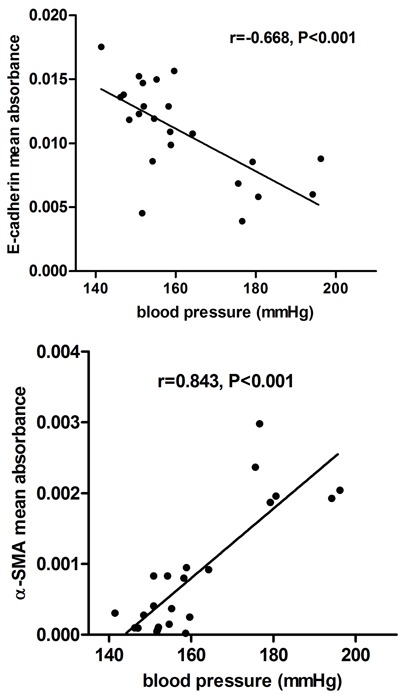
Relationship among EMT, blood pressure, and renal fibrosis
There was a significant negative correlation of blood pressure with E-cadherin immunostaining, and a significant positive correlation with immunostaining of renal interstitial α-SMA in all groups (P<0.001; Figure 6). Renal interstitial fibrosis was negatively related to the expression of E-cadherin (r=-0.778, P<0.001), and positively related to renal interstitial α-SMA (r=0.879, P<0.001) in all groups.
Figure 6. Correlation between blood pressure and positive immunostaining of E-cadherin and α-SMA in rat kidneys from all groups.
Discussion
In the present study, we found that after salt loading, when the blood pressure in SS rats rose to 184 mmHg we could observe the occurrence of EMT. However, high salt did not induce tubular EMT in SS-13BN rats when the blood pressure was only 155 mmHg at 4 weeks. In addition, SS rats on the normal diet for 4 weeks did not demonstrate EMT when blood pressure was 151 mmHg, and tubular EMT was closely correlated with blood pressure. These observations suggest that dietary salt intake could induce EMT of tubular epithelial cells in SS rats, and this relationship might depend on the corresponding increase in blood pressure.
Increasing evidence from multiple clinical studies shows that excess salt intake is related to cardiovascular organ damage, independent of blood pressure (2). Arterial pressure-independent profibrotic effects of salt in the heart and kidneys have been shown in SHR and WKY rats (16). Moreover, according to a recent study (14), dietary salt intake by itself induces EMT, as identified by colocalization of cytokeratin and α-SMA in the submesothelial layer in the kidneys of Wistar rats. However, the relationship between high salt and EMT in SS rats has not been fully established. In the present study, we showed that salt intake induced tubular EMT in SS rats, but this relationship might be dependent on elevated blood pressure. Our findings are consistent with a previous study illustrating that elevations of renal perfusion pressure contributed significantly to EMT found in the early phases of hypertension in SS rats (17). In that study, after 2 weeks of a 4% salt diet, DNA microarray and real-time PCR were used to identify some genes related to EMT in the kidneys exposed to hypertension (17).
Previous evidence has shown that tubular epithelial cells (18), endothelial cells (19), podocytes (20), and glomerular epithelial cells (21) in the kidney can undergo phenotypic transformation. Kalluri and Neilson (22) reported that, during the course of kidney fibrosis in mice, about 30% of fibroblasts were derived via EMT from the tubular epithelial cells of the kidney. Iwano et al. (13) demonstrated that, in unilateral ureteral obstructive rats, more than one-third of renal interstitial fibroblasts, the main mediators of renal interstitial fibrosis, were derived from renal tubular epithelium via EMT. In addition, tubular EMT was also associated with tubulointerstitial fibrosis in chronic allograft nephropathy and kidney stone disease (11,12). In accordance with these previous studies, we found that the severity of renal interstitial fibrosis in SS rats was strongly related to tubular EMT (r=-0.778 for E-cadherin; r=0.879 for α-SMA).
The precise mechanism(s) whereby elevations in blood pressure can induce tubular EMT in SS rats remains to be determined. In wound healing, it has been found that mechanical forces stimulate transforming growth factor β1 (TGF-β1)-associated EMT (22). TGF-β1 was also shown to induce all the basic steps of EMT. Yang and Liu (18) demonstrated that, when HKC-8 cells were treated with TGF-β1, loss of E-cadherin expression occurred and α-SMA expression was induced after 36 h. The study further demonstrated that TGF-β1 was able to induce α-SMA at a concentration of 0.1 ng/mL. TGF-β1-induced EMT also played a major role in progression of renal fibrosis in a mouse model of diabetic nephropathy (23). In addition, Tamaki et al. (4) reported that TGF-β1 significantly increased in kidneys of SS rats that were kept on a high-salt diet for 4 weeks. On the basis of these studies, we speculated that high blood pressure might induce EMT mainly through a TGF-β1 pathway in SS rats. We cannot exclude the possibility that other factors may be involved in pressure-induced EMT in SS rats, and this needs further testing.
In our study, we found that dietary salt intake significantly increased albuminuria and levels of renal fibrosis in SS rats. This phenomenon is consistent with the results of previous studies suggesting that renal function worsened in rat models of chronic allograft nephropathy on a diet of 8.0% salt (24). However, the mechanisms underlying the progression of salt-induced kidney damage were not elucidated in our study. Several mechanisms may be responsible for this: i) high salt intake could lead to increased blood pressure, which may result in renal dysfunction (17); ii) salt loading upregulated TGF-β1 production in the renal medulla (4), as long-term administration of an anti-TGF-β1 antibody has also been found to lower blood pressure and attenuate kidney damage in SS rats (25), and iii) oxidative stress may be elevated in the kidneys of SS rats (26).
In conclusion, this study demonstrated that salt intake induced tubular EMT in SS rats, and this relationship might depend on the increase in blood pressure.
Acknowledgments
Research supported by the National Program on Key Basic Research Project of China (973 Program, grant #2012CB517804) and grants #81200512 and #81070218 from the National Science Foundation of China.
Footnotes
First published online March 18, 2014.
References
- 1.Haddy FJ. Role of dietary salt in hypertension. Life Sci. 2006;79:1585–1592. doi: 10.1016/j.lfs.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 2.Frohlich ED, Varagic J. Sodium directly impairs target organ function in hypertension. Curr Opin Cardiol. 2005;20:424–429. doi: 10.1097/01.hco.0000175519.34933.a5. [DOI] [PubMed] [Google Scholar]
- 3.Sanders PW. Salt intake, endothelial cell signaling, and progression of kidney disease. Hypertension. 2004;43:142–146. doi: 10.1161/01.HYP.0000114022.20424.22. [DOI] [PubMed] [Google Scholar]
- 4.Tamaki K, Okuda S, Nakayama M, Yanagida T, Fujishima M. Transforming growth factor-beta 1 in hypertensive renal injury in Dahl salt-sensitive rats. J Am Soc Nephrol. 1996;7:2578–2589. doi: 10.1681/ASN.V7122578. [DOI] [PubMed] [Google Scholar]
- 5.Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S, et al. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet. 1997;350:1734–1737. doi: 10.1016/S0140-6736(97)05189-1. [DOI] [PubMed] [Google Scholar]
- 6.Heimann JC, Drumond S, Alves AT, Barbato AJ, Dichtchekenian V, Marcondes M. Left ventricular hypertrophy is more marked in salt-sensitive than in salt-resistant hypertensive patients. J Cardiovasc Pharmacol. 1991;17(Suppl 2):S122–S124. doi: 10.1097/00005344-199117002-00028. [DOI] [PubMed] [Google Scholar]
- 7.Bihorac A, Tezcan H, Ozener C, Oktay A, Akoglu E. Association between salt sensitivity and target organ damage in essential hypertension. Am J Hypertens. 2000;13:864–872. doi: 10.1016/S0895-7061(00)00253-3. [DOI] [PubMed] [Google Scholar]
- 8.Campese VM, Parise M, Karubian F, Bigazzi R. Abnormal renal hemodynamics in black salt-sensitive patients with hypertension. Hypertension. 1991;18:805–812. doi: 10.1161/01.HYP.18.6.805. [DOI] [PubMed] [Google Scholar]
- 9.Rapp JP, Dene H. Development and characteristics of inbred strains of Dahl salt-sensitive and salt-resistant rats. Hypertension. 1985;7:340–349. [PubMed] [Google Scholar]
- 10.Chen PY, St John PL, Kirk KA, Abrahamson DR, Sanders PW. Hypertensive nephrosclerosis in the Dahl/Rapp rat. Initial sites of injury and effect of dietary L-arginine supplementation. Lab Invest. 1993;68:174–184. [PubMed] [Google Scholar]
- 11.Bedi S, Vidyasagar A, Djamali A. Epithelial-to-mesenchymal transition and chronic allograft tubulointerstitial fibrosis. Transplant Rev. 2008;22:1–5. doi: 10.1016/j.trre.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boonla C, Krieglstein K, Bovornpadungkitti S, Strutz F, Spittau B, Predanon C, et al. Fibrosis and evidence for epithelial-mesenchymal transition in the kidneys of patients with staghorn calculi. BJU Int. 2011;108:1336–1345. doi: 10.1111/j.1464-410X.2010.10074.x. [DOI] [PubMed] [Google Scholar]
- 13.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI0215518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pletinck A, Consoli C, Van Landschoot M, Steppan S, Topley N, Passlick-Deetjen J, et al. Salt intake induces epithelial-to-mesenchymal transition of the peritoneal membrane in rats. Nephrol Dial Transplant. 2010;25:1688–1696. doi: 10.1093/ndt/gfq036. [DOI] [PubMed] [Google Scholar]
- 15.Moriyama T, Kawada N, Nagatoya K, Takeji M, Horio M, Ando A, et al. Fluvastatin suppresses oxidative stress and fibrosis in the interstitium of mouse kidneys with unilateral ureteral obstruction. Kidney Int. 2001;59:2095–2103. doi: 10.1046/j.1523-1755.2001.00724.x. [DOI] [PubMed] [Google Scholar]
- 16.Yu HC, Burrell LM, Black MJ, Wu LL, Dilley RJ, Cooper ME, et al. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation. 1998;98:2621–2628. doi: 10.1161/01.CIR.98.23.2621. [DOI] [PubMed] [Google Scholar]
- 17.Mori T, Polichnowski A, Glocka P, Kaldunski M, Ohsaki Y, Liang M, et al. High perfusion pressure accelerates renal injury in salt-sensitive hypertension. J Am Soc Nephrol. 2008;19:1472–1482. doi: 10.1681/ASN.2007121271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 2008;172:299–308. doi: 10.2353/ajpath.2008.070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bariety J, Bruneval P, Hill GS, Mandet C, Jacquot C, Meyrier A. Transdifferentiation of epithelial glomerular cells. J Am Soc Nephrol. 2003;14(Suppl 1):S42–S47. doi: 10.1097/01.ASN.0000067651.34743.B0. [DOI] [PubMed] [Google Scholar]
- 22.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI200320530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hills CE, Squires PE. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am J Nephrol. 2010;31:68–74. doi: 10.1159/000256659. [DOI] [PubMed] [Google Scholar]
- 24.Sanders PW, Gibbs CL, Akhi KM, Millan-Crow LA, Zinn KR, Chen YF, et al. Increased dietary salt accelerates chronic allograft nephropathy in rats. Kidney Int. 2001;59:1149–1157. doi: 10.1046/j.1523-1755.2001.0590031149.x. [DOI] [PubMed] [Google Scholar]
- 25.Dahly AJ, Hoagland KM, Flasch AK, Jha S, Ledbetter SR, Roman RJ. Antihypertensive effects of chronic anti-TGF-beta antibody therapy in Dahl S rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R757–R767. doi: 10.1152/ajpregu.00098.2002. [DOI] [PubMed] [Google Scholar]
- 26.Taylor NE, Glocka P, Liang M, Cowley AW., Jr NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats. Hypertension. 2006;47:692–698. doi: 10.1161/01.HYP.0000203161.02046.8d. [DOI] [PubMed] [Google Scholar]