

Abstract
Studies have indicated that early-life or early-onset depression is associated with a 2- to 4-fold increased risk of developing Alzheimers disease (AD). In AD, aggregation of an abnormally phosphorylated form of the tau protein may be a key pathological event. Tau is known to play a major role in promoting microtubule assembly and stabilization, and in maintaining the normal morphology of neurons. Several studies have reported that stress may induce tau phosphorylation. The main aim of the present study was to investigate possible alterations in the tau protein in the hippocampus and frontal cortex of 32 male Sprague-Dawley rats exposed to chronic unpredictable mild stress (CUMS) and then re-exposed to CUMS to mimic depression and the recurrence of depression, respectively, in humans. We evaluated the effects of CUMS, fluoxetine, and CUMS re-exposure on tau and phospho-tau. Our results showed that a single exposure to CUMS caused a significant reduction in sucrose preference, indicating a state of anhedonia. The change in behavior was accompanied by specific alterations in phospho-tau protein levels, but fluoxetine treatment reversed the CUMS-induced impairments. Moreover, changes in sucrose preference and phospho-tau were more pronounced in rats re-exposed to CUMS than in those subjected to a single exposure. Our results suggest that changes in tau phosphorylation may contribute to the link between depression and AD.
Keywords: Alzheimers disease, Depression, Tau, Chronic unpredictable mild stress (CUMS), Re-exposure to CUMS, Fluoxetine
Introduction
Previous studies have shown an association between depression and dementia, and it has been suggested that lifetime depression is associated with a 2- to 4-fold increased risk of developing Alzheimers disease (AD) (1-4). For example, a cross-sectional (case-controlled) study suggested that a history of depression, even 25 years prior to AD onset, was significantly associated with an almost 2-fold increase in the likelihood of developing AD, and that depression symptoms within the year before the onset of AD were associated with an almost 5-fold increased risk (4).
The association between depression and AD suggested by the epidemiologic data may be partially explained by several biological mechanisms, including alterations in glucocorticoid steroids, hippocampal atrophy, increased deposition of -amyloid plaques, inflammatory changes, deficits in nerve growth factors, and altered neuronal plasticity (1,5,6). Aggregation of abnormally phosphorylated tau, the main constituent of neurofibrillary tangles, is most likely a key pathological event of AD (7). Interestingly, research has shown that tangles accumulate in greater numbers in the hippocampus of AD patients with depression than in those without depression (8,9).
Growing evidence indicates that the cytoskeleton, a structural system including microtubules and microtubule-associated proteins (MAPs), may play an important role in stress-induced impairments of neuronal structural plasticity (10). Accounting for more than 80 of the MAPs, tau is known to promote microtubule assembly, stabilize existing microtubules, and to maintain the normal morphology of neurons (7). Developmental and functional regulation of phosphorylation modulates the function of tau proteins in controlling microtubule dynamics during normal neurite outgrowth and maturation. Proper regulation of microtubule dynamics (assembly and disassembly) is essential for normal cell morphology, function, and viability. Accumulating evidence suggests that hyperphosphorylated tau sequesters normal MAPs and disrupts microtubule dynamics (7). Furthermore, acute stresses, such as ether exposure, starvation, forced swimming in cold water, or restraint stress, induce tau phosphorylation in mice (11).
Based upon these studies, we speculated that chronic stress in depression could produce an increase in tau phosphorylation similar to that observed after acute stress. Thus, antidepressant drugs could possibly attenuate the impact of depression on development of AD. It would be interesting to explore what role tau might play (e.g., whether changes in tau in depression are similar to those in AD and what effects antidepressant drugs have on tau). The primary aim of the present study, therefore, was to investigate changes in tau in the rat hippocampus and frontal cortex following stress, fluoxetine (FLX) treatment, and re-exposure to stress.
It is believed that chronic stress is a crucial factor involved in depression onset and relapse (12). Chronic unpredictable mild stress (CUMS)-induced depression is seen as the most promising and valuable depressive model in animals, and it has been widely used for investigating the pathophysiology of depression and its associated therapeutic interventions (13). Therefore, we used CUMS in our study.
Material and Methods
Animals
A total of 32 male Sprague-Dawley rats weighing 200-250g were obtained from the Center for Experimental Animals at Wuhan University for use in this experiment. All animals were housed in groups of four to five in plastic cages with a controlled environment, a 12:12-h light-dark cycle, and ad libitum food and water for 1 week before the start of the investigation. The experimental procedures were conducted in conformity with the instructions for the care of laboratory animals issued by the Ministry of Science and Technology of the Peoples Republic of China in 2006.
FLX treatment
FLX (ShangHai JinHuan, China) was dissolved in physiological saline immediately before each use. Intraperitoneal injections of FLX (10mg/kg) (14) or saline vehicle (V) were administered daily to the rats for 3 weeks.
Experimental design
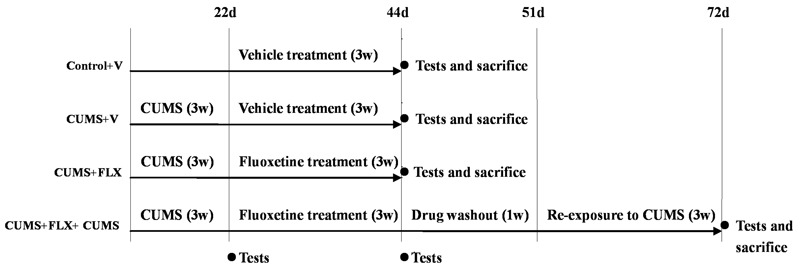
The animals were separated into one of four groups: ControlV (n8), CUMSV (n8), CUMSFLX (n8), or CUMSFLXCUMS (n8). The fourth group included a 1-week drug washout prior to re-exposure to CUMS. Sucrose preference was used to assess potential behavioral abnormalities after CUMS, FLX treatment, and re-exposure to stress. The rats were housed in groups during the experimental period, while the stress conditions, all injections, and behavioral assessments were delivered in isolation. Details of the experimental procedures are given in Figure 1.
Figure 1. Details of the experimental procedure. ControlV, CUMSV, and CUMSFLX rats were tested (via the sucrose preference test) at 22 days and 44 days and sacrificed within 12h of the last test. Rats in the CUMSFLXCUMS group were tested (via the sucrose preference test) at 22, 44, and 72 days, and sacrificed within 12h of the last test. V: vehicle CUMS: chronic unpredictable mild stress FLX: fluoxetine d: days w: weeks.
CUMS
The CUMS procedure was adopted with slight modifications from that described by Luo et al. (15), and some stressors (e.g., empty bottle of water, restricted food) were adapted from Moreau et al. (16). The rats were subjected to various stressors for 3 weeks. These included cage tilting (45) for 24h, damp sawdust (200mL water in a cage) for 24h, predator sounds (recording of an adult cat for 15min), swimming in 25C water for 15min, 24h of food deprivation immediately followed by 1h of restricted access to food (5micro-pellets), 24h of water deprivation immediately followed by 1-h exposure to an empty bottle, tail clamping for 1min, shaking for 15min at 120rpm on a rocking bed, immobilization for 1h in a 258-cm cylindrical plastic rodent restrainer, and alterations of the light-dark cycle. The animals were exposed to one stressor at a time stressors were never presented simultaneously.
Sucrose preference test
In the present study, we used a sucrose preference test to assess anhedonia. All animals were first trained to consume a palatable, weak (1) sucrose solution. Before each test, the animals were deprived of water for 12h (8:00 pm-8:00 am). The rats were presented with a bottle of 1 sucrose solution and a bottle of water during a 1-h window (8:00-9:00 am) as originally described by Willner et al. (17). The sucrose preference was calculated according to the following formula: sucrose preference ()(sucrose intake/total fluid intake)100.
Drug washout period
Although FLX and norfluoxetine have half-lives of several days in humans, in rats the elimination half-lives are approximately 9 and 15h, respectively (18). Thus, it is likely that tissue concentrations of this selective serotonin reuptake inhibitor (SSRI) fall substantially after 1 day (19). The washout period for FLX is reported to be different in different articles. We adopted 1 week as the washout period in the present study.
Western blot analysis of tau
At the end of the last test, the animals were anesthetized with 20 ethylcarbamate (7mL/kg) at room temperature and decapitated. The brain was rapidly separated from the skull, and the hippocampus and frontal cortex were dissected from both hemispheres while the brain was on an ice-chilled plate, and they were then immediately frozen on dry ice and kept at 80C until analysis.
Western blot analysis was performed as previously reported (20), with slight modifications. Briefly, frozen tissue samples were thawed and homogenized in buffer. Homogenates were cleared by centrifugation (12,000 g, 30min, 4C) and boiled for 5min in Laemmli buffer. The protein samples were separated at 70V by 10 sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The amount of protein loaded was 10g for the total tau analysis and 40g for phospho-tau (Ser356 and Thr231) analyses. After electrophoresis, the proteins were transferred to nitrocellulose membranes (0.45-m pore size Millipore Corp., USA) using a transfer unit run at 200mA for 1h for the analyses of phospho-tau and tau (Bio-Rad, USA). A molecular size marker (250-11.0kDa range Sigma, USA) was used to localize tau (50-80kDa) and phospho-tau (Ser356, 46-80kDa Thr231, 60kDa). The membranes were blocked for 2h with 10 skim milk powder and incubated (overnight at 4C) with primary antibodies diluted in 5 skim milk powder in Tris-buffered saline as follows: tau (Tau46, Cell Signaling, USA) at a 1:1000 dilution, phospho-tau (Ser356, Santa Cruz, USA) at a 1:100 dilution, phospho-tau (Thr231, Abcam, USA) at a 1:5000 dilution. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (Promega, USA) for 1h. The protein band intensities were determined densitometrically using the CMIASWIN video imaging system (Bio-Rad). The densitometric values are reported as percentages of the control values. Glyceraldehyde phosphate dehydrogenase was used to normalize the quantification in Western blot experiments.
Statistical analyses
The results are reported as meansSD. Data were analyzed with a one-way ANOVA followed by the Bonferroni test for post hoc multiple comparisons. Statistically significant group differences were set at P0.05.
Results
Sucrose preference tests
We used the sucrose preference test to explore the effects of CUMS and re-exposure to CUMS on anhedonia in rats. Anhedonia is defined as the decreased capacity to experience pleasure of any sort. The theoretical rationale for the chronic mild stress model is that this procedure simulates anhedonia, a loss of responsiveness to pleasant events, which is a core symptom of depression and the defining feature of melancholia (13).
Figure 2 shows the sucrose preference in the four groups of rats during the experimental period. As shown, 3 weeks of chronic stress induced a marked decrease in sucrose consumption in the stressed rats compared with the non-stressed rats (Figure 2A). After the first CUMS administration at 3 weeks, the percentage of sucrose consumed by the stressed animals was significantly lower than that of the non-stressed animals F(3,28)8.937, P0.05. These results suggest that CUMS consistently induced an anhedonia.
Figure 2. A, Effects of chronic unpredictable mild stress (CUMS) on sucrose preference in rats. Prior to each test, rats were water-deprived for 12h. They were then exposed to both a 1 sucrose solution and tap water. B, Effects of fluoxetine (FLX) on sucrose preference in rats. C, Effects of re-exposure to CUMS on sucrose preference in rats. Data are reported as meansSD. P0.01 (Panels A and B) and P0.05 (Panel C) (one-way ANOVA followed by the Bonferroni test). V: vehicle d: days.
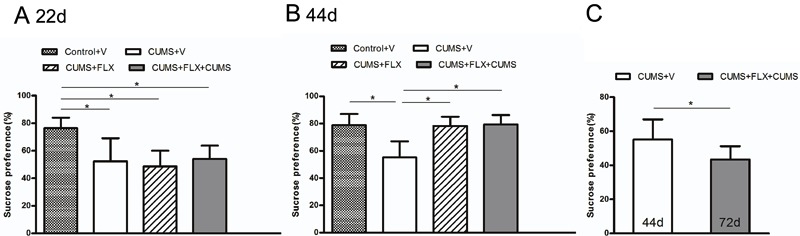
Fluoxetine is a representative SSRI and was chosen as a reference compound for the validation of the chronic mild stress model (21). One-way ANOVA indicated that there were significant differences in the percentage of sucrose consumed among the four groups at the end of FLX treatment F(3,28)14.918, P0.05 (Figure 2B). Post hoc analysis found no differences between the control animals and the animals treated with FLX for 3 weeks. In contrast, the percentage of sucrose consumed was significantly different between the CUMSV group and the other groups (P0.01 for all comparisons). These results suggest that, after 3 weeks of FLX treatment, the sucrose preference changes of the stressed rats were reversed.
After drug washout, the CUMSFLXCUMS group received an additional exposure to CUMS. We compared the results of CUMSV at 44 days and the results of CUMSFLXCUMS at 72 days (Figure 2C). The animals that were re-treated with CUMS consumed significantly less sucrose solution (P0.05) than the CUMSV rats (Figure 2C). This suggests that, after re-exposure to CUMS, the anhedonia in the CUMSFLXCUMS rats became more intense.
Effects of CUMS, FLX, and re-exposure to CUMS on tau and phospho-tau (Thr231 and Ser356) protein levels in the rat hippocampus and frontal cortex
Acute stress has been reported to induce robust hyperphosphorylation of tau in the frontal cortex and hippocampus of animals (22). Furthermore, dysfunction of the frontal cortex has been implicated in the neurobiology of depressive illnesses (23). Thus, to investigate whether depression could promote detrimental tau hyperphosphorylation, we quantified the levels of phospho-tau in both the hippocampus and the frontal cortex of chronic-stressed animals. According to the product information, the tau46 antibody recognizes hippocampal tau regardless of its phosphorylation state. The p-tau (Ser356) and p-tau (Thr231) antibodies stain only the phosphorylated form of rat tau, and only when it is phosphorylated at serine 356 and threonine 231, respectively. The effects of stress and FLX on phospho-tau protein levels in the hippocampus and frontal cortex are reported in Figure 3A and B, respectively.
Figure 3. Effects of chronic unpredictable mild stress (CUMS), fluoxetine (FLX), and re-exposure to CUMS on the levels of tau and phospho-tau in the hippocampi and the frontal cortex of rats. GAPDH was used to normalize the quantification in Western blot experiments. Representative Western blot bands of the hippocampi (A) and the frontal cortex (B) and densitometric analyses of the bands for p-Tau (Ser356) and p-Tau (Thr231) ratio of the hippocampi, and p-Tau (Ser356) and p-Tau (Thr231) ratio of the frontal cortex. Cont: Controlvehicle (V) CUMS: CUMSV FLX: CUMSFLX Re-CUMS: CUMSFLXCUMS. Data are reported as meansSD. P0.01 (one-way ANOVA followed by the Bonferroni test).
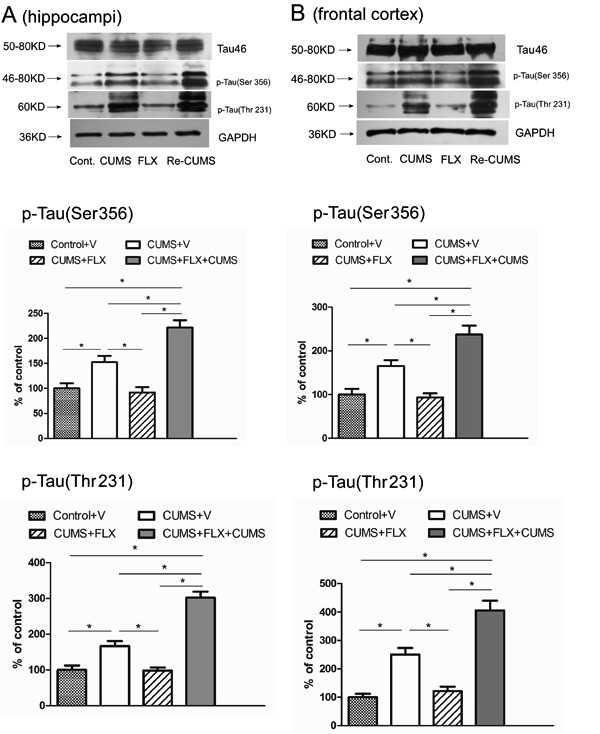
One-way ANOVA revealed that, in the hippocampus and frontal cortex, the staining of tau by tau46 was not significantly altered after stress or re-exposure to stress (Figure 3A and B, first band), but the levels of phospho-tau (Ser356 and Thr231) were significantly altered after stress and re-exposure to stress (Figure 3A and B, middle and bottom panels.
In the hippocampus, densitometric analyses showed that phospho-tau (Ser356) protein levels increased significantly (15212, P0.01) in CUMSV rats subjected to stress once, and also increased significantly (22114, P0.01) following re-exposure to stress in the CUMSFLXCUMS group, compared with the ControlV group. Densitometric analyses of phospho-tau (Thr231) protein levels showed a significant 16614 increase (P0.01) in CUMSV rats subjected to stress once, and a significant 30217 increase (P0.01) in the CUMSFLXCUMS group following re-exposure to stress, compared with the ControlV group. Post hoc analysis showed that the densitometric values of phospho-tau (Ser356 and Thr231) protein levels in the hippocampus were higher in the CUMSFLXCUMS group compared with the CUMSV group (P0.01 for both, Bonferroni test). However, phospho-tau (Ser356 and Thr231) protein levels in the CUMSFLX rats did not differ significantly from the ControlV group (P0.05 for both).
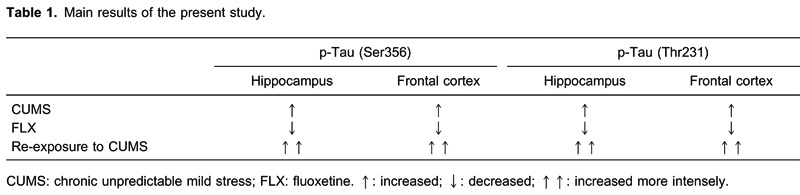
The results in the frontal cortex were very similar to those in the hippocampus. CUMS also increased the levels of phospho-tau (Ser356 and Thr231), and rats that received an additional CUMS exposure after recovering from CUMS-induced depression (i.e., anhedonia) showed higher phospho-tau (Ser356 and Thr231) protein levels. Details of the main results are given in Table 1.
Discussion
The present study revealed elevation of the levels of phosphorylated tau in the hippocampus and frontal cortex of depressive animals, which may indicate a link between depression and AD. In AD brains, tau moves from the axons to the somatodendritic compartment of neurons, where it forms hyperphosphorylated, filamentous aggregates described as neurofibrillary tangles. Postmortem studies report greater neurofibrillary tangle pathology in AD patients with a lifetime history of depression compared with those without such a history (8), and more severe cortical neurofibrillary tangle pathology is found in the brains of AD subjects who suffered from comorbid depression (9). The association between depression and AD may emerge from the impact that depression and depressive symptoms have on the hypothalamic-pituitary-adrenal axis, resulting in impaired negative feedback and chronic elevation of adrenal glucocorticoids (24). In turn, in animal studies, glucocorticoids have been reported to promote amyloid precursor protein expression and tau accumulation (25,26). Thus, longer glucocorticoid exposure augments tau accumulation and might additionally lead to pathological alterations in tau, including alterations in its phosphorylation state. Based upon these studies, we propose that depression leads to elevated tau phosphorylation, which is a key pathological agent in the development of AD, and that glucocorticoids may play a critical role in tau phosphorylation. Thus, our findings may offer an insight into the mechanisms behind the link between depression and AD that is suggested by epidemiologic data showing prior depression to be an etiologic risk factor for AD. However, the currently available data are insufficient to support an exclusive association between depression and AD per se. The precise mechanism remains unclear and requires further research.
The alterations in tau phosphorylation observed here suggest an additional mechanism, by which depression decreases microtubule dynamics. In a previous study (27), we showed that CUMS induced a decrease in Tyr-Tub and an increase in Acet-Tub protein levels, which together suggest decreased microtubule dynamics. The neuronal hyperphosphorylation of tau is a well-known cytoskeletal change related to decreased microtubule dynamics (28). Changes in the dynamic status of the microtubule cytoskeleton are directly related to neuronal morphology (11). The intrinsic dynamic instability of microtubules is fundamental in regulating the axonal trafficking of vesicles (10). Hyperphosphorylated tau is prone to form aggregates, which may block the intracellular trafficking of neurotrophics and other functional proteins. This may result in a loss or decline in axonal or dendritic transport in the neurons (29). Neurons with tau hyperphosphorylation are considered sick because these neurons are no longer competent for axonal transport (30), an early marker of neurodegeneration in neurodegenerative diseases, including AD (30).
The severity of depression has been found to be an important predictor of the risk of dementia. One longitudinal study reported that depression led to as much as a 2-fold increased risk of a diagnosis of dementia over 7 years compared to controls without dysthymia or depression and that there was a dose-effect wherein depression resulted in a higher risk than dysthymia (31). Another study demonstrated a strong association between the number of depressive episodes (i.e., recurrent depression) and the risk of dementia over a median follow-up time of 24 years, suggesting a cumulative, dose-dependent relationship of depression episodes and risk of dementia (32). Our study showed that, after re-exposure to CUMS, sucrose consumption in the CUMSFLXCUMS group was further impaired and was associated with even higher phospho-tau protein levels. We cautiously interpret our findings as a partial explanation of the evidence from clinical studies showing that patients with recurrent depression and more severe episodes have a higher risk of dementia(s) and AD than those with a single episode or multiple, less severe episodes. However, more research is required to determine the mechanisms.
We chose to investigate two phosphorylation sites (Thr231 and Ser356). Phosphorylation of tau at Thr231 plays a critical role in regulating taus ability to bind and stabilize microtubules (33), and increased phosphorylation of Thr231 has been proposed as an early event in AD pathogenesis (34). Ser356 is located in the tau microtubule-binding domain, and hyperphosphorylation at this site is thought to cause a greater decrease in taus promotion of microtubule assembly and stabilization than hyperphosphorylation at other sites (7).
Antidepressants are a rational complementary therapy for the treatment of AD (35). In fact, long-term treatment with SSRIs including FLX improves memory function in AD patients (36). Long-term paroxetine treatment significantly reduces tau immunoreactivity in the hippocampus (37). The results of our study, which showed that FLX decreased tau phosphorylation in the hippocampus and frontal cortex, are consistent with previous paroxetine studies. The possible mechanisms of these antidepressant effects on AD include stimulating neurogenesis, increasing long-term potentiation, anti-inflammatory activity, and inhibiting N-methyl-D-aspartate receptors (30,38). The decrease in tau pathology by FLX that was observed in our study may offer another clue to the understanding of antidepressant action in AD. However, the mechanisms by which paroxetine and FLX suppress tau pathology remain unknown. The antidepressant actions of paroxetine and FLX are thought to be mediated by the inhibition of serotonin reuptake, resulting in enhanced serotonergic signaling and up-regulation of brain-derived neurotrophic factor gene expression (39). Further research is required to gain understanding of these changes, for example, by testing specific serotonin receptor agonists or antagonists.
In conclusion, our results suggest that elevated phosphorylated tau levels following chronic stress may contribute to the mechanisms of depression as an etiologic risk factor for AD, as suggested by epidemiologic data. The efficacy of FLX in modifying tau phosphorylation may offer another clue to why antidepressants are a rational complementary therapy for AD.
Acknowledgments
We sincerely thank the Key Laboratory of HuBei Province for Digestive System Diseases for its support.
Footnotes
First published online March 3, 2014.
References
- 1.Byers AL, Yaffe K. Depression and risk of developing dementia. Nat Rev Neurol. 2011;7:323–331. doi: 10.1038/nrneurol.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geerlings MI, den Heijer T, Koudstaal PJ, Hofman A, Breteler MM. History of depression, depressive symptoms, and medial temporal lobe atrophy and the risk of Alzheimer disease. Neurology. 2008;70:1258–1264. doi: 10.1212/01.wnl.0000308937.30473.d1. [DOI] [PubMed] [Google Scholar]
- 3.Barnes DE, Yaffe K, Byers AL, McCormick M, Schaefer C, Whitmer RA. Midlife vs late-life depressive symptoms and risk of dementia: differential effects for Alzheimer disease and vascular dementia. Arch Gen Psychiatry. 2012;69:493–498. doi: 10.1001/archgenpsychiatry.2011.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green RC, Cupples LA, Kurz A, Auerbach S, Go R, Sadovnick D, et al. Depression as a risk factor for Alzheimer disease: the MIRAGE Study. Arch Neurol. 2003;60:753–759. doi: 10.1001/archneur.60.5.753. [DOI] [PubMed] [Google Scholar]
- 5.Wuwongse S, Chang RC, Law AC. The putative neurodegenerative links between depression and Alzheimers disease. Prog Neurobiol. 2010;91:362–375. doi: 10.1016/j.pneurobio.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Butters MA, Young JB, Lopez O, Aizenstein HJ, Mulsant BH, Reynolds CF, III, et al. Pathways linking late-life depression to persistent cognitive impairment and dementia. Dialogues Clin Neurosci. 2008;10:345–357. doi: 10.31887/DCNS.2008.10.3/mabutters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang JZ, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. 2008;85:148–175. doi: 10.1016/j.pneurobio.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP, Purohit DP, et al. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry. 2006;63:161–167. doi: 10.1001/archpsyc.63.2.161. [DOI] [PubMed] [Google Scholar]
- 9.Rapp MA, Schnaider-Beeri M, Purohit DP, Perl DP, Haroutunian V, Sano M. Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am J Geriatr Psychiatry. 2008;16:168–174. doi: 10.1097/JGP.0b013e31816029ec. [DOI] [PubMed] [Google Scholar]
- 10.Bianchi M, Hagan JJ, Heidbreder CA. Neuronal plasticity, stress and depression: involvement of the cytoskeletal microtubular system. Curr Drug Targets CNS Neurol Disord. 2005;4:597–611. doi: 10.2174/156800705774322012. [DOI] [PubMed] [Google Scholar]
- 11.Cuadrado-Tejedor M, Ricobaraza A, Del Rio J, Frechilla D, Franco R, Perez-Mediavilla A, et al. Chronic mild stress in mice promotes cognitive impairment and CDK5-dependent tau hyperphosphorylation. Behav Brain Res. 2011;220:338–343. doi: 10.1016/j.bbr.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Lee AL, Ogle WO, Sapolsky RM. Stress and depression: possible links to neuron death in the hippocampus. Bipolar Disord. 2002;4:117–128. doi: 10.1034/j.1399-5618.2002.01144.x. [DOI] [PubMed] [Google Scholar]
- 13.Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52:90–110. doi: 10.1159/000087097. [DOI] [PubMed] [Google Scholar]
- 14.Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G, et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry. 2008;64:293–301. doi: 10.1016/j.biopsych.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 15.Luo DD, An SC, Zhang X. Involvement of hippocampal serotonin and neuropeptide Y in depression induced by chronic unpredicted mild stress. Brain Res Bull. 2008;77:8–12. doi: 10.1016/j.brainresbull.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Moreau JL, Jenck F, Martin JR, Mortas P, Haefely WE. Antidepressant treatment prevents chronic unpredictable mild stress-induced anhedonia as assessed by ventral tegmentum self-stimulation behavior in rats. Eur Neuropsychopharmacol. 1992;2:43–49. doi: 10.1016/0924-977X(92)90035-7. [DOI] [PubMed] [Google Scholar]
- 17.Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology. 1987;93:358–364. doi: 10.1007/BF00187257. [DOI] [PubMed] [Google Scholar]
- 18.Caccia S, Cappi M, Fracasso C, Garattini S. Influence of dose and route of administration on the kinetics of fluoxetine and its metabolite norfluoxetine in the rat. Psychopharmacology. 1990;100:509–514. doi: 10.1007/BF02244004. [DOI] [PubMed] [Google Scholar]
- 19.Anthony JP, Sexton TJ, Neumaier JF. Antidepressant-induced regulation of 5-HT(1b) mRNA in rat dorsal raphe nucleus reverses rapidly after drug discontinuation. J Neurosci Res. 2000;61:82–87. doi: 10.1002/1097-4547(20000701)61:182::AID-JNR103.0.CO2-E. [DOI] [PubMed] [Google Scholar]
- 20.Bianchi M, Heidbreder C, Crespi F. Cytoskeletal changes in the hippocampus following restraint stress: role of serotonin and microtubules. Synapse. 2003;49:188–194. doi: 10.1002/syn.10230. [DOI] [PubMed] [Google Scholar]
- 21.Roy M, David N, Cueva M, Giorgetti M. A study of the involvement of melanin-concentrating hormone receptor 1 (MCHR1) in murine models of depression. Biol Psychiatry. 2007;61:174–180. doi: 10.1016/j.biopsych.2006.03.076. [DOI] [PubMed] [Google Scholar]
- 22.Yan J, Sun XB, Wang HQ, Zhao H, Zhao XY, Xu YX, et al. Chronic restraint stress alters the expression and distribution of phosphorylated tau and MAP2 in cortex and hippocampus of rat brain. Brain Res. 2010;1347:132–141. doi: 10.1016/j.brainres.2010.05.074. [DOI] [PubMed] [Google Scholar]
- 23.Anisman H, Merali Z, Hayley S. Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders. Prog Neurobiol. 2008;85:1–74. doi: 10.1016/j.pneurobio.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 24.Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- 25.Budas G, Coughlan CM, Seckl JR, Breen KC. The effect of corticosteroids on amyloid beta precursor protein/amyloid precursor-like protein expression and processing in vivo . Neurosci Lett. 1999;276:61–64. doi: 10.1016/S0304-3940(99)00790-9. [DOI] [PubMed] [Google Scholar]
- 26.Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimers disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang C, Wang G, Wang H, Liu Z, Wang X. Cytoskeletal alterations in rat hippocampus following chronic unpredictable mild stress and re-exposure to acute and chronic unpredictable mild stress. Behav Brain Res. 2009;205:518–524. doi: 10.1016/j.bbr.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 28.Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, et al. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salehi A, Delcroix JD, Mobley WC. Traffic at the intersection of neurotrophic factor signaling and neurodegeneration. Trends Neurosci. 2003;26:73–80. doi: 10.1016/S0166-2236(02)00038-3. [DOI] [PubMed] [Google Scholar]
- 30.Yang Y, Yang XF, Wang YP, Tian Q, Wang XC, Li HL, et al. Inhibition of protein phosphatases induces transport deficits and axonopathy. J Neurochem. 2007;102:878–886. doi: 10.1111/j.1471-4159.2007.04603.x. [DOI] [PubMed] [Google Scholar]
- 31.Byers AL, Covinsky KE, Barnes DE, Yaffe K. Dysthymia and depression increase risk of dementia and mortality among older veterans. Am J Geriatr Psychiatry. 2012;20:664–672. doi: 10.1097/JGP.0b013e31822001c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dotson VM, Beydoun MA, Zonderman AB. Recurrent depressive symptoms and the incidence of dementia and mild cognitive impairment. Neurology. 2010;75:27–34. doi: 10.1212/WNL.0b013e3181e62124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho JH, Johnson GV. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating taus ability to bind and stabilize microtubules. J Neurochem. 2004;88:349–358. doi: 10.1111/j.1471-4159.2004.02155.x. [DOI] [PubMed] [Google Scholar]
- 34.Buerger K, Teipel SJ, Zinkowski R, Blennow K, Arai H, Engel R, et al. CSF tau protein phosphorylated at threonine 231 correlates with cognitive decline in MCI subjects. Neurology. 2002;59:627–629. doi: 10.1212/WNL.59.4.627. [DOI] [PubMed] [Google Scholar]
- 35.Aboukhatwa M, Dosanjh L, Luo Y. Antidepressants are a rational complementary therapy for the treatment of Alzheimers disease. Mol Neurodegener. 2010;5:10–10. doi: 10.1186/1750-1326-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mowla A, Mosavinasab M, Haghshenas H, Borhani HA. Does serotonin augmentation have any effect on cognition and activities of daily living in Alzheimers dementia A double-blind, placebo-controlled clinical trial. J Clin Psychopharmacol. 2007;27:484–487. doi: 10.1097/jcp.0b013e31814b98c1. [DOI] [PubMed] [Google Scholar]
- 37.Nelson RL, Guo Z, Halagappa VM, Pearson M, Gray AJ, Matsuoka Y, et al. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp Neurol. 2007;205:166–176. doi: 10.1016/j.expneurol.2007.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu D, Wang Z, Liu S, Wang F, Zhao S, Hao A. Anti-inflammatory effects of fluoxetine in lipopolysaccharide(LPS)-stimulated microglial cells. Neuropharmacology. 2011;61:592–599. doi: 10.1016/j.neuropharm.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 39.Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–594. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]