

Abstract

LC–MS provides a promising alternative to ligand-binding assays for quantification of therapeutic proteins and biomarkers. As LC–MS methodology is based on the analysis of proteolytic peptides, calibration approaches utilizing various calibrators and internal standards (I.S.) have been developed. A comprehensive assessment of the accuracy and reliability of these approaches is essential but has yet been reported. Here we performed a well-controlled and systematic comparative study using quantification of monoclonal-antibody in plasma as the model system. Method development utilized a high-throughput orthogonal-array-optimization, and two sensitive and stable signature-peptides (SP) from different domains were selected based on extensive evaluations in plasma matrix. With the purities of all protein/peptide standards corrected by quantitative amino acid analysis (AAA), five calibration approaches using stable-isotope-labeled (SIL) I.S. were thoroughly compared, including those at peptide, extended-peptide, and protein levels and two “hybrid” approaches (i.e., protein calibrator with SIL-peptide or SIL-extended-peptide I.S.). These approaches were further evaluated in parallel for a 15 time point, preclinical pharmacokinetic study. All methods showed good precision (CV% < 20%). When examined with protein-spiked plasma QC, peptide-level calibration exhibited severe negative biases (−23 to −62%), highly discordant results between the two SP (deviations of 38–56%), and misleading pharmacokinetics assessments. Extended-peptide calibration showed significant improvements but still with unacceptable accuracy. Conversely, protein-level and the two hybrid calibrations achieved good quantitative accuracy (error < 10%), concordant results by two SP (deviations < 15%), and correct pharmacokinetic parameters. Hybrid approaches were found to provide a cost-effective means for accurate quantification without the costly SIL-protein. Other key findings include (i) using two SP provides a versatile gauge for method reliability; (ii) evaluation of peptide stability in the matrix before SP selection is critical; and (iii) using AAA to verify purities of protein/peptide calibrators ensures accurate quantitation. These results address fundamental calibration issues that have not been adequately investigated in published studies and will provide valuable guidelines for the “fit for purpose” development of accurate LC–MS assays for therapeutic proteins and biomarkers in biological matrices.
Therapeutic proteins, and in particular monoclonal antibodies (mAb), have recently gained enormous success due to their high specificity, efficacy, and lower risks of immunogenicity.1−5 These agents exhibit desired pharmacological characteristics such as long serum half-lives, high potency, and limited off-target toxicity.6,7 However, protein drugs show more complex pharmacokinetic (PK) behaviors than small-molecule drugs.6,7 Studying the pharmacokinetics of therapeutic proteins requires highly accurate quantification methods that enable the correct estimation of drug concentrations in plasma.6,8 Conventionally, ELISA (enzyme-linked immunosorbent assay) is utilized for this purpose owing to its high sensitivity and analytical throughput. However, ELISA methods are often matrix- and species-dependent, and the method development is often time-consuming and costly, which is especially problematic in the early phases of drug discovery and development.9,10 By comparison, liquid chromatography mass spectrometry (LC–MS) using selected reactions monitoring (SRM) is often matrix- and species-independent, and method development is generally faster than that for ELISA; moreover, LC–MS assays can be readily multiplexed, providing multiple potential advantages versus ELISA11−14
Most LC–MS-based methods quantify protein by measuring a selected proteolytic signature peptide (SP) that serves as a surrogate for the intact protein. For this reason, several different calibration approaches exist at the peptide,15,16 extended-peptide,17,18 and protein levels.8,19 The choice of calibrators and stable-isotope-labeled (SIL) internal standards (I.S.) is among the most critical factors governing the reliability and accuracy of the LC–MS-based quantification.17,20−22
Peptide-level calibration is the most widely practiced approach, which employs one synthesized SP as the calibrator and a SIL-analog of the SP as the I.S. (spiked after digestion).16,23 This approach enables a straightforward and facile development of quantitative methods, and both the calibrators and SIL-I.S. are readily available from commercial sources. Nevertheless, the use of an SIL-peptide as I.S. only corrects variations caused by LC–MS analysis but not the upstream steps such as sample preparation and digestion (Figure 1A).21 Moreover, owing to the use of a peptide calibrator, this approach actually derives protein concentrations based on the measured SP concentrations in the digest, with the assumption that the efficiencies of sample preparation and digestion are close to 100%, which may not be true.24 For example, tryptic digestion is rarely complete and can be partially nonspecific.25 Consequently, this approach may result in significant bias that is not readily perceivable when the method is validated by spiking synthesized peptides into matrix digests, a prevalent practice for peptide-level calibration. Our lab and others have observed that for a wide variety of proteins, peptide-level calibration approaches resulted in significant quantitative bias when examined with protein-spiked QC samples.15,19,24,26,27 Furthermore, when two unique SP were selected for the quantification of one protein, the peptide-level calibration approach may give two sets of highly discordant results for the same protein.21,24,26
Figure 1.

Illustrations of various calibration approaches for targeted protein quantification and the study design of the comparative study. (A) The three options of internal standard (I.S.) methods used for targeted protein quantification. Full length stable-isotope-labeled (SIL) protein I.S. is added to the sample before any preparation; SIL-extended-peptide I.S. is added right before digestion, and SIL-peptide I.S. is spiked into the digest mixture after digestion. (B) Scheme of the comparison. The peptide- and extended-peptide-level calibrations and two hybrid calibrations were compared against the protein-level calibration (the Gold Standard), for quantitative performances in both protein-spiked QC samples and a full rat PK study. (C) The calibrators and I.S. employed for each calibration approach for quantification of the anti-HCV mAb in plasma. For each calibration approach, two sets of calibration curves (one for each SP) were independently established and then used for quantification.
More recently, extended-peptide calibration approaches were introduced to address concerns related to digestion efficiency. The calibrator consists of a synthesized extended-peptide containing the SP sequence and (typically) 3–6 flanking residues extended from both the N- and C-termini. A SIL-extended-peptide is used as the I.S., which is spiked prior to digestion.12,18,28,29 With cleavable sites at both ends of the SP sequence, this approach may help to compensate for the bias and variation introduced in the digestion step (e.g., missed cleavage and peptide degradation12). For example, Neubert et al. recently demonstrated that the extended-peptide calibration approach enabled accurate and sensitive quantification of small protein biomarkers, such as nerve growth factors, in plasma.18 Artificial QconCAT proteins that contain multiple extended-peptides have been engineered to enable multiplexed quantification of a large number of targets.30
The protein-level calibration method employs full-length protein as the calibrator and full-length SIL-protein as I.S., which is spiked directly into samples before sample preparation. This method provides high accuracy and precision by correcting the system bias and variation introduced by any of the sample preparation and enzymatic digestions steps19,21−23 and, therefore, is considered the “Gold Standard” for LC–MS-based targeted protein quantification.31−33 However, a drawback of protein-level calibration is that the production of full-length SIL-protein with high isotope purity can be costly and time-consuming23 and is impractical for many classes of proteins.
A systematic, comparative evaluation of the accuracy and precision of these calibration approaches in biological/pharmaceutical matrices will provide valuable information for therapeutic protein quantification. To our knowledge, such an investigation has not been conducted previously. In this study, we comprehensively compared the quantitative performance of five internal-standard calibration approaches, including the three above-mentioned calibration approaches (peptide-calibrator/SIL-peptide-I.S., extended-peptide-calibrator/SIL-extended-peptide-I.S., and protein-calibrator/SIL-protein-I.S.) and two “hybrid” approaches (protein-calibrator/SIL-peptide-I.S. and protein-calibrator/SIL-extended-protein-I.S.). Quantification of a mAb in plasma was used as the model system (illustrated in Figure 1). SP candidates were experimentally discovered, and the most sensitive and stable SP were selected based on evaluations in plasma digest. Two unique SP from different domains of the mAb were chosen to serve as a gauge for quantitative reliability. To avoid possible bias arising from differing purities of calibration standards, the purities of all protein, peptide, and extended-peptide standards were accurately measured by quantitative amino acid analysis (AAA). The accuracy and precision of each approach were assessed using QC samples prepared by spiking pure protein into plasma. Finally, we compared the five calibration approaches in a full pharmacokinetic assessment using a rat model.
Experimental Procedure
Materials and Reagents
A chimeric anti-Hepatitis C virus antibody (HCV-mAb, ∼170 kD) and its full-length stable-isotope-labeled (SIL) cognate were obtained from AbbVie (formerly Abbott Laboratories, North Chicago, IL). Sequencing-grade trypsin was from Promega (Madison WI). Protease, phosphatase, and kinase inhibitor cocktail tablets were from Roche (Basel, Switzerland). Bicinchoninic acid (BCA) protein assay reagents were from Pierce (Rockford, IL). HPLC-grade methanol, acetonitrile, acetone, and water were from B&J (Muskegon, MI). LC–MS-grade formic acid was from Fluka (Buchs, Switzerland). Tris(2-carboxyethyl) phosphine (TCEP), Tris, iodoacetamide (IAA), and phosphate-buffered saline were obtained from Sigma–Aldrich (St. Louis, MO). Signature peptides, extended signature peptides, their isotope-labeled cognates {i.e., GPSVFPLAPSSK[15N,13C] (the GPS peptide), TVAAPSVFIFPPSDEQLK[15N,13C] (the TVA peptide), ASTKGPSVFPLAPSSK[15N,13C]STSG (the extended GPS peptide), and EIKRTVAAPSVFIFPPSDEQLK[15N,13C]SGTA (the extended TVA peptide)}, and their nonlabeled versions were synthesized by New England Peptide (Gardner, MA) with nominal purity >95%. The peptide and extended-peptide stock solutions were prepared in 50% acetonitrile. The actual purities of all standards were calibrated using values measured accurately by quantitative amino acid analysis (AAA). Isotopic purities for synthesized peptide and extended-peptide SIL-standards were >99.9% and ∼97% (for lysine residues) for the SIL-protein produced by SILAC.
Identification of Signature Peptides (SP) via an Experimental Strategy and Development of the LC–MS Method
In this study, the optimal SP were identified using an experimental strategy, which included (i) the discovery of SP candidates by LC-Orbitrap MS analysis; (ii) a high-throughput, on-the-fly orthogonal array optimization to optimize SRM conditions for all candidates in plasma digest, and (iii) selection of two stable and sensitive SP based on the thorough evaluation in the plasma matrix. Details are in the Supporting Information.
Preparation of Plasma Samples
The rat plasma samples were subjected to protein denaturing, reduction/alkylation, and then a precipitation/on-pellet-digestion procedure that provides an efficient sample cleanup and optimal peptide recovery.26,27,34 An aliquot of 2 μL of rat plasma sample was diluted 20-fold by PBS (100 mM, pH7.4) containing 0.5% SDS. Proteins in the diluted sample was reduced by TCEP (2 mM) for 15 min at 37 °C and then alkylated with IAA (100 mM) at 37 °C for 30 min. Both steps were performed under darkness with agitation. The protein content was precipitated in two steps, by adding 1 vol of chilled acetone at −20 °C and vortexing for 1 min to generate fine-sized protein aggregates, followed by adding 5 vol of chilled acetone. The samples were centrifuged at 20000g for 30 min, and the supernatant was removed carefully and the protein pellet was allowed to air-dry. Tris buffer (50 mM, pH 8.2) containing trypsin was added to a final E/S ratio of 1:20 (w/w). The solution was then incubated at 37 °C and vortexed at 500 rpm for 6 h to dissolve the pellet. A second aliquot of trypsin was added at an E/S ratio of 1:15 (w/w), and the mixture was incubated at 37 °C overnight to achieve complete digestion. Formic acid was added to a final concentration of 1% (v/v) to terminate the digestion. The sample was then centrifuged at (20000g) at 4 °C for 30 min, and 2/3 of the supernatant was transferred to the autosampler vial for LC–MS analysis.
LC–SRM–MS Analysis
Quantitative analysis was performed on a microflow-LC system coupled to a triple-quadruple MS.35 An Agillent 1100 capillary HPLC was interfaced to a Thermo Scientific Quantum Ultra EMR triple-quadruple mass spectrometer via an ESI source (San Jose, CA). The separation was performed on a Zorbax-SB-C18 Stablebond column (150 mm × 0.5 mm i.d., 3.5 μm, and 100 Å pore size). A 20 min gradient was employed with a 10 μL injection volume, and the flow rate was 15 μL/min. A and B solvents were mixtures of water:acetonitrile:formic acid at 98:2:0.1 and 15:85:0.1, respectively. The gradient was 80% A to 72.5% A in 3 min and 55% A in 9 min, and then 100% B for 3 min followed by column equilibration at 80% A for 5 min. The column temperature was 50 °C. The spray voltage was 3.5 kV, and the capillary temperature was 320 °C. Quantification was performed using selected reaction monitoring of the transitions m/z 593.8→699.5 for GPS and m/z 973.8→913.4 for TVA. The optimized collision energy and tube-lens voltages were set at 21 eV/100 V and 29 eV/120 V based on the conditions obtained by orthogonal array optimization (OAO). Additional transitions (e.g., m/z 593.8→846.4 and m/z 973.8→1060.0 for GPS and TVA, respectively) were monitored to confirm the accurate assignment of the chromatography peaks. I.S. peptides were monitored using transitions of m/z 597.8→707.4 and m/z 977.8→ 921.2 for GPS and TVA, respectively. The dwell time was 100 ms for each transition. Q1 and Q3 resolution were both set at 0.7 fwhm (full width at half-maximum). The selected reaction monitoring data was processed by LCquan from Thermo Scientific. The signal-to-noise ratios (S/N) were determined as the intensity of the ion-current peak signals of target peptides over the baseline chemical noises. The lower limit of quantification, LLOQ, was determined as at least three folds of the lower limit-of-detection (LOD) of the least sensitive signature peptide of the same protein.
Establishment of the Five Calibration Approaches
The five different calibration approaches (Figure 1B) were prepared across the protein concentration range of 0.1–200 μg/mL (or equal molar concentrations of peptides or extended peptides). The actual purities of all standards were accurately quantified by quantitative amino acid analysis (AAA). Each calibration method was prepared following its own procedure (c.f. Figure 1, panels A and C). For each calibration approach, two calibration curves were prepared independently for the two SP. For each SP, the calibration curve was constructed by plotting the peak area ratios of the SP and I.S. versus the corresponding mAb concentrations. Linear regression with a 1/x2 weighting factor was employed. The procedures for preparing the calibration solutions are specified below.
Protein-Level Calibration
The protein-level calibration solutions were prepared by spiking 0.1, 0.2, 0.5, 1, 5, 20, 40, and 200 μg/mL pure mAb (purity was calibrated by AAA, same below) in pooled blank rat plasma before any preparation procedure; meanwhile, full-length SIL-HCV mAb was spiked at the concentration of 500 ng/mL. All calibration samples were experimentally processed and analyzed by LC-SRM-MS, following the above-mentioned protocols.
Peptide-Level Calibration
Peptide-level calibration solutions were prepared with adding GPS peptide at 0.032, 0.064, 0.16, 0.32, 1.6, 6.4, 12.8, and 64 ng/mL and TVA peptide at 0.052, 0.104, 0.26, 0.52, 2.6, 10.4, 20.8, and 104 ng/mL into digested rat plasma, which had been diluted 50-fold from the original plasma sample before the digestion procedure (c.f. the preparation procedure). The above concentration ranges correspond to the same molarities of 0.1–200 μg/mL mAb protein in plasma. The levels of SIL-GPS and TVA I.S. were 2.00 and 3.25 ng/mL, respectively.
Extended-Peptide Level Calibration
Standard solutions were prepared by spiking 0.052, 0.104, 0.26, 0.52, 2.6, 10.4, 20.8, and 104 ng/mL extended-GPS peptide and 0.076, 0.152, 0.376, 0.752, 3.75, 15, 30, and 150 ng/mL extended-TVA-peptide to the diluted and prepared plasma before digestion. These concentration ranges correspond to the same molarities of 0.1–200 μg/mL mAb protein in plasma. The I.S., SIL-extended-GPS, and SIL-extended-TVA were added at 3.25 and 4.68 ng/mL, respectively.
Hybrid Calibrations
Two varieties of hybrid calibrations were investigated. For both of the hybrid calibration approaches, the protein calibrator was spiked at 0.1, 0.2, 0.5, 1, 5, 20, 40, and 200 μg/mL HCV mAb antibody to blank rat plasma before any preparation step, which is the same as the protein-level calibration approach. For the hybrid approach using SIL-extended-peptide I.S., the SIL-extended-peptide I.S. was added at the same levels as that of the extended-peptide-level calibration approach, after sample preparation but before digestion; for the hybrid approach using SIL-peptide I.S., the protein-spiked plasma samples were prepared and digested, and then the SIL-peptide I.S. was added after digestion at the same levels as that of the peptide-level calibration approach.
Validation and Evaluation of Quantitative Performance Using Protein-Spiked QC Samples
The five calibration approaches were individually validated using the “default” method for each [i.e., spiking blank plasma samples (or sample digest) with the corresponding calibrators (peptides, extended-peptides or protein) of each calibration approach]. Details are shown in the Supporting Information. In order to evaluate the real accuracy of these approaches for the quantification of protein in plasma, another batch of QC samples were prepared by spiking the AAA-certified anti-HCV-mAb (full-length protein) into blank rat plasma at three levels (1.6, 10, and 80 μg/mL). Aliquots of these QC samples were prepared and analyzed by every calibration approach in triplicate for each of two different days (day 1 and day 14, N = 6). For each sample and each calibration approach, two sets of quantitative results were independently obtained by the two SP, and the agreement between the two values was evaluated.
Comparison of the Five Approaches in a Full Preclinical PK Study
Animal protocols were approved by the Institutional Animal Care and Use Committee of the SUNY-Buffalo. Male Sprague–Dawley rats were given a single intravenous dose of 2 mg/kg of the mAb (N = 4), and 50 μL of blood was collected at each of the 15 time points after administration (i.e., 1, 2, 4, 6, 12, 24, 48, 72, 120, 168, 216, 264, 360, 504, and 648 h). The plasma was procured, and after vortexing, five 2 μL aliquots of the same plasma sample were taken, which were quantified with each of the five different calibration methods. Two sets of quantitative values were independently obtained using the two SP. On the basis of the time–concentration curves obtained by these calibration approaches, the PK parameters were assessed with use of WinNonlin (Pharsight Corporation, Palo Alto, CA).
Results and Discussion
Selection of Two SP and Development of the LC–MS Method
In order to conduct an unbiased comparison of the calibration approaches, it is important to select the optimal SP that are stable, sensitive, and specific to the target protein (anti-HCV mAb, sequence shown in Figure 1 of the Supporting Information). To achieve this goal, instead of using an in silico method to predict the optimal SP, we employed an experimental strategy to discover and optimize many SP candidates, and then to evaluate these candidates in target matrices prior to SP selection.24,26,27 Briefly, the pool of SP candidates was generated by a data-dependent nano-LC-LTQ/Orbitrap analysis and a stringent filtering step.26,27 The list of qualified SP candidates is shown in Table 1 of the Supporting Information. To evaluate these candidates, the anti-HCV mAb was spiked to blank plasma and then the samples were prepared and digested; the optimal LC–MS conditions of all SP candidates shown in Table 1 of the Supporting Information were accurately obtained by a high-throughput and on-the-fly orthogonal-array-optimization (OAO)24,26,27 within one single LC–MS run. With the use of the developed LC–MS conditions, all candidates were thoroughly assessed for stability and signal-to-noise ratios (S/N) in the plasma digest.
Selection of unstable peptides as SP carries a significant risk of quantitative bias/variation, which cannot be corrected by SIL-peptide I.S.24,26,36 For this reason, we evaluated the stability of the 15 candidates (Table 1 of the Supporting Information) by continuous incubation of the digest mixture under conditions respectively mimicking tryptic digestion (37 °C, pH 8.5, 24 h) and queuing in a cooled autosampler (4 °C, pH 2.8, 48 h). Details are presented in the Experimental section of the Supporting Information. Interestingly, 5 out of the 15 SP candidates showed considerable degradation (>20%) in at least one of the conditions, and some of these unstable peptides were of high abundance (Figure 2 and Table 1 of the Supporting Information). These observations underscore the potential risks of selecting SP merely based on the signal intensity. Among the stable peptides, two unique SP showing the highest S/N from heavy chain (GPSVFPLAPSSK, referred as the GPS peptide) and light chain (TVAAPSVFIFPPSDEQLK, the TVA peptide) were selected. The SP and their extended peptides (ASTKGPSVFPLAPSSKSTSG and EIKRTVAAPSVFIFPPSDEQLKSGTA) are synthesized in both isotope labeled and nonlabeled forms.
Figure 2.

Evaluation of the stability of the 15 candidates for signature peptides (SP) from the anti-HCV mAb, by continuous incubation of the digest mixture under conditions mimicking (A) prolonged tryptic digestion (37 °C, pH 8.5, 24 h) and (B) queuing in a cooled autosampler (4 °C, pH 2.8, 48 h), respectively. The peptides are represented by the first three residues of their sequence (the full list is in Table 1 of the Supporting Information). GPS and TVA peptides from the heavy and light chains of the mAb, respectively, were finally selected as the SP (shown in red and blue).
To enable an unbiased comparison of the five calibration approaches, the purities of all peptide, extended-peptide, and protein calibrators were quantified by the AAA and the mean quantitative values were corroborated for every amino acid residue to ensure accuracy. The results are shown in Table 2 of the Supporting Information. Clearly, the actual purities of all standards were markedly lower than the nominal values. For example, though the manufacturer-labeled purity for the TVA peptide was >95%, the true purity was only 65%. Manufacturers use HPLC-UV or electrophoresis methods to determine protein/peptide purities, which are unable to detect nonresponsive contaminations (e.g., dried buffer salts) and thus may result in a positive bias. As the standards were produced by highly reputable providers, such biases may be prevalent and thereby performing AAA to verify calibration standards is advisable to ensure quantitative accuracy.
Plasma samples were subjected to precipitation and overnight digestion, a procedure demonstrated to afford high peptide recovery, high reproducibility, and excellent digestion efficiency,24,27,34,37 followed by LC–MS analysis. Typical chromatograms of the two SP in plasma digest are shown in Figure 2 of the Supporting Information. Good selectivity was achieved for both SP. The detection limits were 15 and 30 ng of mAb protein per mL of plasma for GPS and TVA peptides, respectively. In this study, the lower limits of quantification (LLOQ) for both SP were validated at 100 ng protein per milliliters of plasma.
Comparison of the Five Calibration Approaches with Spiked QC Samples
Calibration Approaches at Peptide, Extended-Peptide, and Protein Levels
To ensure an objective comparison, the calibration curves at the peptide, extended-peptide, and protein levels were prepared in the same protein concentration range of 0.1–200 μg/mL in plasma (or equal molarities of peptides/extended-peptides, the same as below). For every approach, two calibration curves were independently prepared for the two SP. All calibration curves showed excellent linearity (R2 ≥ 0.99, Table 3 of the Supporting Information). Then we validated these calibration approaches using QC samples spiked with their corresponding calibrators (e.g. spiking pure synthesized peptides to digested blank plasma for peptide calibration), extended-peptides right before digestion for extended-peptide calibration, and pure protein before sample preparation for protein calibration. With the use of these “by default” validation approaches that have been widely practiced,16 all methods exhibited excellent accuracy and precision at the three QC levels, as shown in Table 3 of the Supporting Information. However, the validation practices used for peptide and extended-peptide calibrations may not correctly reflect the accuracy of protein quantification because the target analyte is the protein rather than a peptide or extended-peptide.
Subsequently, we sought to evaluate the quantitative performance of these three calibration approaches using QC prepared by spiking AAA-certified full-length protein into blank plasma at three levels (1.6, 10, and 80 μg/mL). The results are illustrated in Figure 3A, which plots the quantitative biases by the two SP (on the x- and y-axis) in replicate measurements (N = 6 in two different days). Detailed quantitative data is shown in Table 4 of the Supporting Information. All methods showed excellent precision. However, the peptide-level calibration clearly resulted in severe negative bias with both SP (Figure 3A); moreover, the quantitative values by the two SP differed substantially (Figure 3A and Table 4 of the Supporting Information). These problems may arise from the incomplete peptide recoveries in the preparation and digestion steps, and that the recoveries for the two SP may be substantially different. Incomplete recovery of SP appears to be inevitable, even if thoroughly optimized preparation and digestion strategies were employed.15,19,20,24,26,31 The use of the extended-peptide calibration approach was found to markedly reduce the quantitative biases and discrepancy between the two SP (Figure 3A), probably because the incomplete peptide recovery during digestion was at least partially compensated for.12,18,38 Nevertheless, that accuracy achieved by extended-peptide calibration is still insufficient (Figure 3A). Previously Neubert et al. demonstrated that extended-peptide calibration achieved excellent quantitative accuracy for a small cytokine protein.18 We speculate that the different observations between the two studies may reflect that the extended-peptide could well-resemble the digestion behavior of the SP domain in small proteins, but not so in a much larger protein such as a mAb.39,40
Figure 3.

Two-dimensional representations of the quantitative biases by (A) peptide-, extended-peptide-, and protein-level calibration approaches and (B) the two “hybrid” calibration approaches (c.f., Figure 1). QC samples were prepared by spiking blank plasma with pure protein at three levels: 1.6, 10, and 80 μg/mL. The purities of all standards were accurately measured by the quantitative amino acid analysis method to eliminate bias arising from possible inaccurate purity. Five aliquots of each QC sample were individually prepared and analyzed in replicates by the five calibration approaches. Each sample was analyzed three times in each of two different days (day 1 and day 14, N = 6, shown as individual data points). For every calibration method, the quantitative values were obtained independently using the two signature peptides (SP) (i.e., the GPS and TVA peptides). The two axes represent the quantitative biases by the two SP. The red box in the center of each panel denotes the zone of <20% bias, while the golden box signifies the zone of <10% bias.
Protein-level calibration utilizing full-length protein as calibrator and full-length SIL-protein as I.S. has demonstrated excellent quantitative performance regardless of the efficiencies of the preparation and digestion procedures,19,21,23 and therefore can be used as the “Gold Standard” for targeted protein quantification. Consistent with this notion, in this study, excellent accuracy and precision were observed at all QC levels by the protein-level calibration (Figure 3A). Though protein-level calibration provides high accuracy and precision, SIL-proteins must be produced in high-yields to be cost-effective, limiting their production to those for which optimized cell lines have been developed.20 Additionally, since these proteins are generally produced by the SILAC method,41 achieving very high isotopic purity is often both technically challenging and cost prohibitive. In this study, a 97% isotope purity (i.e., 3% of the mAb molecules is not labeled) was obtained for the SIL-anti-HCV mAb following multiple cycles of metabolic labeling, in comparison with >99.9% isotopic purities that are typically seen in synthesized SIL-peptides. To avoid sample contamination by the I.S., the SIL-protein has to be spiked at a relatively low level (<30-fold of detect limit in this study) and thus may limit the flexibility of method development. Typical chromatograms illustrating this problem are shown in Figure 3 of the Supporting Information.
Hybrid Calibration Approaches
To achieve accurate quantification, one plausible alternative to the protein-level calibration is the “hybrid” calibration methods (i.e., employing the full-length protein as calibrator and SIL-peptide or SIL-extended peptide as I.S.). When reproducible preparation and digestion steps are achieved, the use of full-length proteins as calibrator will offset the peptide losses in these steps even without using SIL-protein I.S. and, therefore, may result in accurate protein quantification. Our previous works as well as these by others showed accurate quantification of liver enzymes and mAb, using protein calibrator and SIL-peptide I.S.12,24,26,27,42 Here we evaluated the quantitative performance of two hybrid calibration methods: protein-calibrator with SIL-peptide I.S. and protein-calibrator with SIL-extended-peptide-I.S., using QC prepared by spiking blank plasma with pure protein. Excellent quantitative accuracy and precision were observed for both methods at the three protein levels (N = 6, Figure 3B and Table 4 of the Supporting Information). The high level of accuracy and precision also suggested high reproducibility of the preparation and digestion procedure used in this study. These results demonstrated that hybrid approaches have a good potential for accurate targeted quantification in the absence of SIL-protein.
Comprehensive Comparison of Calibration Approaches in a Full Pharmacokinetics Study in Rats
The above comparisons were carried out in blank plasma spiked with known amounts of pure protein. In order to examine whether the above observations hold true for “real-world” applications, we further compared the five calibration approaches in parallel for a full pharmacokinetics analysis. Four rats (N = 4) received a single, clinically relevant dose of mAb (2.0 mg/kg), and then plasma samples were collected at 15 time points ranging from 0.5 to 648 h post injection (see Experimental Section). Five 2 μL aliquots of each sample were taken out, and then each aliquot was prepared and analyzed by one of the five calibration approaches. The time courses measured by these approaches are shown in Figure 4 (panels A–E). Protein-level calibration was utilized as the gold standard to estimate the range of true protein concentrations in the time-course samples (Figure 4A) and to define the acceptable error zone (±15%, as indicated by the gray bands in Figure 4 (panels A–F). As a cross validation, results by a parallel ELISA quantification also fell well within this zone (Figure 4F). Consistent with the findings in protein-spiked QC samples, the peptide-level calibration resulted in substantially underestimated concentrations in all time-course samples; moreover, for the same set of plasma samples, quantification independently by the two SP resulted in two substantially different sets of concentration–time curves (Figure 4B). Extended-peptide calibration approach showed a significant improvement over the peptide-level calibration (Figure 4C); however, considerable underestimation was still observed and most of the data points are not in the acceptable range; furthermore, the results by the two SP also differed markedly (Figure 4C). In contrast, both of the hybrid calibration methods afforded accurate measurement in all time-course samples (i.e., all data points are within the acceptable zone, Figure 4, panels D–E), and the results by the two SP agreed well with each other. Time course data at each time points are shown in Table 5 of the Supporting Information. The extents of inconsistency between the two SP in the time course samples are summarized in Figure 5. For protein-level and hybrid calibration approaches, excellent agreements between the SP were observed (deviation <15%), which is in sharp contrast to peptide- and extended-peptide-level calibration methods, where the deviations were >15% in a vast majority of the samples. These observations highlight the benefits of using multiple SP as a gauge for reliability of the quantitative method and results.
Figure 4.

Comparison of the five calibration approaches and ELISA for a full PK study in rats. The time courses of anti-HCV mAb were measured by the five approaches in parallel: (A) protein-level calibration, (B) peptide-level calibration, (C) extended-peptide-level calibration, and two “hybrid approaches” employing protein as a calibrator with (D) SIL-peptides or (E) SIL-extended-peptides as I.S. (F) The results of a parallel ligand binding assay (samples from the four animals were pooled at each time point). PK parameters calculated based on the time courses determined by each calibration approach are shown in (G). Rats (N = 4) were given a single dose (I.V., 2 mg/kg) of anti-HCV mAb, and blood samples were collected at 15 time points post injection. Five aliquots of each sample were individually prepared and analyzed, respectively, by the five calibration approaches. Quantitative data were obtained independently by the two SP. The ±15% error zones were estimated based on the mean values obtained by the protein-level calibration approach (the Gold Standard).
Figure 5.
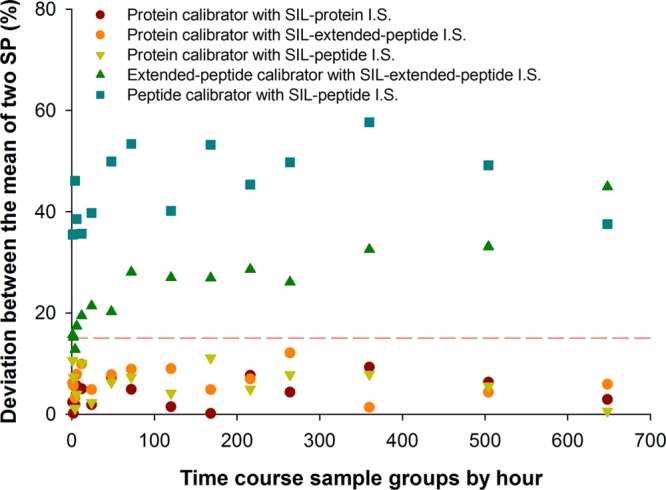
The extent of inconsistency between the two SP in the time course samples. The deviation between quantitative results obtained by the two signature peptides was used as a gauge for the reliability of the quantification method. For each of the calibration approaches, the average deviation of samples within the same time point group was calculated (N = 4 per time point). The threshold of acceptance was deviation < 15% (red line) between the two SP.
Finally, to assess the effects of calibration approaches on pharmacokinetics assessment, we calculated some basic pharmacokinetic parameters based on the time course data (Figure 4G and Table 1). The values measured by the protein-level calibration defined the threshold of correct estimation (±15%, Figure 4G). Both the peptide- and extended-peptide level calibrations severely overestimated the steady state volume of distribution (Vss) and clearance (Cl) of the mAb and underestimated the AUC, while the half-life values (T1/2) were correct because this parameter is not dependent on the absolute level of drug; additionally, two discordant sets of PK parameters were obtained by the two SP (Table 1). Per contra, the hybrid methods achieved correct estimation of all parameters by both SP (Table 1). Interestingly, the PK parameters estimated by the two SP are more concordant using the hybrid approach with SIL-peptide I.S. (hybrid#1), than using SIL-extended-peptide I.S. (hybrid#2). This observation probably rooted from the possible different digestion rates between the extended-peptides and the corresponding domains in the protein.
Table 1. Pharmacokinetic Parameters Calculated Based on the Quantitative Results Obtained by Different Calibration Approaches and Signature Peptides.
| AUC (μg h/mL) | Cl (mL/h) | Vss (mL) | T1/2 (h) | |
|---|---|---|---|---|
| protein calibration, GPS | 7274 | 0.083 | 28.9 | 260.7 |
| protein calibration, TVA | 7113 | 0.085 | 28.8 | 260.8 |
| aprotein calibration, mean of two SP | 7194 | 0.084 | 28.9 | 260.9 |
| protein calibration, deviation between the two SP | 2% | 2% | 0% | 0% |
| hybrid no. 2 (SIL-extended-peptide I.S.), GPS | 7551 | 0.080 | 30.6 | 298.5 |
| hybrid no. 2 (SIL-extended-peptide I.S.), TVA | 6763 | 0.089 | 32.3 | 263.9 |
| hybrid no. 2, mean of two SP | 7155 | 0.084 | 31.4 | 281.2 |
| hybrid no. 2, deviation between the two SP | 8% | 8% | 4% | 9% |
| hybrid no. 1 (SIL-peptide I.S.), GPS | 7244 | 0.083 | 27.9 | 252.5 |
| hybrid no. 1 (SIL-peptide I.S.), TVA | 7125 | 0.085 | 28.9 | 261.1 |
| hybrid no. 1, mean of two SP | 7212 | 0.084 | 28.3 | 252.5 |
| hybrid no. 1, deviation between the two SP | 1% | 1% | 2% | 2% |
| extended-peptide calibration, GPS | 5774 | 0.105 | 38.2 | 271.1 |
| extended-peptide calibration, TVA | 3696 | 0.163 | 49.5 | 239.2 |
| extended-peptide calibration, mean of two SP | 4721 | 0.128 | 43.2 | 255.6 |
| extended-peptide calibration, deviation between SP | 31% | 31% | 18% | 9% |
| peptide calibration, GPS | 5640 | 0.107 | 35.9 | 252.5 |
| peptide calibration, TVA | 2995 | 0.202 | 74.7 | 280.7 |
| peptide calibration, mean of two SP | 4293 | 0.141 | 49.0 | 260.4 |
| peptide calibration, deviation between the two SP | 43% | 43% | 50% | 7% |
Parameters were calculated based on the mean of the concentrations obtained by the two SP.
Conclusions
In biomedical/pharmaceutical research and industry, accurate measurement of protein, drug, or biomarker in biological matrices is often critical. To facilitate the development of accurate LC–MS-based methods, we systematically evaluated the quantitative accuracy achieved by various calibration approaches. For each approach, the target mAb protein was independently quantified by two SP that were stringently selected based on a thorough evaluation of all candidates for stability and sensitivity. Under the sample preparation, digestion, and LC–MS methods developed here, all calibration approaches exhibited excellent quantitative precision.
Though peptide-level calibration achieved excellent accuracy when validated with peptide-spiked QC (the “default” method for validation of peptide-level calibration), severe negative bias, and discordant results by the two SP were observed when this approach was examined with protein-spiked QC, suggesting that reliance on peptide calibration may carry a “hidden” but significant risk of quantitative error. Extended-peptide calibration showed noticeable improvement but negative biases remained, probably reflecting the fact that a short extended-peptide may not resemble the behavior of a large protein such as the mAb. Conversely, good quantitative accuracy was observed for the three approaches using full-length protein as the calibrator, including the protein-level calibration (the gold standard) and two hybrid approaches. While the protein-level calibration requires full-length SIL-protein which is expensive and may have isotope purity issues, the hybrid methods circumvent this need by employing SIL-peptide or SIL-extended-peptide as I.S. without perceivably compromising quantitative accuracy. Therefore, when reproducible sample preparation is achieved, the hybrid methods may provide a cost-effective alternative to protein-level calibration for accurate quantification.
This study also demonstrated that the peptide- and extended-peptide level calibration approaches may result in misleading biological/pharmaceutical information, such as the incorrect pharmacokinetic assessments observed in the thorough pharmacokinetic study. Again the hybrid calibration approaches resulted in accurate pharmacokinetic assessments. This work also underscored the benefits of using two SP from different domains of a protein to gauge the reliability of quantification, which is capable of perceiving quantitative errors arising from such factors as methodological bias and truncation or modification of the target protein. Also highlighted in this study are the importance of using AAA-certified standards for accurate absolute quantification and the necessity of evaluating peptide stabilities prior to the selection of SP, two issues that are often overlooked.
This study addressed important calibration issues in the developing field of LC–MS-based protein quantification, which has not been adequately addressed in published studies. The observations from this study should be of considerable value for practitioners as they develop “fit for purpose” quantitative assays. Preclinical and clinical PK studies require accurate quantification of therapeutic proteins and biomarkers. However, while the peptide-level calibration approach may not accurately reveal absolute protein concentrations in biological matrices, it is straightforward to develop at low cost and high throughput and will perform well for relative quantification; for example, it is an excellent and powerful tool for early estimation of T1/2 or multiplexed evaluation of protein biomarkers in preclinical species.13
Acknowledgments
This work was supported by a Center of Protein Therapeutics Industrial Award (JQ), by the NIH Grants U54HD071594 (J.Q.) and HL103411 (J.Q.), and by the American Heart Association (AHA) Award 12SDG9450036 (J.Q.).
Supporting Information Available
Three figures, five tables, and one experimental file. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ The authors contributed equally in the preparation of the current manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Weiner L. M.; Surana R.; Wang S. Nat. Rev. Immunol. 2010, 10, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A. C.; Carter P. J. Nat. Rev. Immunol. 2010, 10, 301–316. [DOI] [PubMed] [Google Scholar]
- Leader B.; Baca Q. J.; Golan D. E. Nat. Rev. Drug Discovery 2008, 7, 21–39. [DOI] [PubMed] [Google Scholar]
- Nelson A. L.; Dhimolea E.; Reichert J. M. Nat. Rev. Drug Discovery 2010, 9, 767–774. [DOI] [PubMed] [Google Scholar]
- Reichert J. M. mAbs 2012, 4, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Wang E. Q.; Balthasar J. P. Clin. Pharmacol. Ther. 2008, 84, 548–558. [DOI] [PubMed] [Google Scholar]
- Lobo E. D.; Hansen R. J.; Balthasar J. P. J. Pharm. Sci. 2004, 93, 2645–2668. [DOI] [PubMed] [Google Scholar]
- Huillet C.; Adrait A.; Lebert D.; Picard G.; Trauchessec M.; Louwagie M.; Dupuis A.; Hittinger L.; Ghaleh B.; Le Corvoisier P.; Jaquinod M.; Garin J.; Bruley C.; Brun V.. Mol. Cell. Proteomics 2012, 11, 10.1074/mcp.M111.008235. [DOI] [PMC free article] [PubMed]
- Hoofnagle A. N.; Wener M. H. J. Immunol. Method 2009, 347, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezan E.; Dubois M.; Becher F. Analyst 2009, 134, 825–834. [DOI] [PubMed] [Google Scholar]
- Palandra J.; Finelli A.; Zhu M.; Masferrer J.; Neubert H. Anal. Chem. 2013, 85, 5522–5529. [DOI] [PubMed] [Google Scholar]
- Fernandez Ocana M.; James I. T.; Kabir M.; Grace C.; Yuan G.; Martin S. W.; Neubert H. Anal. Chem. 2012, 84, 5959–5967. [DOI] [PubMed] [Google Scholar]
- Shi T.; Sun X.; Gao Y.; Fillmore T. L.; Schepmoes A. A.; Zhao R.; He J.; Moore R. J.; Kagan J.; Rodland K. D.; Liu T.; Liu A. Y.; Smith R. D.; Tang K.; Camp D. G.; Qian W.-J. J. Proteome Res. 2013, 12, 3353–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T.; Fillmore T. L.; Sun X.; Zhao R.; Schepmoes A. A.; Hossain M.; Xie F.; Wu S.; Kim J. S.; Jones N.; Moore R. J.; Pasa-Tolic L.; Kagan J.; Rodland K. D.; Liu T.; Tang K.; Camp D. G. 2nd; Smith R. D.; Qian W. J. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 15395–15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshishian H.; Addona T.; Burgess M.; Kuhn E.; Carr S. A. Mol. Cell. Proteomics 2007, 6, 2212–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S.; Aebersold R.; Chen R.; Rush J.; Goodlett D. R.; McIntosh M. W.; Zhang J.; Brentnall T. A. J. Proteome Res. 2008, 8, 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh M. J. Chromatogr., B 2012, 883–884, 59–67. [DOI] [PubMed] [Google Scholar]
- Neubert H.; Muirhead D.; Kabir M.; Grace C.; Cleton A.; Arends R. Anal. Chem. 2013, 85, 1719–1726. [DOI] [PubMed] [Google Scholar]
- Brun V.; Dupuis A.; Adrait A.; Marcellin M.; Thomas D.; Court M.; Vandenesch F.; Garin J. Mol. Cell. Proteomics 2007, 6, 2139–2149. [DOI] [PubMed] [Google Scholar]
- Bronsema K. J.; Bischoff R.; van de Merbel N. C. J. Chromatogr., B 2012, 893–894, 1–14. [DOI] [PubMed] [Google Scholar]
- Brun V.; Masselon C.; Garin J.; Dupuis A. J. Proteomics 2009, 72, 740–749. [DOI] [PubMed] [Google Scholar]
- Bronsema K. J.; Bischoff R.; van de Merbel N. C. Anal. Chem. 2013, 85, 9528–9535. [DOI] [PubMed] [Google Scholar]
- van den Broek I.; Niessen W. M. A.; van Dongen W. D. J. Chromatogr., B 2013, 929, 161–179. [DOI] [PubMed] [Google Scholar]
- Cao J.; Covarrubias V. M.; Straubinger R. M.; Wang H.; Duan X.; Yu H.; Qu J.; Blanco J. G. Anal. Chem. 2010, 82, 2680–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P.; Aebersold R.; Domon B. Mol. Cell. Proteomics 2007, 6, 1589–1598. [DOI] [PubMed] [Google Scholar]
- Duan X.; Abuqayyas L.; Dai L.; Balthasar J. P.; Qu J. Anal. Chem. 2012, 84, 4373–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X.; Dai L.; Chen S.-C.; Balthasar J. P.; Qu J. J. Chromatogr., A 2012, 1251, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beynon R. J.; Doherty M. K.; Pratt J. M.; Gaskell S. J. Nat. Methods 2005, 2, 587–589. [DOI] [PubMed] [Google Scholar]
- Barnidge D. R.; Hall G. D.; Stocker J. L.; Muddiman D. C. J. Proteome Res. 2004, 3, 658–661. [DOI] [PubMed] [Google Scholar]
- Pratt J. M.; Simpson D. M.; Doherty M. K.; Rivers J.; Gaskell S. J.; Beynon R. J. Nat. Protoc. 2006, 1, 1029–1043. [DOI] [PubMed] [Google Scholar]
- Kuhn E.; Whiteaker J. R.; Mani D. R.; Jackson A. M.; Zhao L.; Pope M. E.; Smith D.; Rivera K. D.; Anderson N. L.; Skates S. J.; Pearson T. W.; Paulovich A. G.; Carr S. A.. Mol. Cell. Proteomics 2012, 11, 10.1074/mcp.M111.013854. [DOI] [PMC free article] [PubMed]
- Heudi O.; Barteau S.; Zimmer D.; Schmidt J.; Bill K.; Lehmann N.; Bauer C.; Kretz O. Anal. Chem. 2008, 80, 4200–4207. [DOI] [PubMed] [Google Scholar]
- Li H.; Ortiz R.; Tran L.; Hall M.; Spahr C.; Walker K.; Laudemann J.; Miller S.; Salimi-Moosavi H.; Lee J. W. Anal. Chem. 2012, 84, 1267–1273. [DOI] [PubMed] [Google Scholar]
- Duan X.; Young R.; Straubinger R. M.; Page B.; Cao J.; Wang H.; Yu H.; Canty J. M.; Qu J. J. Proteome Res. 2009, 8, 2838–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J.; Jusko W. J.; Straubinger R. M. Anal. Chem. 2006, 78, 4543–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilffert D.; Reis C. R.; Hermans J.; Govorukhina N.; Tomar T.; de Jong S.; Quax W. J.; van de Merbel N. C.; Bischoff R. Anal. Chem. 2013, 85, 10754–10760. [DOI] [PubMed] [Google Scholar]
- Tu C.; Li J.; Young R.; Page B. J.; Engler F.; Halfon M. S.; Canty J. M.; Qu J. Anal. Chem. 2011, 83, 4802–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocaña M. F.; Neubert H. Anal. Biochem. 2010, 399, 202–210. [DOI] [PubMed] [Google Scholar]
- Yuan L.; Arnold M. E.; Aubry A.-F.; Ji Q. C. Bioanalysis 2012, 4, 2887–2896. [DOI] [PubMed] [Google Scholar]
- Ouyang Z.; Furlong M. T.; Wu S.; Sleczka B.; Tamura J.; Wang H.; Suchard S.; Suri A.; Olah T.; Tymiak A.; Jemal M. Bioanalysis 2011, 4, 17–28. [DOI] [PubMed] [Google Scholar]
- Ong S.-E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Zeng J.; Titsch C.; Voronin K.; Akinsanya B.; Luo L.; Shen H.; Desai D. D.; Allentoff A.; Aubry A. F.; Desilva B. S.; Arnold M. E. Anal. Chem. 2013, 85, 9859–9867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.