

Smurf2 is an E3 ubiquitin ligase that regulates TGF-β/Smad signaling and is implicated in a wide variety of cellular responses. miR-322 and miR-503 repress Smurf2 translation and thus modulate TGF-β/Smad2 signaling and intestinal epithelial homeostasis.
Abstract
Smad ubiquitin regulatory factor 2 (Smurf2) is an E3 ubiquitin ligase that regulates transforming growth factor β (TGF-β)/Smad signaling and is implicated in a wide variety of cellular responses, but the exact mechanisms that control Smurf2 abundance are largely unknown. Here we identify microRNA-322 (miR-322) and miR-503 as novel factors that regulate Smurf2 expression posttranscriptionally. Both miR-322 and miR-503 interact with Smurf2 mRNA via its 3′-untranslated region (UTR) and repress Smurf2 translation but do not affect total Smurf2 mRNA levels. Studies using heterologous reporter constructs reveal a greater repressive effect of miR-322/503 through a single binding site in the Smurf2 3′-UTR, whereas point mutation of this site prevents miR-322/503–induced repression of Smurf2 translation. Increased levels of endogenous Smurf2 via antagonism of miR-322/503 inhibits TGF-β–induced Smad2 activation by increasing degradation of phosphorylated Smad2. Furthermore, the increase in Smurf2 in intestinal epithelial cells (IECs) expressing lower levels of miR-322/503 is associated with increased resistance to apoptosis, which is abolished by Smurf2 silencing. These findings indicate that miR-322/503 represses Smurf2 translation, in turn affecting intestinal epithelial homeostasis by altering TGF-β/Smad2 signaling and IEC apoptosis.
INTRODUCTION
Ubiquitin modification is implicated in many aspects of cellular physiology by tagging proteins for proteasomal degradation or incorporation into other regulatory complexes (Hershko and Ciechanover, 1998; Hoeller et al., 2006). E3 ubiquitin ligases play a central role in the ubiquitination process, catalyzing the attachment of ubiquitin moieties to target proteins. Smad ubiquitin regulatory factor 2 (Smurf2), a HECT-domain E3 ubiquitin ligase, was initially identified as a negative regulator of transforming growth factor β (TGF-β) signaling by targeting receptor-regulated Smads, including Smad1, Smad2, and Smad3, and the type I receptor (Zhang et al., 2001; Izzi and Attisano, 2004; Lönn et al., 2009). After ligand binding, Smad proteins are phosphorylated by TGF-β receptors and translocate to the nucleus, where they control the transcription of target genes (Ten Dijke and Hill, 2004; Massague et al., 2005; Kamato et al., 2013). The TGF-β–activated Smads must be removed through ubiquitin-dependent degradation in order to exert the biological effects of different ligands in a timely manner. Smurf2 interacts with phosphorylated Smads and induces their rapid ubiquitination and degradation (Lo and Massague, 1999; Gao et al., 2009; David et al., 2013). Subsequent studies extended the repertoire of Smurf2 substrates and show a broader spectrum of Smurf2 biological functions (Li and Seth, 2004; Schwamborn et al., 2007). Induction in Smurf2 expression is linked to telomere attrition, and forced expression of Smurf2 enhances senescence in human fibroblasts (Zhang et al., 2004). Smurf2 also promotes p53 degradation by stabilizing the E3 ligase MDM2 (Nie et al., 2010) and represses tumor growth by controlling the chromatin landscape and genome stability through RNF20 (Blank et al., 2012).
MicroRNAs (miRNAs) are a class of small noncoding RNAs that posttranscriptionally repress the expression of target genes and regulate a variety of cellular processes (Kedde et al., 2007; Omer et al., 2009; Mendell and Olson, 2012). The regulation of mRNA stability and translation by miRNAs is a major mechanism by which mammalian cells control gene expression in response to changes in environmental conditions (Garneau et al., 2007; Mendell and Olson, 2012), but little is known about their roles in the regulation of the ubiquitin system. Generally, miRNAs function by binding to the 3′-untranslated regions (UTRs) of target mRNAs, destabilizing them and/or inhibiting their translation (Eulalio et al., 2008; Leung and Sharp, 2010). Although the exact functions of miRNAs in human development and physiology are not fully elucidated, differential expression of some miRNAs has been linked to several human pathologies. Recently miRNAs have also emerged as major regulators of homeostasis of the intestinal epithelium (Xiao et al., 2011; Ye et al., 2011; Cui et al., 2012; Zhuang et al., 2013), the tissue with the most rapid turnover rate in the body (Sato and Clevers, 2013). For example, miR-503 represses translation of CUG-binding protein 1 (CUGBP1), in turn modulating intestinal epithelial cell (IEC) proliferation and apoptosis (Cui et al., 2012). Other important miRNAs implicated in modulating intestinal epithelial homeostasis are 1) the antiproliferative miRNAs miR-222 (Xiao et al., 2011) and miR-29b (Xiao et al., 2013), 2) the proproliferative miRNAs miR-21 and miR-155 (Bakirtzi et al., 2011), 3) the cell migration–regulatory miR-195 (Zhuang et al., 2013), and 4) the epithelial barrier–regulatory miRNA miR-122b (Ye et al., 2011).
miR-322 is an abundant miRNA that is conserved among many species and is clustered with miR-503 on chromosome X (Griffiths-Jones et al., 2006). miR-322 and miR-503 were studied in myogenesis, where they were shown to promote cell cycle quiescence and differentiation by down-regulating Cdc25 (Sarkar et al., 2010). Two recent studies show that miR-322 regulates the vascular smooth muscle cell phenotype and neointimal formation by inhibiting expression of cyclin D1- and Ca2+-regulating proteins calumenin and stromal-interacting molecule 1 (Stim1; Merlet et al., 2013) and that miR-322 and its target, Tob2, control osteogenesis through modulation of Osx mRNA degradation (Gamez et al., 2013). Our previous studies show that small intestinal mucosal atrophy after polyamine depletion or fasting is associated with a decrease in the levels of miR-322/503 (Xiao et al., 2011, 2013; Cui et al., 2012). The results reported here further indicate that miR-322/503 regulates Smurf2 expression and modulates intestinal epithelial homeostasis by altering IEC apoptosis. miR-322/503 directly interacts with Smurf2 mRNA via its 3′-UTR and represses Smurf2 translation, thus positively modulating the TGF-β/Smad2 signaling pathway. Moreover, the elevated levels of Smurf2 achieved by silencing miR-322/503 increase the resistance of IECs to apoptosis.
RESULTS
Smurf2 mRNA is a novel target of miR-322/503
Using standard online software (TargetScan and RNA22), we predicted three binding sites for miR-322/503 within the 3′-UTR of the Smurf2 mRNA (Figure 1A and Supplemental Table S1), suggesting that the Smurf2 mRNA is a potential target of miR-322/503. To elucidate the involvement of miR-322/503 in the regulation of Smurf2 expression, we determined whether miR-322/503 associated with the Smurf2 mRNA by RNA pull-down assays, using biotin-labeled miR-322 or miR-503 (Figure 1, Ba and Ca). As shown in Figure 1Bb, 24 h after transfection with biotin-labeled miR-322, cells exhibited elevated miR-322 levels but displayed no changes in the abundance of the housekeeping noncoding RNA U6 (unpublished data). The levels of Smurf2 mRNA were highly enriched in the materials from cells transfected with the biotin-labeled miR-322 but not in the pull-down materials from cells transfected with scrambled control miRNA (Figure 1Bc). The enrichment of the Cugbp1 mRNA product was also examined and served as a positive control, since the Cugbp1 mRNA is a target of miR-322/503 (Cui et al., 2012). In contrast, increasing the levels of biotin-miR-322 did not increase its interaction with the mRNAs encoding c-Myc and cyclin-dependent kinase 2 (Cdk2). In addition, transfection with biotin-labeled miR-322 failed to alter the steady-state levels of total Smurf2, Cugbp1, c-Myc, and Cdk2 mRNAs (Figure 1Bd). Consistently, the abundance of Smurf2 and Cugbp1 mRNAs was also highly enriched in the materials from cells transfected with the biotin-labeled miR-503, but there were no changes in the levels of c-Myc and Cdk2 mRNAs between cells transfected with biotin-labeled miR-503 and cells transfected with scrambled miRNA (Figure 1C). These results strongly suggest that the Smurf2 mRNA is a novel target of miR-322/503 in IECs.
FIGURE 1:

miR-322/503 directly interacts with the Smurf2 mRNA. (A) Schematic representation of Smurf2 mRNA depicting predicted target sites for miR-322/503 in its 3′-UTR. BS, predicted miR-322/503-binding site. (B) Association of biotinylated miR-322 with mRNAs encoding Smurf2, Cugbp1, c-Myc, and Cdk2. (a) Schematic representation of biotinylated miR-322; (b) levels of biotinylated miR-322 as measured by Q-PCR analysis 24 after transfection; (c) levels of Smurf2, Cugbp1, c-Myc, and Cdk2 mRNAs in the materials pulled down by biotin-miR-322; and (d) levels of total input mRNAs. Values are means ± SEM from three separate experiments. *p < 0.05 compared with cells transfected with control scramble oligomer. (C) Binding of biotinylated miR-503 toSmurf2, Cugbp1, c-Myc, and Cdk2 mRNAs as measured by biotin pull-down assays used in studies described in B. *p < 0.05 compared with cells transfected with scramble oligomer.
miR-322/503 represses Smurf2 translation by interacting with Smurf2 3′-UTR
To examine the functional consequences of the [miR-322/503-Smurf2 mRNA] association, we designed the first set of experiments to investigate whether increasing the levels of miR-322/503 through transfection with precursors (pre–miR-322 or pre–miR-503) repressed Smurf2 expression. As shown, increased levels of either miR-322 by pre–miR-322 transfection or miR-503 by pre–miR-503 transfection decreased Smurf2 protein levels (Figure 2, A and B), although they did not reduce the levels of total Smurf2 mRNA (Figure 2C). To determine whether miR-322/503 inhibited Smurf2 expression by repressing its translation, we examined changes in the levels of new Smurf2 protein synthesis after ectopic overexpression of miR-322 or miR-503 and demonstrated that newly synthesized Smurf2 protein decreased significantly in cells transfected with pre–miR-322 or pre–miR-503 compared with cells transfected with scrambled oligomer (Figure 2D). Inhibition of Smurf2 protein synthesis by miR-322/503 induction was specific, since there was no change in nascent glyceraldehyde-3-phosphate dehydrogenase (GAPDH) synthesis after transfection with pre–miR-322 or pre–miR-503. To further define the roles of miR-322/503 in the regulation of Smurf2 mRNA translation, we examined the relative distribution of Smurf2 mRNA in individual fractions from polyribosome gradients after ectopic overexpression of pre–miR-322 or pre–miR-503. Although increasing the levels of miR-322 or miR-503 did not affect global polysomal profiles (unpublished data), the association of Smurf2 mRNA with actively translating fractions (fractions 8–10) decreased dramatically, shifting to low-translating fractions (fractions 5–7; Figure 2E, top). In contrast, housekeeping Gapdh mRNA distributed similarly in both groups (Figure 2E, bottom).
FIGURE 2:

Ectopic overexpression of miR-322/503 inhibits Smurf2 translation. (A) Levels of miR-322 (a) and miR-503 (b) 48 h after transfection of pre–miR-322 or pre–miR-503. Values are means ± SEM from three separate experiments. *p < 0.05 compared with scramble. (B) Levels of Smurf2 protein after ectopic miR-322/503 overexpression. Whole-cell lysates were prepared for Western blotting; equal loading was monitored by assessing β-actin levels. (C) The levels of Smurf2 mRNA as examined by Q-PCR analysis. Left, cells transfected with pre–miR-322; right, cells transfected with pre–miR-503. (D) New synthesis of Smurf2 protein. After cells were exposed to l-azidohomoalanine (AHA), cell lysates were incubated with the reaction buffer containing biotin/alkyne reagent; the biotin-alkyne/azide–modified protein complex was pulled down by paramagnetic streptavidin-conjugated Dynabeads. (E) Distributions of Smurf2 (top) and Gapdh (bottom) mRNAs in each gradient fraction of polysomal profile after ectopic miR-322/503 overexpression. After fractionation through sucrose gradients, total RNA was isolated from different fractions; the levels of Smurf2 and Gapdh mRNAs were measured by Q-PCR analysis and plotted as percentage of total Smurf2 or Gapdh mRNA level in the samples. (F) Levels of reporter activities after ectopic overexpression of miR-322/503. Results expressed as means ± SEM data from three separate experiments. *p < 0.05 compared with cells transfected with scrambled RNA.
To determine whether this inhibitory effect was mediated through the Smurf2 CR, we subcloned 3′-UTR or both fractions of the Smurf2 CR and 3′-UTR into the pmirGLO dual-luciferase miRNA target expression vector to generate pmirGLO-smurf2-CR and pmirGLO-smurf2-3′UTR reporter constructs (Figure 2F, schematic). Overexpression of miR-322 or miR-503 by transfection with either pre–miR-322 or pre–miR-503 selectively decreased the levels of Smurf2-3′UTR luciferase reporter activity (Figure 2F, right) but failed to inhibit the activity of Smurf2-CR reporter activity (Figure 2F, left), indicating that miR-322/503 represses Smurf2 mRNA translation through interaction with its 3′-UTR rather than its CR. We also found that increasing the levels of both miR-322 and miR-503 by cotransfection with pre–miR-322 and pre–miR-503 (Supplemental Figure S1) did not synergistically repress Smurf2 translation, as the levels of Smurf2 protein and Smurf2-3′UTR luciferase reporter activity in cells cotransfected with pre–miR-322 and pre–miR-503 were similar to those observed in cells transfected with pre–miR-322 or pre–miR-503 alone (Supplemental Figure S2).
The second set of experiments examined the influence of decreasing the level of miR-322/503 by transfecting the corresponding antisense oligomer (antagomir) targeting miR-322 (anti–miR-322) or miR-503 (anti–miR-503) on Smurf2 expression. Transfection with anti–miR-322 or anti–miR-503 decreased the levels of miR-322 and miR-503 (Figure 3A) and increased the level of Smurf2 protein (Figure 3B), although there were no significant changes in the levels of total Smurf2 mRNA (Figure 3C). Supporting the notion that the antagomirs were specific, anti-miR322 oligo only decreased miR-322 levels and did not alter miR-503 expression, whereas anti–miR-503 silenced miR-503 expression without affecting miR-322 levels (Supplemental Figure S3). The results presented in Figure 3D further show that silencing miR-322/503 induced the synthesis of new Smurf2 protein; this stimulatory effect was mediated via the interaction with the Smurf2 3′-UTR, since the levels of luciferase reporter activity were increased in miR-322– or miR-503–silent population of cells only when cells were transfected with the construct of pmirGLO-Smurf2-3′UTR (Figure 3E). These results indicate that decreasing the levels of miR-322/503 enhances Smurf2 translation by reducing formation of the [miR-322/503-Smurf2 mRNA] complex.
FIGURE 3:

miR-322/503 silencing enhances Smurf2 translation. (A) At 48 h after transfecting IECs with oligomers targeting miR-322 (anti–miR-322) or miR-503 (anti–miR-503) or with a control oligo (C-oligo), the levels of miR-322 (a) and miR-503 (b) were measured by Q-PCR. Values are means ± SEM from three separate experiments. *p < 0.05 compared with cells transfected with C-oligo. (B) Levels of Smurf2 protein as measured by Western immunoblotting analysis in cells described in A. (C) Levels of Smurf2 mRNA as examined by Q-PCR. (D) New synthesis of Smurf2 protein as measured by Click-iT protein analysis using l-azidohomoalaine (AHA). (E) Reporter activities after miR-322/503 silencing. Results expressed as means ± SEM data from three separate experiments. *p < 0.05 compared with cells transfected with C-oligo.
The third set of experiments aimed at further characterizing the specific binding site of miR-322/503 in the Smurf2 3′-UTR. To do so, we prepared various reporter constructs that expressed chimeric RNA containing the luciferase and partial transcripts spanning the Smurf2 3′-UTR with or without the potential binding site, as indicated in the schematic of Figure 4A. Ectopic overexpression of miR-322 or miR-503 was found to decrease the levels of luciferase reporter gene activity when cells were transfected with plasmid FL-Luc (containing full-length Smurf2 3′-UTR) or F3-Luc (containing a predicted binding site located at positions 3974–3998) but not with the F1-Luc (in which the potential binding sites were deleted) or F2-Luc and F4-Luc (which also contains a predicted binding site). Point mutation of the site located at the F3 of the Smurf2 3′-UTR was also performed, in which three nucleotides of F3-Luc of the Smurf2 3′-UTR were mutated (Figure 4B, schematic). Of interest, Smurf2 repression by miR-322/503 was completely prevented when this specific binding site was mutated from the Smurf2 3′-UTR. Taken together, these results indicate that miR-322/503 interacts with Smurf2 mRNA predominantly via the specific binding site at the position spanning 3974–3998 and represses Smurf2 mRNA translation.
FIGURE 4:
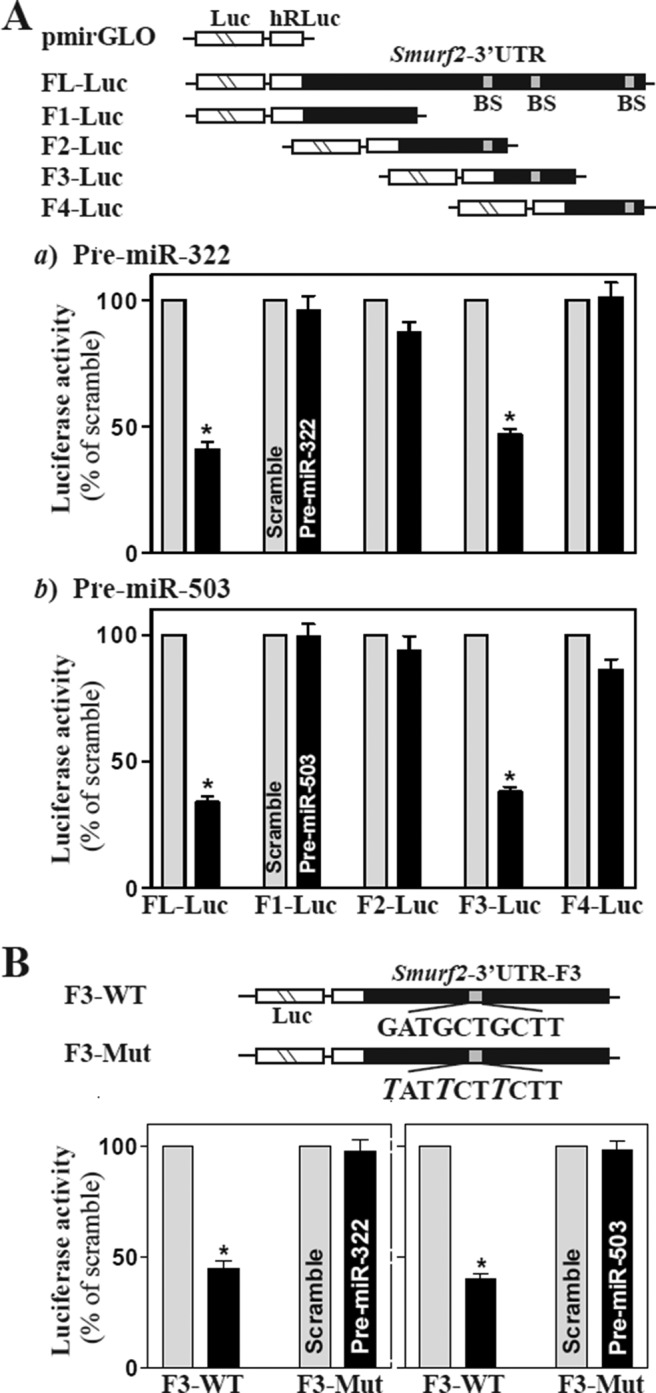
Changes in activities of Smurf2 3′-UTR luciferase reporters after deletion of miR-322/503–binding site. (A) Effect of 5′ deletion of Smurf2 3′-UTR on its luciferase reporter activity in cells transfected with pre–miR-322 (a) or pre–miR-503 (b). Top, schematic of plasmids of different chimeric firefly luciferase Smurf2 3′-UTR reporters. BS, predicted miR-322/503–binding site. Bottom, Smurf2 3′-UTR luciferase reporter activity. At 24 h after transfection with pre–miR-322 or pre–miR-503, cells were cotransfected with Smurf2 F-Luc constructs and a Renilla luciferase reporter. Firefly and Renilla luciferase activities were assayed 24 h later. Results were normalized to the Renilla luciferase activity and expressed as means ± SEM of data from three separate experiments. *p < 0.05 compared with cells transfected with control scrambled oligomer. (B) Effect of point mutation of specific miR-322/503–binding site (schematic) in Smurf2 3′-UTR on luciferase reporter activities after ectopic overexpression of miR-322 (left) or miR-503 (right). *p < 0.05 compared with cells transfected with control scrambled oligomer.
miR-322/503–modulated Smurf2 expression regulates TGF-β/Smad2 signaling
To investigate the implication of miR-322/503–modulated Smurf2 expression in the regulation of TGF-β/Smad2 signaling pathway, we examined changes in the levels of phosphorylated Smad2 (p-Smad2; the activated form of Smad2 and target for ubiquitin-dependent degradation) in miR-322/503–silenced cells and cells overexpressing green fluorescent protein (GFP)–tagged Smurf2 after treatment with TGF-β. As expected, exposure of normal IECs to TGF-β induced Smad2 activation rapidly, as indicated by increase in the accumulation of p-Smad2 protein (Figure 5A), although it did not affect total Smad2 (T-Smad2) or Smurf2 levels. On the other hand, ectopic overexpression of GFP-tagged Smurf2 decreased the basal level of p-Smad2 in the absence of TGF-β (Figure 5B, left) and prevented TGF-β–induced Smad2 activation. There were no significant differences in the levels of p-Smad2 after treatment with TGF-β in cells overexpressing GFP-tagged Smurf2 (Figure 5B, right). Moreover, the increased levels of endogenous Smurf2 after antagonizing miR-322 through transfection with anti–miR-322 were also associated with a decrease in the levels of p-Smad2; this effect was prevented by silencing Smurf2 (Figure 5C). In miR-322–silenced cells, increased Smurf2 suppressed TGF-β–induced Smad2 activation by enhancing p-Smad2 degradation (Figure 5D). In fact, the levels of p-Smad2 protein in miR-322–silenced cells treated with TGF-β were much lower than those observed in miR-silenced cells without TGF-β treatment. Similarly, the increase in levels of endogenous Smurf2 by antagonism of miR-503 had an identical stimulatory effect on p-Smad2 degradation after treatment with TGF-β (Figure 5, E and F). In addition, increased levels of Smurf2, by either ectopic gene expression or silencing miR-322/503, failed to alter the cellular abundance of T-Smad2. These results indicate that increased Smurf2 by miR-322/503 silencing inhibits TGF-β/Smad2 activation by enhancing p-Smad2 degradation.
FIGURE 5:
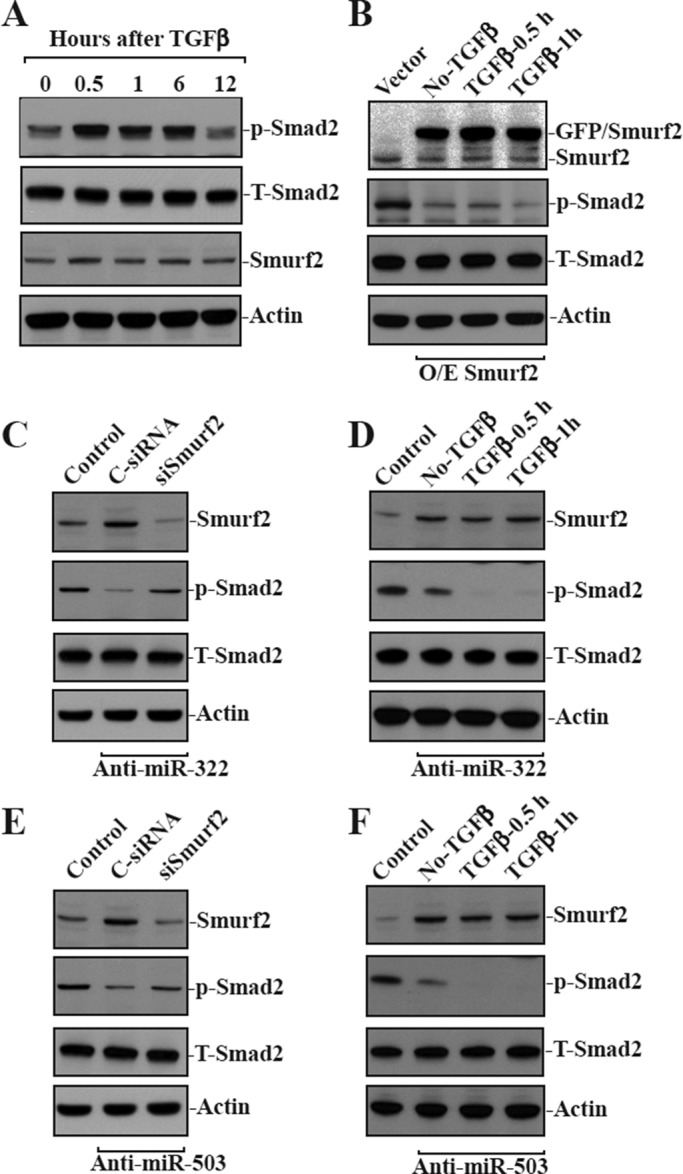
miR-322/503–modulated expression of Smurf2 regulates the degradation of phosphorylated Smad2. (A) Levels of phosphorylated Smad2 (p-Smad2) and total Smad2 (T-Smad2) in cells treated with TGF-β (1 ng/ml) for different times. The levels of p-Smad2 and T-Smad2 were measured by Western blotting analysis using anti–p-Smad2 or anti-Smad2 antibody. (B) Changes in the levels of p-Smad2 and T-Smad2 in cells overexpressing GFP-tagged Smurf2 after treatment with TGF-β. After cells were transfected with the Smurf2 expression vector or control vector for 24 h, they were exposed to TGF-β for 0.5 or 1 h. (C) Levels of p-Smad2 and T-Smad2 48 h after cotransfecting cells with anti–miR-322 and control siRNA (C-siRNA) or siRNA targeting Smurf2 mRNA (siSmurf2). (D) Changes in the levels of p-Smad2 and T-Smad2 in miR-322–silenced cells exposed to TGF-β. Cells were initially transfected with anti–miR-322 for 48 h and then exposed to TGF-β for 0.5 or 1 h. (E) Levels of p-Smad2 and T-Smad2 48 h after cotransfecting cells with anti–miR-503 and siSmurf2 for 48 h. (F) Changes in the levels of p-Smad2 and T-Smad2 in miR-503–silenced cells exposed to TGF-β for different times.
Induced Smurf2 by miR-322/503 silencing protects IECs against apoptosis
To investigate the biological significance of miR-322/503–modulated Smurf2 expression, we examined their possible involvement in the control of intestinal epithelial homeostasis by measuring changes in apoptosis in miR-322/503–silenced cells. Increased levels of endogenous Smurf2 resulting from anti–miR-322 transfection (Figure 3) did not directly induce apoptosis (Figure 6A, a and b, left). There were no apparent differences in cell viability between miR-322–antagonized populations and control cells, including no morphological features of apoptosis and no detectable levels of active caspase-3 regardless of treatment with or without the miR-322 antagomir. To determine whether increased Smurf2 by antagonizing miR-322 altered the susceptibility of IECs to apoptosis, we exposed cells to treatment with tumor necrosis factor-α (TNF-α) plus cycloheximide (CHX). When control cells were exposed to TNF-α/CHX for 4 h, we observed morphological features characteristic of programmed cell death, elevated annexin V staining, a classic indicator of apoptotic cells (Figure 6, Aa, right, and B, left), and increased levels of active (cleaved) caspase-3 (Figure 6C). Increasing Smurf2 by miR-322 silencing protected IECs against TNF-α/CHX–induced apoptosis, as indicated by a decrease in the percentages of apoptotic cells (Figure 6, Ab and B). This protective effect remained intact when cells were transfected with control siRNA (Figure 6Ac), but it was lost when Smurf2 expression was silenced by siRNA targeting Smurf2 (siSmurf2). The percentage of apoptotic cells (Figure 6, Ad and B, right) and level of active caspase-3 protein (Figure 6C) in miR-322–silenced cells transfected with siSmurf2 were higher than those in miR-322–antagonized cells transfected with C-siRNA after exposure to TNF-α/CHX. Consistently, the increase in Smurf2 levels by miR-503 silencing also elicited antiapoptotic effects after exposure to TNF-α/CHX, and this protective effect was prevented by Smurf2 silencing (Supplemental Figure S4). Furthermore, ectopic overexpression of GFP-tagged Smurf2 protected IECs against TNF-α/CHX–induced apoptosis, because the percentage of apoptotic cells and the level of active caspase-3 in cells overexpressing GFP/Smurf2 decreased compared with those observed in control cells after treatment with TNF-α/CHX (Figure 7). These results indicate that Smurf2 is an antiapoptotic factor in IECs, whereas miR-322/503 silencing protects cells against apoptosis by increasing Smurf2.
FIGURE 6:

miR-322 silencing protects IECs against apoptosis by induction in the levels of endogenous Smurf2. (A) TNF-α/CHX–induced apoptosis after various treatments. Cells were transfected with the anti–miR-322 or control oligo (control); 48 h after transfection, apoptosis was measured after 4 h of treatment with TNF-α/CHX. (a) Cells transfected with Con-oligo; (b) cells transfected with anti-miR-322; (c) cells transfected with anti–miR-322 and control siRNA (C-siRNA); (d) cells transfected with anti–miR-322 and siRNA targeting Smurf2 (siSmurf2). Original magnification, ×150. (B) Percentages of apoptotic cells after different treatments as described in A. Values are means ± SEM of data from three experiments. *p < 0.05 compared with cells untreated with TNF-α/CHX; +p < 0.05 compared with cells exposed to TNF-α/CHX; #p < 0.05 compared with cells cotransfected with anti–miR-322 and C-siRNA and then exposed to TNF-α/CHX. (C) Changes in levels of caspase-3 in cells described in A. Whole-cell lysates were harvested, and the levels of procaspase-3 and caspase-3 were assessed by Western blot analysis. β-Actin immunoblotting was performed as an internal control for equal loading.
FIGURE 7:
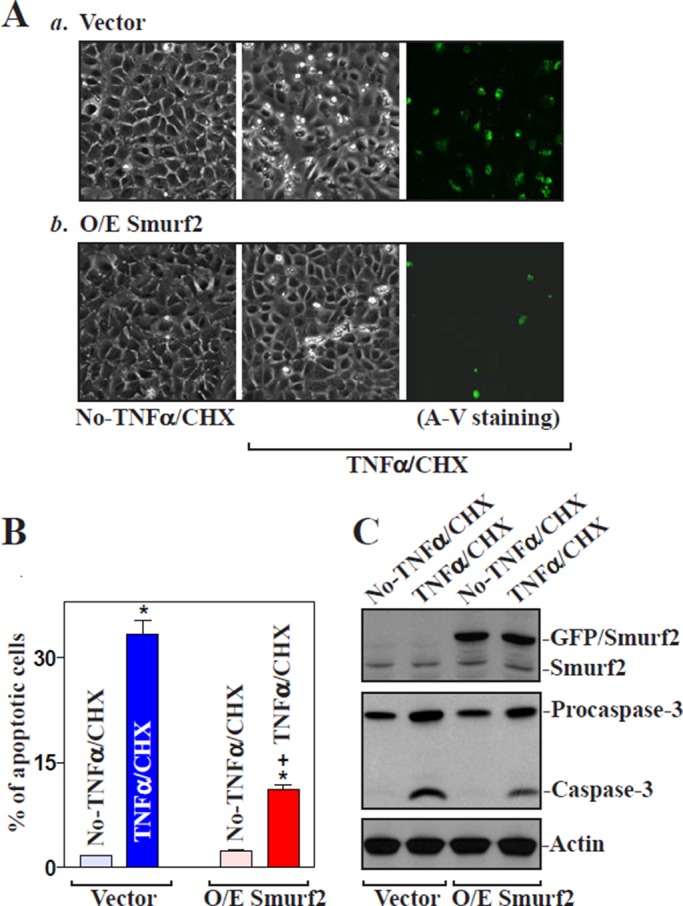
Ectopic overexpression of Smurf2 protects IECs against apoptosis. (A) Images of apoptotic cell death after treatment without (left) or with (middle and right) TNFα/CHX for 4 h. (a) Cells transfected with control vector (vector) and (b) cells transfected with the vector expressing GFP-tagged Smurf2 protein (Smurf2) for 24 h. Original magnification, ×150. (B) Percentage of apoptotic cells in study as described in A. Values are means ± SEM of data from three experiments. *p < 0.05 compared with No-TNFα/CHX. +p < 0.05 compared with cells that were transfected with control vector and then treated with TNFα/CHX for 4 h. (C) Representative immunoblots for GFP/Smurf2, procaspase-3, and caspase-3 in cells described in A, as measured by Western immunoblotting analysis.
DISCUSSION
Smurf2 is an important E3 ubiquitin ligase and plays a critical role in defining substrate specificity and subsequent protein degradation by the 26S proteasomes (Lin et al., 2000; Ten Dijke and Hill, 2004; Xu et al., 2012), but the exact mechanisms that control Smurf2 abundance have not been identified. The present study demonstrates a novel function of miR-322/503 in the regulation of Smurf2 translation and provides insight into the control of Smurf2 expression at the posttranscriptional level. Both miR-322 and miR-503 interact with the Smurf2 mRNA via its 3′-UTR and repress Smurf2 translation, but they do not appear to affect Smurf2 mRNA stability, since miR-322/503 overexpression specifically reduced Smurf2 nascent translation and anti–miR-322 and anti–miR-503 antagomirs elevated it, but neither intervention affected total Smurf2 mRNA levels. Our study further shows direct evidence of the site of miR-322/503 in the Smurf2 3′-UTR, as point mutation of this site within the Smurf2 3′-UTR prevented miR-322/503–induced repression of Smurf2 translation. These findings advance our understanding of the molecular mechanisms underlying the regulation of Smurf2 gene expression and indicate that association of miR-322/503 with the Smurf2 mRNA controls its translation and therefore modulates the Smurf2-dependent ubiquitin system. Furthermore, our findings strongly suggest that control of Smurf2 mRNA translation by miR-322/503 is relevant for maintaining intestinal epithelial homeostasis by altering IEC apoptosis.
Our results also indicate that miR-322/503 interacted with the Smurf2 3′-UTR but not its CR in IECs. Studies using various ectopic reporters bearing partial transcripts spanning the Smurf2 3′-UTR with or without the miR-322/503 binding site further demonstrated that the 3974-3998 site of the Smurf2 3′-UTR was the predominant and functional site through which miR-322/503 interacted with and repressed Smurf2 translation, whereas two other predicted sites spanning positions 3721–3774 and 4919–4921 in the Smurf2 3′-UTR exhibited a lesser effect. Consistent with our results, several previous studies showed that miR-322/503 directly interact with the 3′-UTRs of Cdc25, Tob2, Ccnd1 (encoding cyclin D1), and Cal (encoding calumenin) mRNAs (Sarkar et al., 2010; Gamez et al., 2013; Merlet et al., 2013) and thus destabilizes them and/or represses their translation. In some instances, miR-322/503 was also found to associate with the CRs of target mRNAs for their regulatory actions; for example, we recently reported that miR-503 represses CUGBP1 translation by directly interacting with the Cugbp1 CR rather than the 3′-UTR (Cui et al., 2012).
Although the specific molecular mechanism by which miR-322/503 association with Smurf2 mRNA represses Smurf2 translation is unknown, our recent studies show that miR-503 inhibits CUGBP1 translation in normal IECs by enhancing the recruitment of Cugbp1 mRNA to processing bodies (P-bodies; Cui et al., 2012), where mRNAs are sorted for translation repression and/or degradation (Buchan and Parker, 2009; Kulkarni et al., 2010; Yu et al., 2012). Silencing P-body–resident proteins such as Ago2 or RCK prevents miR-503–induced repression of CUGBP1 translation, whereas Ago2 overexpression enhances the inhibitory effect of miR-503. In support of this notion, our previous studies also demonstrated that miR-222–induced repression of CDK4 translation and miR-195–induced Stim1 mRNA destabilization are directly linked to increased translocation of Cdk4 and Stim1 mRNAs to P-bodies (Xiao et al., 2011; Zhuang et al., 2013). It is unclear, however, whether miR-322/503 alters the subcellular localization of the Smurf2 mRNA to P-bodies in normal IECs and whether an increase in the recruitment of Smurf2 mRNA to P-bodies affects its translation rate. These possibilities are being investigated in our ongoing studies.
Our results presented here also indicate that the miR-322/503–modulated expression of Smurf2 regulates TGF-β/Smad2 signaling pathway. Increased levels of endogenous Smurf2 by miR-322/503 silencing not only decreased basal levels of p-Smad2 (activated Smad2 protein) in no-TGF-β–stimulated cells but also prevented TGF-β–induced Smad2 activation by increasing p-Smad2 degradation. Smad proteins consist of two conserved globular domains, MH1 and MH2, connected by a linker region (Shi and Massague, 2003). The MH1 domain binds DNA, whereas the MH2 domain binds membrane receptors for activation, nucleoporins for nuclear translocation, and other members of the Smad family to form transcriptional complexes. On the other hand, the linker region of Smads functions as a critical regulatory domain, and its phosphorylation activates Smad proteins and then induces their degradation. In Smad2 the linker region contains four SP phosphorylation sites for proline-directed kinases and also has a PPAY motif that binds Smurf2 ubiquitin ligase (Zhu et al., 1999; Sapkota et al., 2007). Exposure to TGF-β induces phosphorylation of Smad2 at the C-terminus, as well as at an interdomain linker region. TGF-β–induced linker phosphorylation marks the activated Smad proteins for proteasome-mediated destruction, and this process is tightly regulated by numerous factors, including Nup214 and Nedd4L (Sapkota et al., 2007; Gao et al., 2009). To the best of our knowledge, the present study provides the first evidence that miR-322/503 regulates TGF-β/Smad2 signaling by altering Smurf2 expression. Because the lowering of miR-322/503 by transfection with their respective antagomirs was similar to that observed in IECs exposed to cell stimuli such as polyamine depletion (Cui et al., 2012), the induced expression of Smurf2 by miR-322/503 silencing likely reflects biologically relevant events.
Finally, our findings show that miR-322/503–modulated Smurf2 expression has cellular function, playing a role in maintaining homeostasis of the intestinal epithelium by regulating IEC apoptosis. The epithelium of the intestinal mucosa is subject to rapid and dynamic self-renewal, and its homeostasis is preserved through strict regulation of cell proliferation, migration, differentiation, and apoptosis (Wang et al., 1991; Sato and Clevers, 2013). IECs continuously replicate within the crypts of intestine, and this process is counterbalanced by apoptosis (Liu et al., 2003; Rao and Wang, 2011). Apoptosis occurs in the crypt area, where it maintains a critical balance in cell number between newly divided and surviving cells, and at the luminal surface of the intestinal mucosa, where differentiated cells are lost. Our previous studies and studies from other laboratories (Seiler and Raul, 2007) demonstrated that NF-κB (Li et al., 2001), Akt kinase (Zhang et al., 2004), ATF-2 (Xiao et al., 2007), XIAP (Zhang et al., 2009), MEK1 (Wang et al., 2010), and CUGBP1 (Cui et al., 2012) are involved in the control of IEC apoptosis, and these signals are highly regulated by cellular polyamines. Of interest, polyamines also regulate expression of miR-322/503 in IECs, and depletion of cellular polyamines decreases the level of miR-322/503 (Xiao et al., 2011; Cui et al., 2012). Our present studies provide additional evidence that the miR-322/503–modulated protein Smurf2 is a new member of this family of regulators of the control intestinal epithelial homeostasis, since increasing endogenous Smurf2 by silencing miR-322/503 protected cells against apoptosis, and this effect was abolished when Smurf2 expression was silenced. Taken together, our findings suggest that miR-322/503–mediated Smurf2 repression increases the susceptibility of IECs to apoptosis under pathological conditions, thus representing a novel therapeutic target for patients with intestinal mucosal atrophy and/or impaired epithelial integrity.
MATERIALS AND METHODS
Chemicals and cell culture
Tissue culture medium and dialyzed fetal bovine serum were from Invitrogen (Carlsbad, CA), and biochemicals were from Sigma-Aldrich (St. Louis, MO). The antibodies recognizing Smurf2, Smad2, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Cell Signaling (Danvers, MA), and the secondary antibody conjugated to horseradish peroxidase was from Sigma-Aldrich. Pre–miR miRNA precursors and anti-miR miRNA inhibitors of miR-322 and miR-503 were purchased from Ambion (Austin, TX). Biotin-labeled miR-322 and miRNA-503 were custom made by Dharmacon (Lafayette, CO).
The IEC-6 cell line, derived from normal rat intestinal crypt cells (Quaroni et al., 1979), was purchased from the American Type Culture Collection (Manassas, VA) at passage13 and was maintained in T-150 flasks in DMEM supplemented with 5% heat-inactivated fetal bovine serum. IEC-6 cells at passages 15–20 exhibited no significant changes in biological function or characterization (Wang et al., 1995; Liu et al., 2009) and were used in all experiments.
Plasmid construction
The chimeric firefly luciferase reporter construct of the Smurf2 CR or 3′-UTR was generated as described previously (Zou et al., 2010). The full-length Smurf2 CR or its 3′-UTR and different 3′-UTR fragments with or without predicted miR-322/503–binding site were amplified and subcloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Madison, WI) to generate the pmirGLO-Luc-Smurf2-CR and pmirGLO-Smurf2-3′UTR. The sequence and orientation of the fragment in the luciferase reporter were confirmed by DNA sequencing and enzyme digestion. Transient transfections were performed using the Lipofectamine Reagent as recommended by the manufacturer (Invitrogen). The luciferase reporter constructs were transfected into cells along with phRL-null, a Renilla luciferase control reporter vector from Promega, to monitor transfection efficiencies as described previously (Liu et al., 2009). Luciferase activity was measured using the Dual Luciferase Assay System, and the levels of pmirGLO-Luc-Smurf2-CR or Smurf2-3′UTR luciferase activity were normalized to Renilla luciferase activity and further compared with the levels of luciferase mRNA in every experiment. All of the primer sequences for generating these constructs are list in Supplemental Table S2. The vector expressing GFP-tagged Smurf2 protein was obtained from OriGene (Rockville, MD).
Reverse transcription and quantitative real-time PCR analyses
Total RNA was isolated by using the RNeasy mini kit (Qiagen, Valencia, CA) and used in reverse transcription (RT) and PCR amplification reactions as described (Yu et al., 2011). The levels of Gapdh PCR product were assessed to monitor the evenness in RNA input in RT-PCR samples. Real-time quantitative PCR (Q-PCR) analysis was performed using 7500-Fast Real-Time PCR Systems with specific primers, probes, and software (Applied Biosystems, Foster City, CA). For miRNA studies, levels of miR-322 and miR-503 were also quantified by Q-PCR by Taqman MicroRNA assay; levels of small nuclear RNA U6 were measured as endogenous control.
Western blotting analysis
Whole-cell lysates were prepared using 2% SDS, sonicated, and centrifuged (12,000 rpm) at 4°C for 15 min. The supernatants were boiled for 5 min and size fractionated by SDS–PAGE (7.5% acrylamide). After transferal of proteins onto nitrocellulose filters, the blots were incubated with primary antibodies recognizing Smurf2, Smad2, or phosphorylated Smad2; after incubations with secondary antibodies, immunocomplexes were developed by using chemiluminescence.
Analysis of newly translated protein
New synthesis of nascent Smurf2 protein was detected by Click-iT protein analysis detection kit (Life Technologies, Grand Island, NY) and performed following the manual with minor modification (Xiao et al., 2013). Briefly, cells were incubated in methionine-free medium and then exposed to l-azidohomoalanine. After mixing of cell lysates with the reaction buffer containing biotin/alkyne reagent and CuSO4 for 20 min, the biotin-alkyne/azide–modified protein complex was pulled down using paramagnetic streptavidin-conjugated Dynabeads. The pull-down material was resolved by 10% SDS–PAGE and analyzed by Western immunoblotting analysis using the antibody against Smurf2 or GAPDH.
Polysome analysis was performed as described (Yu et al., 2012). Briefly, cells at ∼70% confluence were incubated for 15 min in 0.1 mg/ml cycloheximide, lifted by scraping in 1 ml of polysome extraction lysis buffer, and lysed on ice for 10 min. Nuclei were pelleted, and the resulting supernatant was fractionated through a 10–50% linear sucrose gradient to fractionate cytoplasmic components according to their molecular weights. The eluted fractions were prepared with a fraction collector (Brandel, Gaithersburg, MD), and their quality was monitored at 254 nm using a UV-6 detector (ISCO, Louisville, KY). After RNA in each fraction was extracted, the levels of each individual mRNA were quantified by Q-PCR in each of the fractions.
Biotin-labeled miR-322/503 pull-down assays
Binding of miR-322 or miR-503 to target mRNAs was examined by biotin-labeled miR-322 or miR-503 as described previously (Orom and Lund, 2007; Nonne et al., 2010). Briefly, biotin-labeled miR-322 or miR-503 was transfected into cells for 24 h, and then whole-cell lysate was collected. Cell lysates were mixed with streptavidin-Dynal beads (Invitrogen) and incubated at 4°C with rotation overnight. After the beads were washed thoroughly, the bead-bound RNA was isolated and subjected to RT followed by Q-PCR analysis. Input RNA was extracted and served as control.
Determination of apoptosis
Apoptosis was induced by treatment with TNF-α in combination with CHX as described previously (Li et al., 2001). After various experimental treatments, cells were photographed with a Nikon (Melville, NY) inverted microscope before fixation. Annexin V staining of apoptosis was carried out by using a commercial apoptosis kit (Clontech Laboratories, Palo Alto, CA) and performed according to the protocol recommended by the manufacturer. Briefly, cells were rinsed with 1× binding buffer and resuspended in 200 μl of 1× binding buffer. A 5-μl amount of annexin V was added on slides and incubated at room temperature for 10 min in the dark. Annexin-stained cells were visualized and photographed under fluorescence microscope using a dual filter set for fluorescein isothiocyanate and rhodamine, and the percentage of apoptotic cells was determined.
Statistics
Values are means ± SEM from three to six samples. Immunoblotting results were repeated three times. The significance of the difference between means was determined by analysis of variance. The level of significance was determined by using Duncan's multiple-range test (Harter, 1960).
Supplementary Material
Acknowledgments
We thank Jennifer L. Martindale for technical support in studies of polysome analysis. This work was supported by Merit Review Awards (to J.Y.W., D.J.T., and J.N.R.) from the Department of Veterans Affairs and by National Institutes of Health Grants DK-57819, DK-61972, and DK-68491 (to J.Y.W.). J.Y.W. is a Senior Research Career Scientist, Medical Research Service, U.S. Department of Veterans Affairs. M.G. is supported by the National Institute on Aging–Intramural Research Program, National Institutes of Health.
Abbreviations used:
- Cdk2
cyclin-dependent kinase 2
- CHX
cycloheximide
- CR
coding region
- CUGBP1
CUG-binding protein 1
- GFP
green fluorescent protein
- IEC
intestinal epithelial cell
- miRNAs
microRNAs
- Q-PCR
real-time quantitative PCR
- Smurf2
smad ubiquitin regulatory factor 2
- TGF-β
transforming growth factor-β
- UTRs
untranslated regions
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-09-0560) on February 19, 2014.
REFERENCES
- Bakirtzi K, Hatziapostolou M, Karagiannides I, Polytarchou C, Jaeger S, Iliopoulos D, Pothoulakis C. Neurotensin signaling activates microRNAs-21 and -155 and Akt, promotes tumor growth in mice, and is increased in human colon tumors. Gastroenterology. 2011;141:1749–1761. doi: 10.1053/j.gastro.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank M, Tang Y, Yamashita M, Burkett SS, Cheng SY, Zhang YE. A tumor suppressor function of Smurf2 associated with controlling chromatin landscape and genome stability through RNF20. Nat Med. 2012;18:227–234. doi: 10.1038/nm.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell. 2009;36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui YH, Xiao L, Rao JN, Zou T, Liu L, Chen Y, Turner DJ, Gorospe M, Wang JY. miR-503 represses CUG-binding protein 1 translation by recruiting CUGBP1 mRNA to processing bodies. Mol Biol Cell. 2012;23:151–162. doi: 10.1091/mbc.E11-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D, Nair SA, Pillai MR. Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochim Biophys Acta. 2013;1835:119–128. doi: 10.1016/j.bbcan.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Huntzinger E, Izaurralde E. Getting to the root of miRNA-mediated gene silencing. Cell. 2008;132:9–14. doi: 10.1016/j.cell.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Gamez B, Rodriguez-Carballo E, Bartrons R, Rosa JL, Ventura F. MicroRNA-322 (miR-322) and its target protein Tob2 modulate Osterix (Osx) mRNA stability. J Biol Chem. 2013;288:14264–14275. doi: 10.1074/jbc.M112.432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Alarcon C, Sapkota G, Rahman S, Chen PY, Goerner N, Macias MJ, Erdjument-Bromage H, Tempst P, Massague J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-β signaling. Mol Cell. 2009;36:457–468. doi: 10.1016/j.molcel.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harter JL. Critical values for Duncan's new multiple range tests. Biometric. 1960:671–685. [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer. 2006;6:776–788. doi: 10.1038/nrc1994. [DOI] [PubMed] [Google Scholar]
- Izzi L, Attisano L. Regulation of the TGF-β signalling pathway by ubiquitin-mediated degradation. Oncogene. 2004;23:2071–2078. doi: 10.1038/sj.onc.1207412. [DOI] [PubMed] [Google Scholar]
- Kamato D, Burch ML, Piva TJ, Rezaei HB, Rostam MA, Xu S, Zheng W, Little PJ, Osman N. Transforming growth factor-β signaling: role and consequences of Smad linker region phosphorylation. Cell Signal. 2013;25:2017–2024. doi: 10.1016/j.cellsig.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Kedde M, et al. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell. 2007;131:1273–1286. doi: 10.1016/j.cell.2007.11.034. [DOI] [PubMed] [Google Scholar]
- Kulkarni M, Ozgur S, Stoecklin G. On track with P-bodies. Biochem Soc Trans. 2010;38:242–251. doi: 10.1042/BST0380242. [DOI] [PubMed] [Google Scholar]
- Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Seth A. An RNF11: Smurf2 complex mediates ubiquitination of the AMSH protein. Oncogene. 2004;23:1801–1808. doi: 10.1038/sj.onc.1207319. [DOI] [PubMed] [Google Scholar]
- Li L, Rao JN, Bass BL, Wang JY. NF-κB activation and susceptibility to apoptosis after polyamine depletion in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2001;280:G992–G1004. doi: 10.1152/ajpgi.2001.280.5.G992. [DOI] [PubMed] [Google Scholar]
- Lin X, Liang M, Feng XH. Smurf2 is an ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-β signaling. J Biol Chem. 2000;275:36818–36822. doi: 10.1074/jbc.C000580200. [DOI] [PubMed] [Google Scholar]
- Liu L, Rao JN, Zou T, Xiao L, Wang PY, Turner DJ, Gorospe M, Wang JY. Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation. Mol Biol Cell. 2009;20:4885–4898. doi: 10.1091/mbc.E09-07-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Santora R, Rao JN, Guo X, Zou T, Zhang HM, Turner DJ, Wang JY. Activation of TGF-β-Smad signaling pathway following polyamine depletion in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1056–G1067. doi: 10.1152/ajpgi.00151.2003. [DOI] [PubMed] [Google Scholar]
- Lo RS, Massague J. Ubiquitin-dependent degradation of TGF-β-activated smad2. Nat Cell Biol. 1999;1:472–478. doi: 10.1038/70258. [DOI] [PubMed] [Google Scholar]
- Lönn P, Moren A, Raja E, Dahl M, Moustakas A. Regulating the stability of TGF-β receptors and Smads. Cell Res. 2009;19:21–35. doi: 10.1038/cr.2008.308. [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlet E, Atassi F, Motiani RK, Mougenot N, Jacquet A, Nadaud S, Capiod T, Trebak M, Lompre AM, Marchand A. miR-424/322 regulates vascular smooth muscle cell phenotype and neointimal formation in the rat. Cardiovasc Res. 2013;98:458–468. doi: 10.1093/cvr/cvt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie J, Xie P, Liu L, Xing G, Chang Z, Yin Y, Tian C, He F, Zhang L. Smad ubiquitylation regulatory factor 1/2 (Smurf1/2) promotes p53 degradation by stabilizing the E3 ligase MDM2. J Biol Chem. 2010;285:22818–22830. doi: 10.1074/jbc.M110.126920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonne N, Ameyar-Zazoua M, Souidi M, Harel-Bellan A. Tandem affinity purification of miRNA target mRNAs (TAP-Tar) Nucleic Acids Res. 2010;38:e20. doi: 10.1093/nar/gkp1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omer AD, Janas MM, Novina CD. The chicken or the egg: microRNA-mediated regulation of mRNA translation or mRNA stability. Mol Cell. 2009;35:739–740. doi: 10.1016/j.molcel.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Orom UA, Lund AH. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods. 2007;43:162–165. doi: 10.1016/j.ymeth.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Quaroni A, Wands J, Trelstat RL, Isselbacher KJ. Epithelial cell cultures from rat small intestine. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JN, Wang JY. Regulation of gastrointestinal mucosal growth. In: Granger DN, Granger JP, editors. Molecule to Function to Disease. Princeton, NJ: Morgan & Claypool; 2011. pp. 11–114. [Google Scholar]
- Sapkota G, Alarcon C, Spagnoli FM, Brivanlou AH, Massague J. Balancing BMP signaling through integrated inputs into the Smad1 linker. Mol Cell. 2007;25:441–454. doi: 10.1016/j.molcel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Dey BK, Dutta A. MiR-322/424 and -503 are induced during muscle differentiation and promote cell cycle quiescence and differentiation by down-regulation of Cdc25A. Mol Biol Cell. 2010;21:2138–2149. doi: 10.1091/mbc.E10-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340:1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- Schwamborn JC, Muller M, Becker AH, Puschel AW. Ubiquitination of the GTPase Rap1B by the ubiquitin ligase Smurf2 is required for the establishment of neuronal polarity. EMBO J. 2007;26:1410–1422. doi: 10.1038/sj.emboj.7601580. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Seiler N, Raul F. Polyamines and the intestinal tract. Crit Rev Clin Lab Sci. 2007;44:365–411. doi: 10.1080/10408360701250016. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Ten Dijke P, Hill CS. New insights into TGF-β-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Wang JY, McCormack SA, Viar MJ, Johnson LR. Stimulation of proximal small intestinal mucosal growth by luminal polyamines. Am J Physiol Gastrointest Liver Physiol. 1991;261:G504–G511. doi: 10.1152/ajpgi.1991.261.3.G504. [DOI] [PubMed] [Google Scholar]
- Wang JY, Wang H, Johnson LR. Gastrin stimulates expression of protooncogene c-myc through a process involving polyamines in IEC-6 cells. Am J Physiol Cell Physiol. 1995;269:C1474–C1481. doi: 10.1152/ajpcell.1995.269.6.C1474. [DOI] [PubMed] [Google Scholar]
- Wang PY, Rao JN, Zou T, Liu L, Xiao L, Yu TX, Turner DJ, Gorospe M, Wang JY. Post-transcriptional regulation of MEK-1 by polyamines through the RNA-binding protein HuR modulating intestinal epithelial apoptosis. Biochem J. 2010;426:293–306. doi: 10.1042/BJ20091459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Cui YH, Rao JN, Zou T, Liu L, Smith A, Turner DJ, Gorospe M, Wang JY. Regulation of cyclin-dependent kinase 4 translation through CUG-binding protein 1 and microRNA-222 by polyamines. Mol Biol Cell. 2011;22:3055–3069. doi: 10.1091/mbc.E11-01-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Rao JN, Zou T, Liu L, Cao S, Martindale JL, Su W, Chung HK, Gorospe M, Wang JY. miR-29b represses intestinal mucosal growth by inhibiting translation of cyclin-dependent kinase 2. Mol Biol Cell. 2013;24:3038–3046. doi: 10.1091/mbc.E13-05-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Rao JN, Zou T, Liu L, Marasa BS, Chen J, Turner DJ, Zhou H, Gorospe M, Wang JY. Polyamines regulate the stability of activating transcription factor-2 mRNA through RNA-binding protein HuR in intestinal epithelial cells. Mol Biol Cell. 2007;18:4579–4590. doi: 10.1091/mbc.E07-07-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Liu J, Derynck R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 2012;586:1871–1884. doi: 10.1016/j.febslet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141:1323–1333. doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TX, Rao JN, Zou T, Liu L, Xiao L, Ouyang M, Cao S, Gorospe M, Wang JY. Competitive binding of CUGBP1 and HuR to occludin mRNA controls its translation and modulates epithelial barrier function. Mol Biol Cell. 2012;24:85–99. doi: 10.1091/mbc.E12-07-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TX, Wang PY, Rao JN, Zou T, Liu L, Xiao L, Gorospe M, Wang JY. Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function. Nucleic Acids Res. 2011;39:8472–8487. doi: 10.1093/nar/gkr567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci USA. 2001;98:974–979. doi: 10.1073/pnas.98.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HM, Rao JN, Guo X, Liu L, Zou T, Turner DJ, Wang JY. Akt kinase activation blocks apoptosis in intestinal epithelial cells by inhibiting caspase-3 after polyamine depletion. J Biol Chem. 2004;279:22539–22547. doi: 10.1074/jbc.M314337200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zou T, Rao JN, Liu L, Xiao L, Wang PY, Cui YH, Gorospe M, Wang JY. Stabilization of XIAP mRNA through the RNA binding protein HuR regulated by cellular polyamines. Nucleic Acids Res. 2009;37:7623–7637. doi: 10.1093/nar/gkp755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang R, Rao JN, Zou T, Liu L, Xiao L, Cao S, Hansraj NZ, Gorospe M, Wang JY. miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration. Nucleic Acids Res. 2013;41:7905–7919. doi: 10.1093/nar/gkt565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400:687–693. doi: 10.1038/23293. [DOI] [PubMed] [Google Scholar]
- Zou T, Rao JN, Liu L, Xiao L, Yu TX, Jiang P, Gorospe M, Wang JY. Polyamines regulate the stability of JunD mRNA by modulating the competitive binding of its 3′ untranslated region to HuR and AUF1. Mol Cell Biol. 2010;30:5021–5032. doi: 10.1128/MCB.00807-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.