

Abstract
The ability to acquire highly accurate quantitative data is an increasingly important part of any proteomics experiment, whether shotgun or top-down approaches are used. We recently developed a quantitation strategy for peptides based on neutron encoding, or NeuCode SILAC, which uses closely spaced heavy isotope-labeled amino acids and high-resolution mass spectrometry to provide quantitative data. We reasoned that the strategy would also be applicable to intact proteins and could enable robust, multiplexed quantitation for top-down experiments. We used yeast lysate labeled with either 13C615N2-lysine or 2H8-lysine, isotopologues of lysine that are spaced 36 mDa apart. Proteins having such close spacing cannot be distinguished during a medium resolution scan, but upon acquiring a high-resolution scan, the two forms of the protein with each amino acid are resolved and the quantitative information revealed. An additional benefit NeuCode SILAC provides for top down is that the spacing of the isotope peaks indicates the number of lysines present in the protein, information that aids in identification. We used NeuCode SILAC to quantify several hundred isotope distributions, manually identify and quantify proteins from 1:1, 3:1, and 5:1 mixed ratios, and demonstrate MS2-based quantitation using ETD.
Top-down proteomics methodologies continue to evolve and can provide a useful alternative to the more commonly used shotgun strategies for protein characterization by mass spectrometry.1 Top-down experiments offer many advantages, including the ability to characterize the entire primary sequence of a given protein and identify combinatorial patterns of post-translational modifications.2−4 The analysis of intact protein samples is inherently more complex, complicating experimental outcomes such as quantitation.5,6 Although many of the same methods used for quantifying peptides have been used in top-down workflows, there are unique challenges to quantifying intact proteins.
Label-free methods are the most accessible form of quantitation for top-down experiments. Here, quantitative information is acquired through the use of spectral counts or extracted ion chromatograms.7 These experiments, however, require many technical and biological replicates to account for run-to-run variability.8 Label-free approaches also lack the ability to multiplex several samples in a single run, a feature of some label-based quantitation methods that can considerably reduce instrument run-time requirements.9,10
Chemical labeling techniques such as acrylamide labeling and tandem mass tags have been used, but the complexity of intact protein samples often leads to incomplete labeling and side reactions that complicate the data analysis.11,12 Stable Isotope Labeling of Amino acids in Cell culture (SILAC), which is a metabolic labeling technique, is considered the gold standard for quantitative proteomics, and has been used with affinity-purified proteins with some success.10,13,14 A drawback of this method, however, is that even a small amount of incomplete labeling results in broad isotope distributions that challenge quantitation.15 Furthermore, the spectral complexity from multiple isotopic distributions for each protein hinders the ability to multiplex.
We have recently introduced a new strategy for protein quantification: neutron encoding (NeuCode).16 Although initially applied to shotgun approaches, NeuCode SILAC has the potential to address many of the difficulties that the aforementioned quantitative strategies have when used for top-down experiments. NeuCode SILAC is similar in structure to a traditional SILAC experiment, except that the distance between forms of the protein is greatly compressed, such that they are indistinguishable during a medium-resolution scan. Only upon using a very high resolution (>120k) scan are the separate peaks revealed. NeuCode provides quantitative accuracy similar to SILAC, but permits considerably higher multiplexing, because the quantitative channels do not add to spectral complexity.16 In addition, comparisons between heavy labels, rather than between light and heavy as in SILAC, alleviates the need to correct for isotopic broadening and minimizes concerns about subunity reagent purities of the heavy labels. Here, we present a first look at NeuCode quantification of proteins from a top-down perspective in yeast.
Experimental Section
Yeast Samples
For lysine NeuCode SILAC, Saccharomyces cerevisiae strain BY4741 Lys1Δ was grown in defined, synthetic-complete (SC, Sunrise Science) drop-out media supplemented with either “K602” 13C6/15N2 lysine (+8.0142 Da, Cambridge Isotopes), or “K080” 2H8 lysine (+8.0502 Da, Cambridge Isotopes). Cells were allowed to propagate for a minimum of 10 doublings to ensure complete lysine incorporation. Upon reaching midlog phase, cells were harvested by centrifugation at 3000g for 3 min and washed three times with chilled doubly distilled (dd) H2O. Cell pellets were resuspended in 5 mL of lysis buffer (50 mM Tris pH 8, 8 M urea, 75 mM sodium chloride, 100 mM sodium butyrate, 1 mM sodium orthovanadate, protease and phosphatase inhibitor tablet), and total protein was extracted by glass bead milling (Retsch). Protein concentration of yeast lysate was measured by BCA (Pierce). The 1:1, 3:1, and 5:1 (K602:K080) yeast samples were mixed in the defined ratios based on the BCA and all yeast lysates were desalted via a tC2 sep-pak (Waters). The samples were dried down and then resuspended in 0.2% formic acid.
Online reverse-phase chromatography was performed using a Nano-Acuity UPLC system (Waters, Milford, MA). Proteins were eluted over an analytical column (75 μm ID, packed with 30 cm of 5 μm, 300 Å Magic C4 particles, Bruker, Michrom) at 300 nL/min using a 96 min gradient of solvent A (94.8% water, 5% DMSO, 0.2% formic acid) and solvent B (99.8% acetonitrile, 0.2% formic acid): 5% to 8% B from 0 to 1 min, 8% to 22% B from 1 to 45 min, 22% to 30% B from 45 to 60 min, and 30% to 90% B from 60 to 96 min, followed by a 4 min wash at 90% B.
Data were collected on an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The nitrogen flow to the Orbitrap chamber was altered such that the increase in pressure (as measured by a Penning ionization gauge) was ∼0.15 × 10–10 Torr, compared to the pressure in the absence of nitrogen. Medium-resolution survey scans (30 000 resolving power; 4 microscans) were used to guide data-dependent sampling of the most intense peaks. Before acquiring the MS2 scans, a high resolution (240 000 or 480 000, 4 microscans) MS1 was acquired for quantitation purposes. Precursors were fragmented with ETD (reaction time = 50 ms) and detected in the orbitrap at 120 000 resolution with 6 microscans. Target ion accumulation values were set to 3 × 106 and 5 × 105 for MS1 and MS2 scans, respectively. For all scan functions, the precursor ions were isolated ±2.5 Th and peaks with assigned charge states of 1–3 were excluded from analysis. Dynamic exclusion was turned off for the duration of the run.
Data Analysis
Quantification of unidentified isotopic clusters was performed by constructing a list of unique MS1 features with the corresponding peak apex retention time. This was accomplished by considering each peak in each MS1 in order of decreasing intensity. Peaks above a signal-to-noise ratio of 15 and assigned a charge state were saved for future use. To ensure unique clusters within an MS1, after the peak was saved all peaks ±5 Da of the saved peak were excluded. Then, the apex retention time for each saved peak was calculated by assessing the intensity of the peak within a window of ±15 MS1 scans. All peaks representing unique clusters from each MS1 were then ranked in order of increasing apex retention time. Starting with the earliest eluting peak, peaks were saved to a final list of unique clusters. To ensure clusters were unique, a ±5 Da exclusion window was applied for all precursors with an apex retention time within 30 s of the peak that was added to the final unique cluster list.
For each unique cluster, the precursor intensity for the K602 partner was calculated using five high-resolution MS1 scans proceeding and following the apex retention time. The m/z values for potential NeuCode SILAC partners were calculated assuming the number of lysine residues in the protein was less than or equal to the charge state. The intensity of the peak with the smallest deviation from the expected mass within a 10 ppm window was summed for each potential partner over the same MS1 range as the light species. Due to the lack of sequence information, confidence in assigning the correct partners was increased by ranking partners by the deviation from the expected ratio (e.g., 1:1, 3:1, or 5:1). This automated algorithm was manually validated to ensure accurate quantitation and partner assignment.
Results and Discussion
As we did with peptides, we first explored our ability to resolve NeuCode SILAC-labeled intact proteins using a recently published dataset.17 A set of 1206 top-down protein identifications (Kelleher) were examined to calculate the theoretical number of resolvable NeuCode SILAC proteins at different label spacings. Briefly, for each protein identification the number of lysine residues (N) were counted and the expected m/z difference between NeuCode pairs with 12, 18, and 36 mDa label spacing (Δm) at the identified m/z value and charge state (z) were computed as follows:
Next, assuming two Gaussian peaks of equal intensities, we calculated the theoretical m/z spacing needed to resolve them at a full-width 10% (f) maximum (FWTM) for resolving powers (RP) between 15 000 and 1 000 000 (steps of 1000, RP defined at 400 m/z and scaled for identified m/z). For each protein identification and its observed m/z value, the theoretical m/z spacing needed to resolve a NeuCode pair was calculated by
The percentage of quantifiable proteins (i.e., Δ(m/z)exp ≥ Δ(m/z)theo) was plotted as a function of resolving power in Figure 1. Using the largest label spacing (36 mDa), over 84% of the proteins are theoretically resolvable, and thus quantifiable, at 480 000, a level achievable on commercial Orbitrap Elite mass spectrometers. Lysine isotopologues spaced 36 mDa apart enable duplex quantitation. We plotted all of the proteins, as well as the resolved proteins, as a histogram of molecular weight, demonstrating that we can theoretically resolve proteins even over 100 kDa (see Figure 1 in the Supporting Information). More closely spaced lysine forms require higher resolving powers. Approximately 77% of the proteins with NeuCode SILAC peaks spaced 12 mDa apart in the dataset would be separated at a resolution of 960 000. This resolution can be achieved on both Orbitraps18 and FT-ICR instruments. Furthermore, ICR mass spectrometers are a common platform for top-down proteomics, and many are capable of resolutions in excess of 1 million, permitting routine use of the 12 mDa spaced lysines and allowing 4-plex quantitative comparisons.19 We conclude that NeuCode SILAC quantitation of intact proteins is obtainable for a large percentage of the identified top-down proteome.
Figure 1.
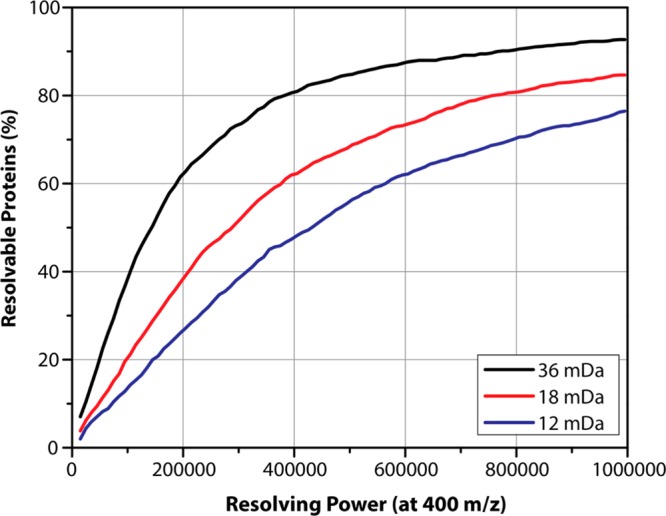
Theoretical resolvability of intact proteins incorporating lysine NeuCode pairs from a top-down dataset. Using a top-down dataset of 1206 proteins, we calculated the fraction of resolvable peptides assuming NeuCode spacings of 12, 18, and 36 mDa at the resolution required to resolve the two peaks at full-width 10% maximum (FWTM), taking into account the decrease in resolution as m/z increases.
We next used K602 and K080 lysine to label yeast from which we prepared top-down samples at mixing ratios of 1:1, 3:1, and 5:1 (K602:K080). At present, NeuCode SILAC-labeled intact protein spectra are incompatible with existing top-down search algorithms, complicating data analysis. To circumvent this, we also prepared unlabeled yeast that we could search with ProSight. Figure 2 illustrates a protein that was identified from an unlabeled yeast sample and then matched to the NeuCode SILAC sample, using the number of lysines and the retention time. The +9 charge state of the 1–47 fragment of ribosomal protein L26A was identified, yielding a protein isotope distribution at an m/z value of 570 and consisting of ∼9 peaks at a resolution of 30 000. At this resolution, there was no indication of the multiple forms of the protein that convey quantitative information. However, upon acquisition of a 240 000 resolution scan, each single peak of the isotope distribution was revealed to consist of two peaks corresponding to the two forms of lysine used (Figure 2A). The spacing of the isotopologue pairs revealed the number of lysines in the sequence, aiding in the identification of the protein (Figure 2B).20 In this case, the distance is 19.8 mTh and the charge state is +9, indicating five lysines. This information, when matched with the retention time to an unlabeled yeast sample, indicates that it is a fragment of ribosomal protein L26A.
Figure 2.

Quantitation of the 1–47 fragment of Ribosomal Protein L26A from yeast using NeuCode. (A) The +9 charge state of the protein (Uniprot B3RHL4) was analyzed first at a resolution of 30 000, which shows one distinct isotope distribution. However, a scan at a resolution of 240 000 reveals the presence of two forms of the protein. (B) The spacing between the isotopologue peaks can be used to calculate the number of lysines present in the protein. The protein is carrying 9 charges and the peaks are spaced 19.8 Th apart, indicating 5 lysines. (C) Annotated fragmentation spectrum of the L26A precursor. The fragment ions that contain a lysine also show pairs of peaks that can be used to calculate the number of lysines. Two fragments (c5 and z18, ppm errors of 6.40 and 6.56, respectively) were used as examples for quantitation in all three samples, demonstrating excellent quantitative accuracy.
As shown in Figure 2C, the spacing in Th between the isotopologue peaks in a fragmentation spectrum collected at sufficient resolution aids in annotation by revealing the number of lysine molecules present in the sequence, as well as providing another avenue through which to acquire quantitative information.21 Figure 2C presents several examples of fragments that contain two or three lysine molecules. This information verifies the protein identification from the MS1 scan, as well as matching the quantitation of the intact protein. We also used this information to aid in the identification of Elongin-C from yeast in a previous experiment, as well as annotate an ETD MS/MS spectrum of the +18 charge state precursor of histone H2B (see Figures 2 and 3 in the Supporting Information).
We observed several hundred intact protein isotope distributions in the defined ratio yeast samples. Due to the aforementioned difficulties with database searching, we only identified a few of the MS/MS scans by hand. However, this does not prevent us from extracting quantitative information from the unknown distributions. Using a list of unique isotopic clusters and the predicted number of lysine residues we calculated the ratio of partners in each known ratio sample. Figure 3A summarizes the quantitation of all of the m/z peaks extracted from each sample, demonstrating median ratios of 1.01, 3.02, and 4.58 for the 1:1, 3:1, and 5:1 samples, respectively. Encouragingly, our simple quantitative algorithm achieved excellent accuracy, even though we did not have the benefit of the protein sequence. More than 500 species were quantified in the 1:1 and 3:1 samples; however, fewer clusters were quantified in the 5:1 sample where the effect of stringent filters employed during lysine prediction limited the number of detected NeuCode SILAC partners. Future implementations of protein quantification will utilize identified protein sequences removing the requirement to predict the number of lysines and enabling the development of more-advanced partner-picking algorithms. Using the same identification strategy as above, we were able to identify the +12 charge state of ubiquitin, based on its retention time and the number of lysines present. Shown in Figure 3B, summing together the isotopologue peaks yields ratios of 0.77, 2.09, and 4.27 for the 1:1, 3:1 and 5:1 samples, in relatively good agreement with the box plots above.
Figure 3.

Quantitation of isotopic distributions from yeast lysate. (A) Overall quantitation of the distributions (after filtering for a minimum signal/noise of 15) in yeast lysates that were mixed in ratios of 1:1, 3:1, and 5:1 (K602:K080). Measured (box and whiskers) and true (dotted lines) ratios for all three samples is displayed with the median (stripe), mean (square), interquartile range (25th to 75th, box), and 1.5X interquartile range (whiskers). (B) Quantitation of the +12 charge state of ubiquitin (Uniprot P0CG63) from the same mixtures, demonstrating ratios of 0.77, 2.09, and 4.27, in good agreement with the mixing ratios of 1:1, 3:1, and 5:1.
Conclusions
We demonstrate the use of NeuCode SILAC to enable multiplexed-quantitation for top-down experiments. First, we demonstrated that the various lysine isotopologues for NeuCode SILAC incorporate sufficiently (at the intact protein level) in two disparate cellular systems. Second, we provide evidence that NeuCode SILAC works identically to that observed in our prior shotgun experiments—i.e., the multiplexed signals are concealed under low to medium resolution scans and only revealed upon analysis under high resolution. We conclude that these benefits will circumvent the problem of MS1 spectral complication that occurs with traditional SILAC for intact protein analysis. This work provides a basis from which to continue NeuCode SILAC development for top-down methodology. A current bottleneck is that top-down spectral searching routines must be modified to accommodate the mass differences and isotopic envelope shifts imparted by NeuCode SILAC labels; note this lack of searching capability limited the current study to only examples that we could manually identify. With these examples, we demonstrate that the embedded NeuCode SILAC signals can be fully resolved, even in the context of a protein isotopic distribution. We also note that the high-resolution MS requirement of NeuCode SILAC is a condition that is typically met in top-down workflows. In summary, NeuCode SILAC has solid potential as a robust method for obtaining quantitative data in top-down experiments and bypasses many of the drawbacks of current top-down quantitative strategies.
Acknowledgments
We are grateful to Adam Catherman and Neil Kelleher for the details of the top-down dataset we used for the theoretical calculations. We also thank Rebecka Manis for critical proofreading of the manuscript. This work was supported by the National Institutes of Health (through Grant No. R01GM080148 to J.J.C., Grant No. R01DK098672 to D.J.P., an Innovator Grant (No. DP2OD007447) to B.A.G., and a training grant (No. 5T32GM007215-37) to A.J.S.). The authors also acknowledge support from the National Science Foundation, through an NSF graduate fellowship (No. DGE-1256259) to A.J.S. and a National Science Foundation Early Faculty Career grant from the Office of the Director to B.A.G. C.M.R. was funded by an NSF Graduate Research Fellowship and NIH Traineeship (No. T32GM008505).
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
○ These authors contributed equally to this work.
Author Contributions
∥ A.E.M., L.M.S., A.S.H., M.S.W., D.J.P., C.M.R., N.M.R., D.J.B., T.W.R., and B.A.G. designed the experiments. A.J.N., N.M.R., R.C.M., T.W.R., C.M.R., and D.J.B. performed the experiments. T.W.R., C.M.R., D.J.B., N.M.R., and J.J.C. wrote the paper.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Siuti N.; Kelleher N. L. Nat. Methods 2007, 4, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansong C.; Wu S.; Meng D.; Liu X.; Brewer H. M.; Deatherage Kaiser B. L.; Nakayasu E. S.; Cort J. R.; Pevzner P.; Smith R. D.; Heffron F.; Adkins J. N.; Pasa-Tolic L. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10153–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia B. A. J. Am. Soc. Mass Spectrom. 2010, 21, 193–202. [DOI] [PubMed] [Google Scholar]
- Cui W.; Rohrs H. W.; Gross M. L. Analyst 2011, 136, 3854–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell J. D.; Scalf M.; Book A. J.; Ladror D. T.; Vierstra R. D.; Smith L. M.; Coon J. J. PLoS One 2013, 8, e58157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon E. F.; Mansoori B. A.; Carroll C. F.; Muddiman D. C. J. Mass Spectrom. 1999, 34, 1055–1062. [DOI] [PubMed] [Google Scholar]
- Asara J. M.; Christofk H. R.; Freimark L. M.; Cantley L. C. Proteomics 2008, 8, 994–999. [DOI] [PubMed] [Google Scholar]
- Elliott M. H.; Smith D. S.; Parker C. E.; Borchers C. J. Mass Spectrom. 2009, 44, 1637–1660. [DOI] [PubMed] [Google Scholar]
- Thompson A.; Schafer J.; Kuhn K.; Kienle S.; Schwarz J.; Schmidt G.; Neumann T.; Johnstone R.; Mohammed A. K.; Hamon C. Anal. Chem. 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Mol. Cell. Proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- Du Y.; Parks B. A.; Sohn S.; Kwast K. E.; Kelleher N. L. Anal. Chem. 2006, 78, 686–694. [DOI] [PubMed] [Google Scholar]
- Hung C. W.; Tholey A. Anal. Chem. 2012, 84, 161–170. [DOI] [PubMed] [Google Scholar]
- Waanders L. F.; Hanke S.; Mann M. J. Am. Soc. Mass Spectrom. 2007, 18, 2058–64. [DOI] [PubMed] [Google Scholar]
- Collier T. S.; Sarkar P.; Rao B.; Muddiman D. C. J. Am. Soc. Mass Spectrom. 2010, 21, 879–89. [DOI] [PubMed] [Google Scholar]
- Collier T. S.; Hawkridge A. M.; Georgianna D. R.; Payne G. A.; Muddiman D. C. Anal. Chem. 2008, 80, 4994–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert A. S.; Merrill A. E.; Bailey D. J.; Still A. J.; Westphall M. S.; Strieter E. R.; Pagliarini D. J.; Coon J. J. Nat. Methods 2013, 10, 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman A. D.; Durbin K. R.; Ahlf D. R.; Early B. P.; Fellers R. T.; Tran J. C.; Thomas P. M.; Kelleher N. L. Mol. Cell. Proteomics 2013, 12, 3465–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov E.; Damoc E.; Lange O.; Makarov A. Int. J. Mass Spectrom. 2012, 325, 80–85. [Google Scholar]
- Bogdanov B.; Smith R. D. Mass Spectrom. Rev. 2005, 24, 168–200. [DOI] [PubMed] [Google Scholar]
- Rose C. M.; Merrill A. E.; Bailey D. J.; Hebert A. S.; Westphall M. S.; Coon J. J. Anal. Chem. 2013, 85, 5129–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A. L.; Vincent C. E.; Guthals A.; Rose C. M.; Westphall M. S.; Bandeira N.; Coon J. J. Mol. Cell. Proteomics 2013, 12, 3812–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.