

Abstract
An absolute quantitation method for measuring free human milk oligosaccharides (HMOs) in milk samples was developed using multiple reaction monitoring (MRM). To obtain the best sensitivity, the instrument conditions were optimized to reduce the source and postsource fragmentation prior to the quadrupole transmission. Fragmentation spectra of HMOs using collision-induced dissociation were studied to obtain the best characteristic fragments. At least two MRM transitions were used to quantify and identify each structure in the same run. The fragment ions corresponded to the production of singly charged mono-, di-, and trisaccharide fragments. The sensitivity and accuracy of the quantitation using MRM were determined, with the detection limit in the femtomole level and the calibration range spanning over 5 orders of magnitude. Seven commercial HMO standards were used to create calibration curves and were used to determine a universal response for all HMOs. The universal response factor was used to estimate absolute amounts of other structures and the total oligosaccharide content in milk. The quantitation method was applied to 20 human milk samples to determine the variations in HMO concentrations from women classified as secretors and nonsecretors, a phenotype that can be identified by the concentration of 2′-fucosylation in their milk.
Multiple reaction monitoring (MRM) on a triple quadrupole instrument (QqQ) is valued for its ability to quantify low-abundant compounds in complicated mixtures. As a result, it is widely used for quantitation of small molecules such as drugs1,2 and metabolites,3,4 and even larger biomolecules such as proteins, which are digested to peptides.5−7 However, its application to complicated and labile biomolecules such as oligosaccharides and glycoconjugates is still limited.8−10 One challenge is that the masses of oligosaccharides can be large, up to 5000 Da. The quadrupoles typically have limited transmission mass ranges such that large m/z ions are not effectively transmitted. Large peptides of that size are typically neglected in MRM in favor of smaller peptides from the same proteins; however, this option is not possible with glycans. A more practical concern is that commercial QqQ are most often optimized for peptides and small robust analytes, while oligosaccharides tend to fragment more readily during ionization and may not be well-represented by their quasimolecular ions.
Human milk is an important source for bioactive oligosaccharides and glycoconjugates. Human milk oligosaccharides (HMOs) are the third most abundant group of compounds after lactose and fat, with as much as 5–23 g/L in milk.11−13 Interestingly, HMOs are produced by mothers not for direct nourishment of their infants,14 but instead have other biological functions such as inhibiting the binding of pathogens, stimulating the growth of commensal intestinal bacterial, and promoting postnatal brain development.13,15−17 To date, over 200 HMOs have been reported,18−21 and it has been found that HMO structures are related to their biological functions. For example, lacto-N-tetraose (LNT) and lacto-N-fucopentaose II and III (LNFP-II, III) were found to reduce Entamoeba histolytica attachment and cytotoxicity, while lacto-N-fucopentaose I (LNFP-I), an isomer of LNFP-II and LNFP-III, had no such effects.22 It has also been found that specific bifidobacterial strains, commensal bacteria found in the gut of healthy infants, selectively consumed distinct HMO structures.23 For these reasons, accurate and sensitive quantitation methods capable of monitoring HMO structures would be very useful for functional studies.
The absolute quantitation of HMOs is challenging due to the lack of standards and the difficulty in achieving complete chromatographic resolution of individual structures. High-performance anion-exchange chromatography (HPAEC) coupled with a pulsed amperometric detector (PAD) is a common technique for oligosaccharide quantitation.24−27 HPLC separation in combination with spectrophotometric detection is also available for derivatized oligosaccharides.28−31 However, none of these detection methods are structural selective, and the quantitation depends on effective chromatographic separation. Multiple reaction monitoring on a QqQ provides the possibility of a robust label-free method for quantitation. Its detection can also be structure-selective. However, it has not been widely used to monitor free oligosaccharides in milk. One exception is a very recent example employing negative mode MRM for the absolute quantitation for six acidic bovine milk oligosaccharides (BMO) using hydrophilic interaction chromatography (HILIC).32 Low-femtomole detection limits, which are 3 orders of magnitude lower than those of the PAD detector, illustrated the value of MRM for quantifying oligosaccharides. The only other example employed negative mode MRM to determine eight soluble milk oligosaccharides in rat serum using normal phase liquid chromatography.33
In this study, we present a comprehensive MRM analysis of HMOs. HMOs differ from other milk oligosaccharides such as BMOs in terms of their structural complexity and diversity and in their natural abundances.19,20,34 HMOs are more abundant than BMOs and have a much higher degree of fucosylation than BMOs. Conversely, BMOs have a much higher degree of sialylation. Furthermore, the mass spectrometry conditions were optimized particularly for free oligosaccharides to achieve high MRM sensitivity, thus enabling the detection of HMO over a wider dynamic range. The optimized method was then applied to study the variation in HMO concentrations in milk collected from healthy mothers. It is known that HMO profiles are greatly affected by the mother’s secretor phenotype, which is encoded by the fucosyltransferase 2 (FUT2) gene and causes the secretion of α(1–2) fucosylation.35 This MRM method was then used to examine the absolute concentrations of individual structures to determine the variation in α(1–2) fucosylation and the total HMO content between mothers of different secretor phenotypes.
Experimental Procedures
A brief description of the experimental procedures is given here. A more detailed description of the materials and methods used can be found in the Supporting Information.
Samples and Materials
Milk oligosaccharide standards, lacto-N-tetraose (LNT), lacto-N-fucopentaose I (LNFP-I), sialyllacto-N-tetraose c (LSTc), 3′-sialyllactose (3′-SL), and 6′-sialyllactose (6′-SL), 2′-fucosyllactose (2′-FL), and lacto-N-hexaose (LNH) were purchased to build the standard calibration curves. Milk samples were obtained from 20 healthy women who gave birth to healthy full term infants enrolled in the UC Davis Foods for Health Institute lactation study. The participants’ secretor statuses were determined by measuring specific fucosylated HMO structures in their milk as previously described.36 Ten secretor and 10 nonsecretor participants were chosen for this study.
Sample Preparation
HMOs were extracted from milk samples and reduced using the method described previously.18,36 Briefly, 100 μL of raw milk was defatted via centrifuge followed by an ethanol precipitation. The defatted samples were reduced using NaBH4 at 65 °C water bath for 1.5 h followed by a solid phase extraction (SPE) cleanup using graphitized carbon cartridges. The lyophilized samples were reconstituted in 100 μL of Nanopure water and diluted by 20-folds for MS analysis. Oligosaccharide standards were also reduced using NaBH4. The lyophilized reduced oligosaccharide standards were weighed using a microbalance (Mettler Toledo, XP26), reconstituted, and diluted to create standard calibration curves. A standard addition experiment was performed for one of the milk samples by adding varying amount of HMO standards into the 20× diluted extracted milk oligosaccharide sample.
LC–ESI-MS Analysis
Human milk oligosaccharides were quantified using an Agilent 6490 triple quadrupole equipped with an Agilent 1290 infinity LC system, and a Thermo 100 × 2.1 mm Hypercarb column with a 10 × 2.1 mm Hypercarb precolumn (particle size of 3 μm for both columns). A 55 min LC separation was performed using a binary gradient at 0.2 mL/min flow rate: solvent A of 3% acetonitrile, 0.1% formic acid; solvent B of 90% acetonitrile, 0.1% formic acid in Nanopure water (v/v). For the initial optimization of the mass analyzer, pure standards were ran on fast LC gradients (10 min) employing the same solvent composition as described.
The MS was operated in positive mode. The first and third quadrupoles were operated at unit resolution. The following parameters were optimized for oligosaccharide analysis: drying gas temperature and sheath gas temperature 150 °C, drying gas flow rate 11 L/min, sheath gas flow rate 7 L/min, nebulizer pressure 25 psi, capillary voltage 1800 V, fragmentor voltage 250 V; rf voltage amplitudes of high-pressure and low-pressure ion funnels are 100 and 60 V, respectively.
Statistical Analysis
The box-and-whisker plot and statistic tests were performed in JMP 10.0 statistical software. One-tailed Student t test (α = 0.05) was used to compare the means of secretor and nonsecretor groups. Data sets were tested for homoscedasticity using Bartlett tests, with an α value of 0.05. Nonhomoscedastic distributions were transformed using logarithmic functions to meet assumptions for parametric testing. Distributions that were not normal or homoscedastic were analyzed by a nonparametric Mann–Whitney–Wilcoxon method.
Results and Discussion
A method for the accurate absolute quantitation of free oligosaccharides using MRM was developed by first optimizing ionization conditions for oligosaccharides. The fragmentation conditions under collision-induced dissociation (CID) were also optimized, and the common fragments were determined. The MS instrument parameters required further optimization for HMOs because they are structurally labile compared to metabolites and peptides. Chromatographic separation of reduced HMOs was performed with porous graphitized carbon (PGC). Each HMO compound was identified on the basis of its characteristic fragments and its elution time using a previously developed annotated HMO library.19,20
Method Optimization
Minimization of In-Source and Postsource Fragmentation
Oligosaccharides are biomolecules consisting of monosaccharides linked via glycosidic bonds.19 Triple quadrupole MS (QqQ) has been primarily applied to small metabolites2,37 and peptides,38,39 which tend to be intrinsically more stable than oligosaccharides under most common ionization techniques. Commercial instruments are often optimized for small molecules and peptides but not for oligosaccharides. An MS scan of the LNT standard using the manufacturer default setting (Figure 1a) shows that the quasimolecular ions degrade into fragments prior to and during source extraction resulting in low MRM sensitivity.
Figure 1.
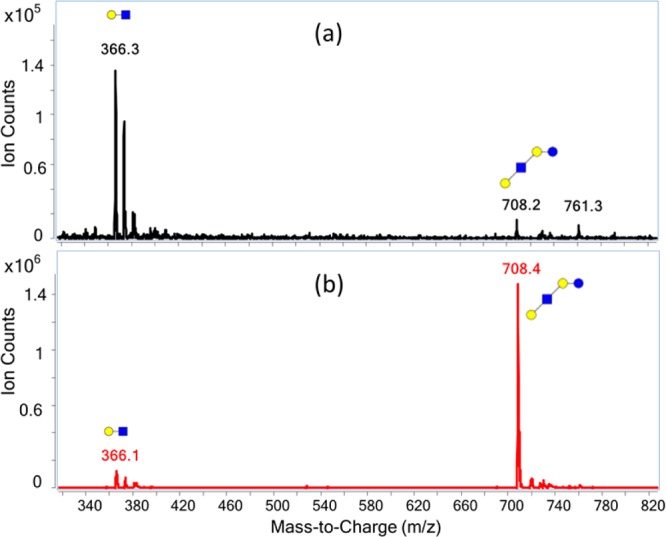
Response of the method to LNT. (a) The manufacturer default conditions for peptides produced a large amount of unintended LNT fragments (m/z 366) in the MS scan mode. (b) After the instrument optimization, the fragment ion signal was diminished and the quasimolecular ion was increased: blue squares, GlcNAc (N-acetylglucosamine); blue circles, Glc (glucose); yellow circles, Gal (galactose).
To increase the specificity and sensitivity of the detection, minimizing fragmentation during ion transmission and prior to the collision-induced dissociation is necessary. While the optimization process is instrument-specific the principle is the same for all instruments, i.e., decrease the fragmentation while maintaining quasimolecular ion abundances. Furthermore, the dual ion funnel feature makes this instrument still unique among triple quadrupole instruments making the discussion of the optimization highly relevant. Several parameters decrease the internal energy of the ion for this instrument including the source gas temperature and flow, nebulizer voltage, capillary voltage, fragmentor voltage, and dual ion funnel voltages. These parameters were optimized using oligosaccharide standards. Both reduced and unreduced compounds were examined and yielded similar responses. The results with LNT are shown as representative of the entire effort. Optimization of the gas temperature, fragmentor voltage, and the dual ion funnel voltage were most effective in increasing the quasimolecular ion as illustrated in Figure 2. Generally, higher gas temperature during electrospray ionization (ESI) decreases the formation of cluster ions, but may increase the internal energy of the ion and result in in-source fragmentation. Figure 2a shows that the optimum temperature is around 150 °C for producing the highest quasimolecular signal.
Figure 2.

Optimization of collision conditions with LNT. (a) The drying gas and sheath gas were maintained at the same temperature. The optimal temperature at 150 °C provides the strongest quasimolecular ion signal. (b) Fragmentor voltage of 250 V provides the lowest degree of fragmentation and the strongest quasimolecular ion signal; 250 V is the minimum setting in this instrument. (c) The rf amplitude of the low-pressure ion funnel at 60 V provides the strongest quasimolecular ion signal. (d) The rf amplitude of the high ion funnel at 100 V provides the lowest degree of fragmentation. The optimum settings are thus 100 and 60 V for the high-pressure and low-pressure ion funnels, respectively. Red triangle: the symbol corresponds to the ratio of the quasimolecular ion (m/z 708) counts to the total ion counts. Blue diamond: the symbol corresponds to the ratio of the fragment ion (m/z 366) counts to the quasimolecular ion (m/z 708) counts. Instrument optimization of the low-pressure ion funnel was performed with rf amplitude at 40 (black triangles), 60 (green circles), and 80 V (red squares), respectively.
The fragmentor voltage is the voltage placed at the exit of the capillary to transmit ions into the mass analyzer. Higher voltages facilitate the transmission, but can also cause in-source CID. As shown in Figure 2b, a fragmentor voltage of 250 V provides the least fragmentation and thus the highest quasimolecular ion signal. However, 250 V is the minimum setting on this instrument and it is possible that lower voltages may still yield higher molecular ion abundances. The triple quadrupole used in this analysis was equipped with two ion funnels placed consecutively to focus and transmit ions into the MS with high efficiency. The ion funnel consists of a series of concentric electrodes with an applied radio frequency (rf) to compress the ion cloud, and a dc electrical field to transmit ions into the mass analyzers.40,41 The ion funnels are used under relatively high pressures. An appropriate level of neutral gas inside the funnels is beneficial for ion confinement through collisional cooling; however, the gas can also increase the potential for unintended CID. The suggested manufacturer conditions involved pressures of 7–14 Torr (first ion funnel, also called high-pressure ion funnel) and 1–3 Torr (second ion funnel, also called low-pressure ion funnel). Under these conditions, large amounts of postsource fragmentation were found using the default rf amplitudes. Decreasing the rf levels was found to yield more abundant oligosaccharide quasimolecular ions. As shown in Figure 2c, the rf amplitude of 100–150 V for the first funnel and 60 V for the second ion funnel gave the strongest quasimolecular ion signal. Consequently, the rf amplitudes of 100 and 60 V, respectively, were used for the analysis. Under the optimized conditions, the instrument yielded the strongest quasimolecular ion with the least amount of fragmentation (Figure 1b).
Multiple Reaction Monitoring of Human Milk Oligosaccharides
MRM Transitions for HMOs
The MRM method requires intense and reproducible fragment ions in order to achieve high specificity and sensitivity. HMO fragmentations employing CID were studied to obtain the optimum MRM transitions for HMO quantitation. The tandem MS of several compounds are provided for illustrative purposes in Supporting Information Figure S-1. The most abundant fragment ions of protonated HMO precursor were small mono-, di-, and some trisaccharide fragments including m/z 366.2 (GlcNAc–Gal), 204.1 (N-acetylglucosamine, GlcNAc), 183.1 (glucose, Glc, reducing end), 512.2 (Fuc–GlcNAc–Gal), 292.1 (N-acetylneuraminic acid, Neu5Ac), and 657.1 (Neu5Ac–GlcNAc–Gal). At least two of these abundant fragment ions were monitored for each compound. The collision energy was optimized for composition to yield the highest response. The collision energy was then applied to a specific mass eluting at a specific time. As listed in Supporting Information Table S-1, 42 HMOs, corresponding to 32 distinct masses were monitored. This represents a large fraction of the total HMOs in individuals corresponding to over 95% of the abundances.36
Dynamic MRMs
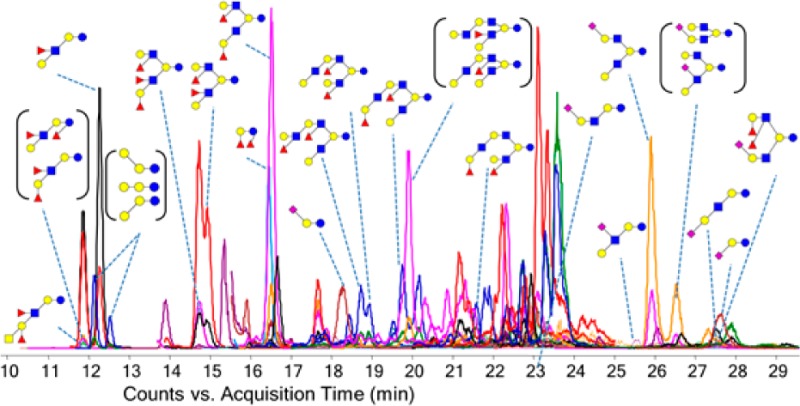
MRM detects multiple transitions sequentially in one duty cycle. Transitions monitored within a single duty cycle are called concurrent transitions. The dwell time and cycle time are two important parameters in an MRM method. The cycle time is the time spent monitoring all transitions during one duty cycle, and the dwell time is the time spent on acquiring one specific MRM transition within each duty cycle. A long cycle time will result in poor LC peak sampling and thus poor data quality, while a short dwell time will result in poor signal-to-noise ratios (S/N). As the number of concurrent MRM transitions increase, either the cycle time needs to be increased resulting in poor data quality or the dwell time needs to be decreased resulting in poor S/N. In order to reduce the number of concurrent transitions, a dynamic MRM method was employed whereby specific transitions are only monitored at specific times, i.e., when the compound elutes. The cycle time of the dynamic MRM method was set in these analyses to 500 ms with the minimum and maximum dwell times of 15.33 and 248.63 ms, respectively, resulting in between 30 and 60 points across a chromatographic peak. Figure 3 shows the dynamic MRM transitions monitored for a pooled HMO sample. The abundant HMOs were annotated in Figure 3a. The lower abundant HMOs were annotated in Figure 3b. The extracted ion chromatograms are provided separately in Supporting Information Figure S-2. The isomers were well-resolved using the PGC stationary phase. Homologues that coelute have different masses and were readily distinguished with MS.
Figure 3.

Extracted MRM chromatogram. The MRM transitions monitored are provided in Supporting Information Table S-1. Peaks are labeled with corresponding structures. Structures in parentheses mean they could not be specifically resolved or identified. (a) The total dynamic MRM chromatogram monitored for a pooled human milk oligosaccharide sample. Nine abundant HMO compounds can be readily identified and annotated. (b) Lower abundant HMOs are identified and annotated: blue circles, Glc (glucose); yellow circles, Gal (galactose); blue squares, GlcNAc (N-acetylglucosamine); yellow squares, GalNAc (N-acetylgalactosamine); red triangles, Fuc (fucose); purple diamonds, Neu5Ac (N-acetylneuraminic acid).
Limits of Detection and Quantitation
Limits of detection and the range of quantitation were studied for the standard samples, which include two neutral (LNT and LNH), two fucosylated (2′-FL and LNFP-I), and three sialylated HMOs (6′-SL, 3′-SL, and LSTc). The limits of detection (LOD, S/N ≥ 3) for these compounds were at low-femtomole levels (10–150 fmol), and the limits of quantitation (LOQ, S/N ≥ 6) were at high-femtomole levels (100–1500 fmol) (Table 1). The LOD for the neutral HMOs were slightly better than for the sialylated HMOs. The reason may be due to the lower ionization efficiency of sialylated species or a greater degree of postsource fragmentation compared to the neutral compounds in positive mode ESI.42 The HMOs show good linearity (R2 > 0.99 for over 2 orders of magnitude); however, quantitation can be performed over 5 orders of magnitude using a quadratic calibration curve (0.001–500 μg/mL, Supporting Information Figures S-3 and S-4).
Table 1. Evaluation of Method Accuracy and Sensitivity.
| concn (mg/mL)a |
|||||
|---|---|---|---|---|---|
| compd | standard calibration | standard addition | diff (%)b | LOD (pmol) | LOQ (pmol) |
| 2′-FL | 1.4 ± 0.1 | 1.6 ± 0.1 | –15 | 0.017 | 0.17 |
| LNFP-I | 0.24 ± 0.05 | 0.26 ± 0.01 | –6.9 | 0.0097 | 0.097 |
| LNT | 1.0 ± 0.1 | 0.95 ± 0.05 | 9.0 | 0.012 | 0.12 |
| LNH | 0.021 ± 0.004 | 0.020 ± 0.001 | 7.5 | 0.047 | 0.47 |
| 3′-SL | 0.25 ± 0.01 | 0.40 ± 0.05 | –39 | 0.13 | 1.3 |
| 6′-SL | 0.18 ± 0.01 | 0.18 ± 0.01 | –2.2 | 0.13 | 1.3 |
| LSTc | 0.063 ± 0.003 | 0.072 ± 0.015 | –12 | 0.083 | 0.83 |
Standard errors from the linear regression were reported.
The difference in concentration obtained from standard calibration curves and from the standard addition.
HMO Response Factor
The standard calibration curves for seven HMO standards, LNT, 2′-FL, LNFP-I, LNH, 6′-SL, LSTc, and 3′-SL, are illustrated in Supporting Information Figure S-3. With the exception of LNT and 3′-SL, the response factors (the slopes of the standard calibration curves) of the HMOs are similar, within a factor of 2 (Table 2). The response factor for LNT is higher compared to 2′-FL, LNFP-I, LNH, 6′-SL, and LSTc, while the response factor of 3′-SL is the lowest. Variations in ionization efficiencies and fragmentation efficiencies are two possible reasons for these differences. However, because HMO standards are very expensive and often unavailable, a universal response factor to quantify each component and the total HMO content would be desirable. To this end, a linear regression equation, y = 6966.4x – 1752.6, was created to correlate the MRM responses (y, ion counts) to the HMO concentration (x, μg/mL) by averaging the slopes and y-intercepts of the linear regression equations using the seven HMO standards.
Table 2. Absolute HMO Concentration for 10 Secretors and 10 Nonsecretors on Day 35 Postpartum Using Standard and Universal Calibration Curves.
| concn
in milk (mg/mL) |
|||||||
|---|---|---|---|---|---|---|---|
| standard calibration curveb |
universal calibration curvec |
||||||
| compda | mass (Da) | RT (min) | nonsec. | sec. | nonsec. | sec. | concn diff (%)d |
| 2′-FL | 490.2 | 13.6 | (4.8 ± 10.5) × 10–1 | 2.5 ± 0.7 | (5.8 ± 12.4) × 10–1 | 3.0 ± 0.8 | 20 |
| 3000 a | 506.2 | 11.7 | (6.2 ± 2.1) × 10–2 | (4.2 ± 1.5) × 10–2 | |||
| 3000 b | 506.2 | 12.1 | (1.2 ± 0.5) × 10–2 | (1.2 ± 0.6) × 10–2 | |||
| 3000 c | 506.2 | 15.8 | (5.6 ± 0.2) × 10–3 | (5.5 ± 0.1) × 10–3 | |||
| 3′-SL | 635.2 | 27.0 | (1.8 ± 0.1) × 10–1 | (1.7 ± 0.4) × 10–1 | (5.3 ± 0.8) × 10–2 | (4.9 ± 1.0) × 10–2 | –71 |
| 6′-SL | 635.2 | 17.5 | (3.2 ± 0.7) × 10–1 | (2.9 ± 1.1) × 10–1 | (2.8 ± 0.6) × 10–1 | (2.5 ± 1.0) × 10–1 | –14 |
| LDFTe | 636.3 | 15.9 | (1.9 ± 3.2) × 10–2 | (9.7 ± 4.2) × 10–2 | |||
| 6′-SLNe | 676.3 | 17.5 | (6.4 ± 0.3) × 10–3 | (6.5 ± 0.5) × 10–3 | |||
| LNT | 709.3 | 15.5 | 1.2 ± 0.7 | (7.5 ± 3.1) × 10–1 | 2.2 ± 1.3 | 1.3 ± 0.6 | 80 |
| LNnT | 709.3 | 15.7 | (2.2 ± 1.5) × 10–1 | (3.6 ± 1.4) × 10–1 | |||
| 3110a | 855.3 | 11.5 | (1.3 ± 0.5) × 10–1 | (1.2 ± 0.3) × 10–1 | |||
| LNFP II | 855.3 | 11.9 | (5.6 ± 2.9) × 10–1 | (1.1 ± 0.7) × 10–1 | |||
| LNFP-I | 855.3 | 14.9 | (8.9 ± 23.8) × 10–2 | (4.2 ± 2.1) × 10–1 | (1.0 ± 2.7) × 10–1 | (4.8 ± 2.4) × 10–1 | 14 |
| LNFP-V | 855.3 | 16.0 | (1.4 ± 0.7) × 10–2 | (2.0 ± 1.0) × 10–2 | |||
| LSTc | 1000.4 | 25.0 | (5.0 ± 1.4) × 10–2 | (5.5 ± 2.2) × 10–2 | (3.8 ± 0.9) × 10–2 | (4.2 ± 1.5) × 10–2 | –24 |
| LSTa | 1000.4 | 26.8 | (2.7 ± 1.0) × 10–2 | (2.1 ± 1.1) × 10–2 | |||
| LNDFH Ie | 1001.4 | 11.4 | (6.6 ± 3.0) × 10–3 | (5.0 ± 1.6) × 10–3 | |||
| LNDFH IIe | 1001.4 | 11.5 | (7.2 ± 7.0) × 10–3 | (1.8 ± 0.8) × 10–2 | |||
| 3120 a | 1001.4 | 12.0 | (9.5 ± 3.8) × 10–3 | (5.4 ± 3.1) × 10–3 | |||
| LNH | 1074.4 | 18.5 | (4.3 ± 2.1) × 10–2 | (5.0 ± 1.8) × 10–2 | (4.8 ± 2.2) × 10–2 | (5.1 ± 1.7) × 10–2 | 9 |
| LNnH | 1074.4 | 19.1 | (2.1 ± 1.4) × 10–2 | (4.0 ± 2.1) × 10–2 | |||
| 3220 a | 1204.5 | 11.9 | (2.7 ± 1.2) × 10–2 | (9.5 ± 2.6) × 10–3 | |||
| 3220 b | 1204.5 | 11.6 | (1.0 ± 0.2) × 10–2 | (1.0 ± 0.2) × 10–2 | |||
| MFLNH IIIe | 1220.5 | 17.1 | (1.6 ± 0.4) × 10–1 | (9.6 ± 2.8) × 10–2 | |||
| MFLNH Ie | 1220.5 | 17.3 | (1.5 ± 3.4) × 10–2 | (5.1 ± 3.1) × 10–2 | |||
| IFLNH IIIe | 1220.5 | 18.0 | (3.9 ± 1.5) × 10–2 | (5.2 ± 2.1) × 10–2 | |||
| 4300 | 1277.5 | 17.1 | (1.6 ± 0.2) × 10–2 | (1.4 ± 0.2) × 10–2 | |||
| S-LNH | 1365.5 | 26.0 | (2.3 ± 0.8) × 10–2 | (3.6 ± 1.6) × 10–2 | |||
| 4201 b | 1365.5 | 25.5 | (3.4 ± 0.9) × 10–2 | (2.6 ± 0.5) × 10–2 | |||
| 4220 a | 1366.5 | 14.3 | (5.9 ± 2.4) × 10–2 | (5.6 ± 2.4) × 10–2 | |||
| DFLNH be | 1366.5 | 14.5 | (1.5 ± 0.9) × 10–1 | (2.6 ± 1.2) × 10–2 | |||
| DFLNH ae | 1366.5 | 16.0 | (2.5 ± 4.8) × 10–2 | (1.0 ± 0.6) × 10–1 | |||
| DFLNH ce | 1366.5 | 20.3 | (5.8 ± 3.7) × 10–3 | (7.9 ± 0.9) × 10–3 | |||
| 4211 a | 1511.6 | 23.1 | (8.4 ± 1.5) × 10–3 | (8.3 ± 1.2) × 10–3 | |||
| 4211 b | 1511.6 | 23.7 | (1.4 ± 0.6) × 10–2 | (6.1 ± 2.1) × 10–3 | |||
| 4211 c | 1511.6 | 24.0 | (6.3 ± 4.9) × 10–3 | (1.3 ± 0.8) × 10–2 | |||
| 4211 d | 1511.6 | 25.5 | (8.0 ± 2.3) × 10–2 | (6.6 ± 1.3) × 10–2 | |||
| TFLNHe | 1512.6 | 14.4 | (1.0 ± 1.4) × 10–2 | (2.3 ± 1.0) × 10–2 | |||
| 4230 a | 1512.6 | 19.8 | (5.6 ± 3.5) × 10–3 | (7.3 ± 3.0) × 10–3 | |||
| 5310 | 1585.6 | 19.2 | (2.8 ± 1.2) × 10–2 | (2.2 ± 0.8) × 10–2 | |||
| 4221 a | 1657.6 | 24.4 | (5.9 ± 4.1) × 10–3 | (7.6 ± 2.7) × 10–3 | |||
| 4221 b | 1657.6 | 27.0 | (5.4 ± 2.9) × 10–3 | (7.5 ± 1.6) × 10–3 | |||
| 5320 a | 1731.6 | 18.1 | (2.9 ± 1.2) × 10–2 | (1.1 ± 0.2) × 10–2 | |||
| 5320 b | 1731.6 | 18.3 | (6.3 ± 5.4) × 10–3 | (1.2 ± 0.4) × 10–2 | |||
| 5320 c | 1731.6 | 19.2 | (8.3 ± 1.3) × 10–3 | (7.9 ± 2.8) × 10–3 | |||
| 5320 d | 1731.6 | 19.4 | (6.9 ± 6.7) × 10–3 | (1.5 ± 0.4) × 10–2 | |||
| 5320 e | 1731.6 | 20.8 | (5.9 ± 4.4) × 10–3 | (7.8 ± 1.6) × 10–3 | |||
| 6410 a | 1950.7 | 22.6 | (6.8 ± 6.5) × 10–3 | (1.2 ± 0.4) × 10–2 | |||
| 6410 b | 1950.7 | 22.9 | (1.1 ± 0.3) × 10–2 | (1.0 ± 0.3) × 10–2 | |||
| total HMO content | 5.1 ± 1.6 | 6.8 ± 0.7 | |||||
Monosaccharide composition, e.g., 5(Hex):3(HexNAc):2(Fuc):1(Neu5Ac) was represented as 5321 (Hex, hexose; HexNAc, N-acetylhexosamine; Fuc, fucose; Neu5Ac, N-acetylneuraminic acid).
Concentration was obtained using specific standard calibration curves. The linear regression equations used were as follows: (2′-FL) y = 8082x + 20297, R2 = 0.996; (LNFP-I) y = 7851x – 452, R2 = 1.000; (LNT) y = 12880x – 14067, R2 = 1.000; (LNH) y = 6710x – 547, R2 = 1.000; (3′-SL) y = 1895x – 564, R2 = 0.999; (6′-SL) y = 5686x – 795, R2 = 1.000; (LSTc) y = 4615x – 218, R2 = 1.000; nonsec., milk from nonsecretor mothers; sec., milk from secretor mothers.
Concentration was obtained using the average linear regression equation, y = 6966.4x – 1752.6, fitted to the 0.8–100 μg/mL concentration window.
The difference in concentration obtained from the standard curves and the average standard curves.
6′-SLN, 6′-sialyllactosamine; DFLNH, difucosyllacto-N-hexaose; IFLNH, isomeric fucosylated lacto-N-hexaose; LDFT, lacto-difucotetraose; LNDFH, lacto-N-difucohexaose; MFLNH, monofucosyllacto-N-hexaose; TFLNH, trifucosyllacto-N-hexaose.
This equation was then used to quantify the total HMO content of a pooled HMO sample. The pooled HMO sample was collected and extracted from milks of several mothers. Milligrams of extracted HMOs were then reduced, lyophilized, weighed using a microbalance, reconstituted in Nanopure water, and analyzed. Using this average linear regression equation, the total HMO content was determined to be 2.2 ± 0.1 (SE) mg/mL compared to the actual sample value of 2.0 mg/mL. The average response factor can therefore be used to obtain the total quantity of HMO in milk. The average response factor was evaluated for each individual compound. For the group of compounds that were used to obtain the average response factor, the difference of concentration using two different linear regression equations was within ±20% for most of the standards. For LNT, the compound was overestimated by 80%, whereas 3′-SL was underestimated by 70% as shown in Table 2. Nonetheless, using the average response factor produces sufficiently accurate result for each individual compound.
Validation of Matrix Effect with Standard Addition
To evaluate the matrix effect in this ESI LC–MS experiment, standard additions were performed by adding varying concentrations of pure HMO standards into a pooled HMO sample. Standard addition takes the matrix effect into account and may provide more accurate values compared to a standard curve. As illustrated in Table 1, the concentrations obtained using these two different methods were found to differ by only ±10% except for 3′-SL (−38%). Ion suppression and ion enhancement are the two major reasons for recovery rate higher and lower than 100%, respectively. Nevertheless, the result shows that the matrix effect is not a major source of error in our experiments.
Quantitation of HMO in Biological Samples
Absolute Quantitation of HMOs in Human Milk
To evaluate the variation in absolute HMO concentrations in a general population, the method was applied to milk samples collected from 20 mothers on day 35 postpartum. The absolute amounts of 2′-FL, LNT, LNFP-I, 3′-SL, 6′-SL, and LSTc in these 20 milk samples were determined (Table 2) using the standard calibration curves depicted in Supporting Information Figure S-3. Average concentrations of LNT and 2′-FL were 0.97 ± 0.56 and 1.5 ± 1.4 μg/mL, respectively, and are very close to previously reported values.43,44 The standard deviation reflects the biological diversity in these 20 samples. The LNFP-I, 3′-SL, 6′-SL, and LSTc concentrations measured by our LC–MS method were 20%–65% lower than that for HPAEC-PAD,44 well within biological variations. The LNFP-I concentration reported previously using single-quadrupole LC–MS using abundances and a calibration curve yielded a range of 0.001–1.80 mg/mL,43 which includes our value 0.26 ± 0.28 mg/mL (mean ± SD).
HMO Abundance and Secretor Status
It is known that the secretor status of the mother largely influences her HMO profile.36,44,45 Milk from secretor mothers contains a higher percentage of α(1–2)-fucose in compounds such as 2′-FL and LNFP-I as a consequence of the fucosyltransferase 2 (FUT2) gene in the epithelial cells. Indeed, we find the abundances of 2′-FL and LNFP-I are significantly higher in secretors (2′-FL, 3.0 mg/mL; LNFP-, 0.48 mg/mL) than in nonsecretors (2′-FL, 0.58 mg/mL; LNFP-I, 0.10 mg/mL) (Figure 4 and Table 2). We have previously suggested to use these compounds to phenotype the mother based on her milk HMO.36 The results depicted in Figure 4, parts a and b, however, show that a single compound may be insufficient to completely separate secretors from nonsecretors. For example, there are two nonsecretor mothers in Figure 4a (mothers D1036 and D1041) who produced 2′-FL abundances of the same magnitude as the secretor mothers; however, the fraction of all HMOs with α(1–2)-fucose is lower than those for secretor mothers. For other abundant oligosaccharides such as LNT, LNH, 3′-SL, 6′-SL, and LSTc, there appears no significant difference between the two phenotypes (Figure 4c–g).
Figure 4.
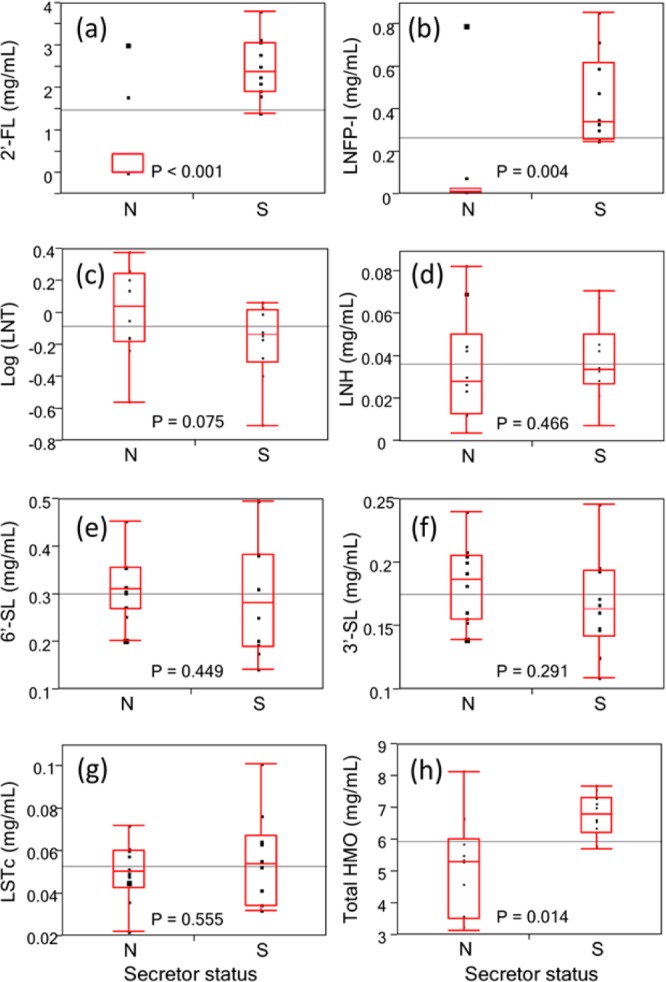
Box-and-whisker diagrams for secretor mothers (S) and nonsecretor mothers (N) reveal that nonsecretor mothers produce lower amounts of 2′-FL (P < 0.001) and LNFP-I (P = 0.004) than secretor mothers. However, there are some outliers who still produce similar amounts of 2′-FL or/and LNFP-I with secretor mothers, while their total α(1–2) fucosylation is still at relatively low level. The total HMO content produced by nonsecretor mothers is also lower (P = 0.014, Mann–Whitney–Wilcoxon test) than that of secretor mothers using the nonparametric test. (a) 2′-FL. (b) LNFP-I. (c) LNT. (d) LNH. (e) 6′-SL. (f) 3′-SL. (g) LSTc. (h) Total HMO content. Reported P-values are from one-tail Student t test unless specified.
The concentrations for the individual compounds, also the total HMO content, from the universal response factors are listed in Table 2. The standard deviations in this table represent the biological variation. The total amounts of HMOs were found to be higher in the secretor (6.8 ± 0.7 mg/mL) compared to the nonsecretor mothers (5.1 ± 1.6 mg/mL). The difference was statistically significant (Figure 4h, P = 0.014 from Mann–Whitney–Wilcoxon test) and consistent with a recent report on Gambian mothers.36 Furthermore, the biological variation in total HMO content among nonsecretor mothers is larger than among secretor mothers, which was also found for Gambian mothers.36
Conclusions
MRM is highly suited for the absolute quantitation of oligosaccharides, specifically human milk oligosaccharides. The limit of quantitation is approximately in the high-femtomole level with the quantitation range spanning 5 orders of magnitude. The small number of standards is typical in for oligosaccharides, because the syntheses of specific structures, even as small as those found in milk, remain a major task. This issue will not soon be solved. We therefore examined the use of a universal response factor based on the average of seven compounds. Accurate absolute quantitation, while desirable, is not often always needed. For many studies such as those for discovering markers of health or diseases states, sample sets are often matched and analyzed simultaneously. Relative quantitation is therefore generally sufficient. The advantage of absolute quantitation is that the value is generally independent of the method. MRM is attractive nonetheless because it provides both relative quantitation and, when standards are available, absolute quantitation. A general response factor for HMO may be helpful because it provides a good approximate value for a component even when standards are not available but provide values that can be directly compared for relative quantitation. A general response factor is also useful for obtaining accurate values for the total amounts of HMOs. This method will be of general use in oligosaccharide analyses but specifically in studying the bioactivity of HMOs.
Acknowledgments
Funding provided by the National Institutes of Health (AT007079, HD061923) is gratefully acknowledged.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bakhtiar R.; Lohne J.; Ramos L.; Khemani L.; Hayes M.; Tse F. J. Chromatogr., B 2002, 768, 325–340. [DOI] [PubMed] [Google Scholar]
- Li A. C.; Alton D.; Bryant M. S.; Shou W. Z. Rapid Commun. Mass Spectrom. 2005, 19, 1943–1950. [DOI] [PubMed] [Google Scholar]
- Ross A. R. S.; Ambrose S. J.; Cutler A. J.; Allan Feurtado J.; Kermode A. R.; Nelson K.; Zhou R.; Abrams S. R. Anal. Biochem. 2004, 329, 324–333. [DOI] [PubMed] [Google Scholar]
- Bennett B. D.; Kimball E. H.; Gao M.; Osterhout R.; Van Dien S. J.; Rabinowitz J. D. Nat. Chem. Biol. 2009, 5, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnidge D. R.; Goodmanson M. K.; Klee G. G.; Muddiman D. C. J. Proteome Res. 2004, 3, 644–652. [DOI] [PubMed] [Google Scholar]
- Pan S.; Aebersold R.; Chen R.; Rush J.; Goodlett D. R.; McIntosh M. W.; Zhang J.; Brentnall T. A. J. Proteome Res. 2008, 8, 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L.; Hunter C. L. Mol. Cell. Proteomics 2006, 5, 573–588. [DOI] [PubMed] [Google Scholar]
- Song E.; Pyreddy S.; Mechref Y. Rapid Commun. Mass Spectrom. 2012, 26, 1941–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda M.; Pompach P.; Brnakova Z.; Wu J.; Makambi K.; Goldman R. Mol. Cell. Proteomics 2013, 12, 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Q.; Lebrilla C. B.; Miyamoto S.; Ruhaak L. R. Anal. Chem. 2013, 85, 8585–8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viverge D.; Grimmonprez L.; Cassanas G.; Bardet L.; Bonnet H.; Solere M. Ann. Nutr. Metab. 1985, 29, 1–11. [DOI] [PubMed] [Google Scholar]
- Coppa G. V.; Pierani P.; Zampini L.; Carloni I.; Carlucci A.; Gabrielli O. Acta Paediatr. 1999, 88, 89–94. [DOI] [PubMed] [Google Scholar]
- Kunz C.; Rudloff S.; Baier W.; Klein N.; Strobel S. Annu. Rev. Nutr. 2000, 20, 699–722. [DOI] [PubMed] [Google Scholar]
- Gnoth M. J.; Kunz C.; Kinne-Saffran E.; Rudloff S. J. Nutr. 2000, 130, 3014–3020. [DOI] [PubMed] [Google Scholar]
- Coppa G. V.; Zampini L.; Galeazzi T.; Facinelli B.; Ferrante L.; Capretti R.; Orazio G. Pediatr. Res. 2006, 59, 377–382. [DOI] [PubMed] [Google Scholar]
- Jantscher-Krenn E.; Zherebtsov M.; Nissan C.; Goth K.; Guner Y. S.; Naidu N.; Choudhury B.; Grishin A. V.; Ford H. R.; Bode L. Gut 2012, 61, 1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcobal A.; Sonnenburg J. L. Clin. Microbiol. Infect. 2012, 18Suppl 412–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninonuevo M. R.; Park Y.; Yin H. F.; Zhang J. H.; Ward R. E.; Clowers B. H.; German J. B.; Freeman S. L.; Killeen K.; Grimm R.; Lebrilla C. B. J. Agric. Food Chem. 2006, 54, 7471–7480. [DOI] [PubMed] [Google Scholar]
- Wu S.; Tao N. N.; German J. B.; Grimm R.; Lebrilla C. B. J. Proteome Res. 2010, 9, 4138–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Grimm R.; German J. B.; Lebrilla C. B. J. Proteome Res. 2011, 10, 856–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobata A. Proc. Jpn. Acad., Ser. B 2010, 86, 731–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode L.; Jantscher-Krenn E. Adv. Nutr. 2012, 3, 383S–391S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoCascio R. G.; Ninonuevo M. R.; Freeman S. L.; Sela D. A.; Grimm R.; Lebrilla C. B.; Mills D. A.; German J. B. J. Agric. Food Chem. 2007, 55, 8914–8919. [DOI] [PubMed] [Google Scholar]
- Kunz C.; Rudloff S.; Hintelmann A.; Pohlentz G.; Egge H. J. Chromatogr. B: Biomed. Sci. Appl. 1996, 685, 211–221. [DOI] [PubMed] [Google Scholar]
- Hardy M. R.; Townsend R. R. Carbohydr. Res. 1989, 188, 1–7. [DOI] [PubMed] [Google Scholar]
- Townsend R. R.; Hardy M. R.; Cumming D. A.; Carver J. P.; Bendiak B. Anal. Biochem. 1989, 182, 1–8. [DOI] [PubMed] [Google Scholar]
- Townsend R. R.; Hardy M. R.; Lee Y. C. Methods Enzymol. 1989, 179, 65–76. [DOI] [PubMed] [Google Scholar]
- Sumiyoshi W.; Urashima T.; Nakamura T.; Arai I.; Saito T.; Tsumura N.; Wang B.; Brand-Miller J.; Watanabe Y.; Kimura K. Br. J. Nutr. 2003, 89, 61–69. [DOI] [PubMed] [Google Scholar]
- Asakuma S.; Urashima T.; Akahori M.; Obayashi H.; Nakamura T.; Kimura K.; Watanabe Y.; Arai I.; Sanai Y. Eur. J. Clin. Nutr. 2008, 62, 488–494. [DOI] [PubMed] [Google Scholar]
- Leo F.; Asakuma S.; Fukuda K.; Senda A.; Urashima T. Biosci., Biotechnol., Biochem. 2010, 74, 298–303. [DOI] [PubMed] [Google Scholar]
- Leo F.; Asakuma S.; Nakamura T.; Fukuda K.; Senda A.; Urashima T. J. Chromatogr., A 2009, 1216, 1520–1523. [DOI] [PubMed] [Google Scholar]
- Fong B.; Ma K.; Mcjarrow P. J. Agric. Food Chem. 2011, 59, 9788–9795. [DOI] [PubMed] [Google Scholar]
- Santos-Fandila A.; Zafra-Gómez A.; Vazquez E.; Navalón A.; Rueda R.; Ramírez M. Talanta 2014, 118, 137–146. [DOI] [PubMed] [Google Scholar]
- Tao N.; DePeters E. J.; German J. B.; Grimm R.; Lebrilla C. B. J. Dairy Sci. 2009, 92, 2991–3001. [DOI] [PubMed] [Google Scholar]
- Oriol R.; Le Pendu J.; Mollicone R. Vox Sang. 1986, 51, 161–171. [DOI] [PubMed] [Google Scholar]
- Totten S. M.; Zivkovic A. M.; Wu S.; Ngyuen U.; Freeman S. L.; Ruhaak L. R.; Darboe M. K.; German J. B.; Prentice A. M.; Lebrilla C. B. J. Proteome Res. 2012, 11, 6124–6133. [DOI] [PubMed] [Google Scholar]
- Xiao J. F.; Zhou B.; Ressom H. W. TrAC, Trends Anal. Chem. 2012, 32, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitteringham N. R.; Jenkins R. E.; Lane C. S.; Elliott V. L.; Park B. K. J. Chromatogr., B 2009, 877, 1229–1239. [DOI] [PubMed] [Google Scholar]
- Gallien S.; Duriez E.; Domon B. J. Mass Spectrom. 2011, 46, 298–312. [DOI] [PubMed] [Google Scholar]
- Kelly R. T.; Tolmachev A. V.; Page J. S.; Tang K.; Smith R. D. Mass Spectrom. Rev. 2010, 29, 294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer S. A.; Tang K.; Anderson G. A.; Prior D. C.; Udseth H. R.; Smith R. D. Rapid Commun. Mass Spectrom. 1997, 11, 1813–1817. [Google Scholar]
- Wheeler S. F.; Harvey D. J. Anal. Chem. 2000, 72, 5027–5039. [DOI] [PubMed] [Google Scholar]
- Bao Y.; Chen C.; Newburg D. S. Anal. Biochem. 2013, 433, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurl S.; Munzert M.; Henker J.; Boehm G.; Muller-Werner B.; Jelinek J.; Stahl B. Br. J. Nutr. 2010, 104, 1261–1271. [DOI] [PubMed] [Google Scholar]
- Thurl S.; Henker J.; Siegel M.; Tovar K.; Sawatzki G. Glycoconjugate J. 1997, 14, 795–799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.